

Abstract
Ang II/JAK/STAT3 signaling pathway is known to be involved in atrial remodeling associated with development and progression of atrial fibrillation. In the present study, we used in vivo animal model and human atrial specimens to further characterize the role of this pathway in the atrial structural remodeling. We observed an elevated level of Ang II in the atrial samples of AF patients. This increase in Ang II was accompanied by increased expression of collagens I and III, MMP1, MMP2 and elevated phosphorylation of STAT3. Using rat models, we demonstrated that Ang II infusion induced profound changes in the level of apoptosis, expression of collagen subtypes I and III, caspase-3, caspase-8, MMP1, MMP2 and redistribution of cytochrome C. The data further support the key role of Ang II in the development of AF and highlight the specific mechanisms and changes associated with this process.
Keywords: Atrial fibrillation, atrial structural remodeling, angiotensin II, STAT, Ang II/JAK/STAT3 pathway
Introduction
Atrial fibrillation (AF) is the most commonly encountered arrhythmia in clinical practice and one of the major causes of mortality [1]. Multiple studies revealed that AF is associated with functional and anatomical obstacles encountered by propagating atrial ectopic beat. These obstacles cause re-entry of the excitation wavefront [2]. “Leading circle” theory and cardiac electric rotors theory are two major schools of thoughts that attempt to explain the phenomena observed in AF [3].
Self-perpetuation of arrhythmia in persistent AF appears to be the result of atrial remodeling, the process that includes multiple structural and electrical changes in cardiomyocytes, such as alterations of ion channels, gap-junctions and extracellular matrix (ECM), and neurohumoral dysregulation, particularly in renin-angiotensin-aldosterone system (RAAS).
Structural remodeling is considered to be the main contributing mechanism and the hallmark in the progression of AF [4]. Changes in cellular and non-cellular components of the atrial tissue lead to heterogeneity in local conduction. Structural remodeling was extensively studied in the recent decade, with particular emphasis on the atrial fibrosis.
Atrial fibrosis occurs as a reparative process aimed at the replacement of the degenerating myocardial parenchima, Tissue fibrosis leads to the accumulation of deposits of fibrillar collagen [5] produced by atrial fibroblasts [6]. Volume and composition of ECM correlates with the persistence of AF [7]. Collagen produced in pathological conditions differs from that in normal myocardium and has altered ratio of subtypes [7,8]. ECM remodeling in atrium is accompanied by down-regulation of TIMP-2 and up-regulation of MMP-2 and CVF-I [7]. Disorganized collagen fibers physically separate myocytes and create barriers for impulse propagation [9].
Atrial fibrosis is a result of complex interaction between several profibrotic signaling pathways, inflammation and oxidative stress [10]. Atrial fibrosis is promoted by RAAS, with angiotensin II (Ang II) acting on AT-1 receptors and aldosterone acting on the mineralocorticoid receptors, and SMAD pathway, which mediates the stimulation of collagen production in response to TGF-β1 [6,11-14].
The molecular processes involved in structural remodeling are not fully understood at present time. Better understanding of these processes may provide the venues for therapeutic interventions and help in designing new strategies for preventing of structural remodeling underlying the AF development and progression.
Tsai and co-authors have demonstrated for the first time that atrial structural remodeling is mediated by Ang II/JAK/STAT3 pathway [15]. They found thatAng II activates STAT3 via Rac1 in both atrial myocytes and fibroblasts, and can be blocked by both losartan (AT-1 receptor antagonist) and simvastatin (HMG-CoA inhibitor). Rats subjected to log-term infusions of Ang II exhibited higher levels of activated Rac1, phospho-STAT3, collagen synthesis, and atrial fibrosis in the atria. The levels of Ang II and phospho-STAT3 were also elevated in human atrial tissues from patients with AF. The authors hypothesized that losartan might be useful in reversing the Ang II-induced structural remodelling in atrium. The analysis of the available data on the prevention of AF recurrence by the inhibitors of renin-angiotensin system do not support this assumption [16], thus emphasizing the need for further research into the mechanism of structural remodelling in AF.
In the present study, we used in vivo animal model and human atrial specimens to further characterize the status of Ang II-STAT signaling pathway in the atrial structural remodeling. We found that Ang II activated STAT3 in atria, induced apoptosis, collagen production and expression of matrix metalloproteinases in atria which may contribute to the structural changes in the atrium.
Materials and methods
Rat models and angiotensin II infusion
All experimental procedures were performed according to the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996) and were in compliance with the guidelines specified by the Chinese Heart Association policy on the use of research animals and the Public Health Service policy on the use of laboratory animals. Wistar rats (weight 300 ± 20 g) were randomly divided into 3 groups (n = 9 in each group): normal control group, Ang II group, Ang II + losartan group. Rats were subcutaneously infused with either 0.9% sodium chloride (control) or Ang II (2 mg•kg-1•d-1) or/and losartan (10 mg•kg-1•d-1) for 3 days, 7 days and 14 days. Upon finishing the Ang II treatment, the rats’ hearts were rapidly excised and retrogradely perfused in a Langendorff apparatus with HEPES-buffered Tyrode solution. The perfusate was oxygenated and maintained at 37 ± 0.2°C. After perfusion for 5 min, the atria were excised for subsequent studies.
Human atrial samples
Consecutive patients with valvular heart disease who underwent mitral or aortic valve replacement were recruited. Patients taking angiotensin converting enzyme inhibitors (ACEIs) and/or angiotensin II-receptor blockers (ARB) were excluded from the study. Overall, a total of 9 patients with chronic persistent atrial fibrillation (AF group) and 9 patients without the history of AF (sinus rhythm, SR group) were recruited. All patients underwent echocardiography before the surgery. Samples of right atrial appendages were obtained during the open heart surgery. The samples were quickly frozen in liquid nitrogen and maintained at -80°C until used for the mRNA and protein analysis. The study protocol was approved by the Ethics Committee of 5th Central Hospital of Tianjin, China. The research protocol was registered through the audit of Chinese Clinical Trial Registry (registration number:ChiCTR-CCC-14004220). The study subjects gave the informed consent.
Western blot
The extraction of cytosolic and cytoskeleton proteins was performed according to the manufacturer’s instructions (Chemicon Compartment Protein Extraction Kit, Millipore, MA, USA). Western blotting was performed according to the standard protocols. Low-molecular-weight marker (Cell Signaling Technology) and 50 µg of proteins from the samples were separated by 10% or 12% SDS-PAGE. Separated proteins were transferred to a PVDF (polyvinylidene difluoride) membrane that was blocked at room temperature for 1 hour in Tris-buffered saline (pH 7.4) with 0.2% Tween 20 (TBS-T) containing 5% skim milk and probed with primary antibodies overnight at 4°C. The diluted concentrations of the primary antibodies (Abcam, Cell Signaling Technology) were as follows: STAT3 (phospho Y705), 1:200; STAT3 (phospho S727), 1:250; STAT3, 1:200; caspase-3, 1:250; caspase-8, 1:250; cytochrome C, 1:200; β-actin, 1:500. Secondary antibodies (Cell Signaling Technology) labeled with horseradish peroxidase were diluted 1:1000 with 0.2% TBS-T and 1% skim milk and incubated for 1 hour at room temperature. Protein bands on Western blots were visualized using ECL Plus (Amersham, Arlington Heights, IL). Relative band densities of proteins in Western blots were normalized against β-actin.
Measurement of Ang II concentration by ELISA
Aliquots of 50 μl of sample or standard (Ang II dissolved in DMSO containing 0.01% Triton X-100-PBS, 1:1, v/v) per well were added. The plates were incubated for 90 min and then washed five times with PBST (phosphate-buffered saline with 0.2% Tween 20). Goat anti-rabbit immunoglobulin G conjugated to horseradish peroxidase (diluted 1:3,000 in PBST, 100 μl per well) was added, and the plates were incubated for 60 min at room temperature. After another washing step with PBST (five times), the substrate solution (TMB, 100 μl per well) was added. The blue color development was stopped by addition of 2 M sulfuric acid (50 μl per well) after 10 to 20 min, and absorbance was measured at 450 nm. Standard curves were obtained by plotting absorbance (A450) against the logarithm of Ang II concentration.
Immunohistochemistry staining for p-STAT3
Paraffin blocks were sectioned at 4 μm onto charged glass slides. The slides were baked, deparaffinized through two changes of xylene, and rehydrated through graded ethanol to water, then treated for 5 min in 3% hydrogen peroxide to block endogenous peroxidase activity. Slides were subjected to antigen retrieval by heating in Target Retrieval Solution, pH 6 (DAKO-Cytomation; Carpinteria, CA), followed by a rinse with distilled H2O and then Tris-buffered saline (TBS). The primary antibodies were diluted appropriately (1:200 for STAT3 (phospho Y705), 1:200 for STAT3 (phospho S727)) in blocking buffer and incubated overnight at 4°C. Following several TBS washes, sections were blocked with 2% NDS before treatment with biotin-conjugated donkey anti-rabbit antibody (5 μg/mL; Jackson ImmunoResearch) in PBS. After rinsing in PBS, sections were treated with Extravidin-peroxidase (0.4-0.5 μg/mL; Sigma, St Louis, MO, USA) and processed with diaminobenzidine tetrahydrochloride (DAB). Sections were then mounted on gelatin-coated slides, air-dried, cleared with xylene, and coversliped with DPX.
Picric acid sirius red staining
Sections were cut at 4 μm and mounted on glass slides. The sections were dewaxed, rehydrated and stained with Weigert’s haematoxylin for 10 min before washing in running tap water. Sections were then stained in Picrosirius Red solution (0.5 g Sirius red F3B in 500 ml saturated aqueous picric acid solution) for 1 h. Sections were washed in two changes of acidified water solution (5 ml acetic acid in 1 L of water). Finally, sections were dehydrated in three changes of 100% ethanol, cleared in xylene and mounted in a resinous medium. The slides were examined in polarized light. Picrosirius Red stains collagen red on a pale yellow background in bright field microscope, whereas, under a polarization microscope, collagen appears bright orange-red and/or bright green.
RNA extraction and real-time reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was prepared by using RNA isolation kit (Isogen) and treated with DNase. First-strand cDNA synthesis was performed according to the manufacturer’s protocol. PCR was carried out with 1.5 mM MgCl2, 1 mM dNTPs, 0.2 mM of each primer and 0.04 U/ml Ex Taq polymerase. The sequences of primers and expected size of amplification products are provided in Table 1. ΔΔCT method was used to determine the relative expression level of the target genes compared with the β-actin gene.
Table 1.
Primers used in RT-PCR
| Gene (size of amplicon) | Primers | |
|---|---|---|
| MMP-1 (297 bp) | forward | 5’ TCTGCCAGGTAAACTTGATGC 3’ |
| reverse | 5’ ATTCCAGGGAAATCTTCTGCT 3’ | |
| MMP-2 (243 bp) | forward | 5’ TGGAAGCATCAAATCGGACTG 3’ |
| reverse | 5’ GAAAGTAGCACCTGGGAGGGA 3’ | |
| Collagen I (175 bp) | forward | 5’ GGTCCCAAAGGTGCTGATGG 3’ |
| reverse | 5’ GACCAGGCTCACCACGGTCT 3’ | |
| Collagen III (336 bp) | forward | 5’ CGAGGTGACAGAGGTGAAAGA 3’ |
| reverse | 5’ AACCCAGTATTCTCCGCTCTT 3’ | |
| β-actin (320 bp) | forward | 5’ GCATCCATGAAACTACATTCA 3’ |
| reverse | 5’ ACAGTCCGCCTAGAAGCATTT 3’ |
TUNEL assay
Apoptosis in the atrial tissue was determined using an in situ apoptosis detection kit (Roche Diagnostics, Indianapolis, IN, USA). Briefly, sections were cut from paraffin blocks at 44-μm thickness and mounted onto slides. Sections were dewaxed in xylene and rehydrated through a graded series of ethanol. Sections were immersed in 0.1% TX-100 in 0.1% sodium citrate buffer for 8 min at room temperature. Following rinsing, sections were incubated with TUNEL buffer (containing 30 mM Tris, 0.7 M sodium cacodylate, CoCl2, 10% BSA in water) for 10 min at room temperature. Sections were then incubated with the reaction mixture as specified by the manufacturer’s instructions. Subsequently, tissue sections were incubated with anti-fluorescein antibody (labeled with alkaline phosphatase) in a humidified chamber for 1 h. Apoptotic cells were visualized with precipitating substrate Fast Red in 0.1 M Tris-HCl (pH 8.2) for 15 min at room temperature. Sections were counterstained with Lillie-Mayer’s haematoxylin and blued in lithium carbonate before being mounted in glycerol. Apoptotic cells were viewed by light microscopy.
Results
Ang II infusion activated STAT3 and induced atrial fibrosis and apoptosis
To verify the results obtained previously from cellular studies [15], we infused Ang II and investigated structural changes and the activation of STAT3 in the atrium using an in vivo rat model.
Infusion with Ang II increased the number of apoptotic cells in atria. This process was inhibited by losartan (Figure 1). Ang II infusion also improved the expression of apoptosis-related factors caspase-3 and caspase-8, and promoted the transfer of cytochrome C from mitochondria to cytoplasm in atria. These processes were also attenuated by losartan (Figure 2).
Figure 1.
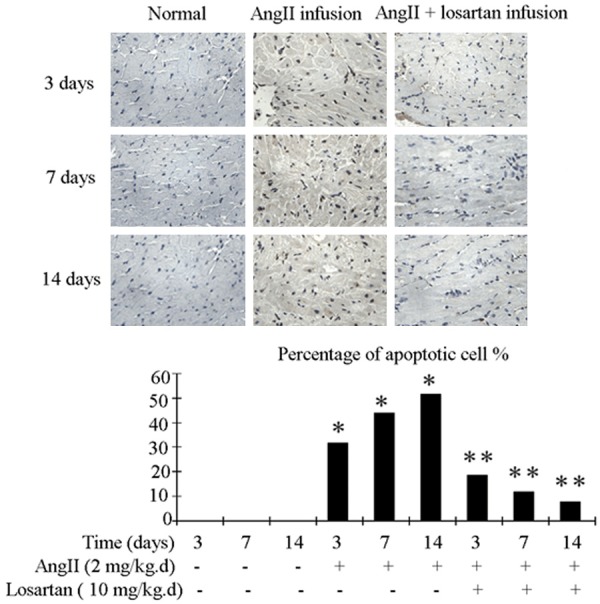
Rat atrial tissue apoptosis detection with the TUNEL procedure. Upper panel show representative photograph of TUNEL. Nucleiof normal cell were stained blue, while nuclei of apoptotic cell were stained brownish black. Bottom panel shows quantification by percentage. Ang II infusion group demonstrated higher level of apoptosis compared to the control group. This increase in apoptosis was attenuated in Ang II + losartan group. n = 3 animals per group; data are mean ± SD. *P < 0.05 vs control group, **P < 0.05 vs Ang II infusion group.
Figure 2.
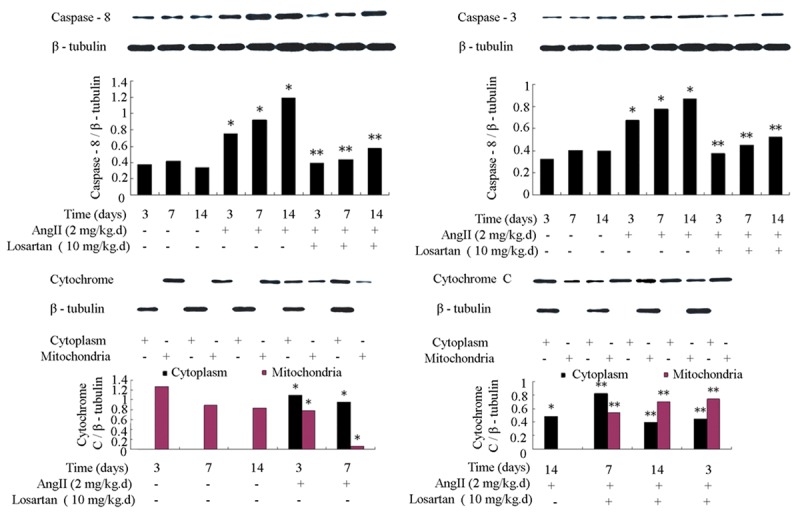
Rat atrial apoptosis-related factors caspase-3, caspase-8 and cytochrome C were measured by Western blot. Ang II infusion increased expression of caspase-3 and caspase-8, and promoted release of cytochrome C from the mitochondria to the cytoplasm in rat atria. These changes were attenuated by losartan infusion. n = 3 animals per group; data are mean ± SD. *P < 0.05 vs control group, **P < 0.05 vs Ang II infusion group.
Ang II infusion enhanced the production of collagen in atria, which was decreased by losartan (Figure 3). Infusion with Ang II also increased the levels of collagen I, collagen III and related metalloproteinases MMP1 and MMP2 in atria. These changes were also attenuated by losartan (Figure 4).
Figure 3.
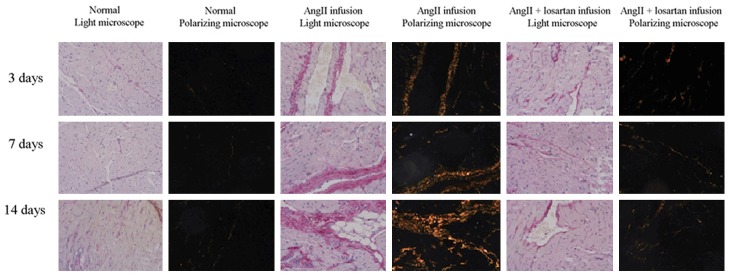
Synthesis of collagen in rat atria was evaluated by Picric acid Sirius red staining. Ang II infusion enhanced the production of collagen in rat atria. The effect was decreased by addition of losartan.
Figure 4.
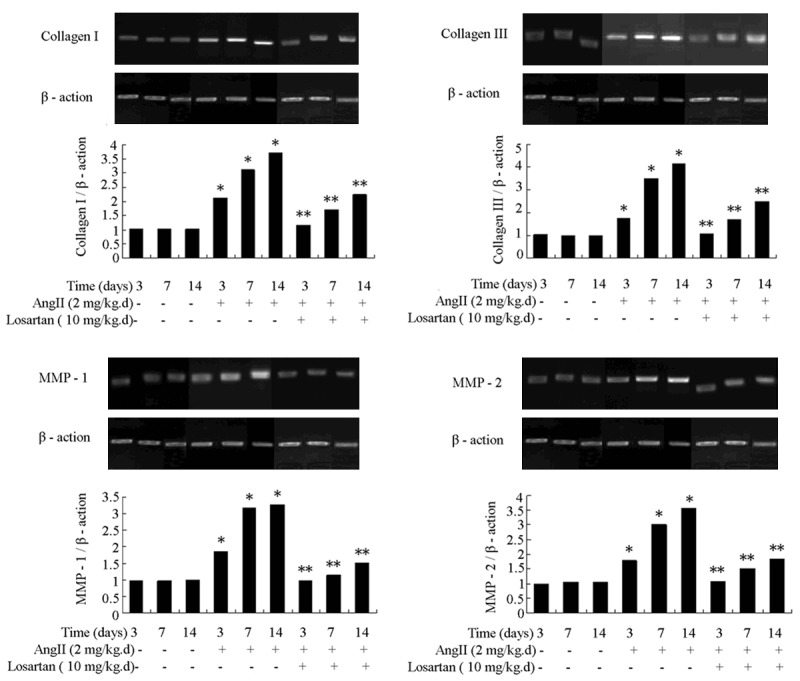
Rat atrial transcripts of collagen I, collagen III, MMP1 and MMP2 were measured by RT-PCR. Ang II infusion increased the rat atrial expression of collagen I, collagen III, MMP1 and MMP2. The level of expression of these genes was attenuated by losartan infusion. n = 3 animals per group; data are mean ± SD. *P < 0.05 vs control group, **P < 0.05 vs Ang II infusion group.
Ang II infusion improved the tyrosine 705 phosphorylation and serine 727 phosphorylation of STAT3. The phosphorylation of STAT3 was inhibited by losartan (Figure 5).
Figure 5.
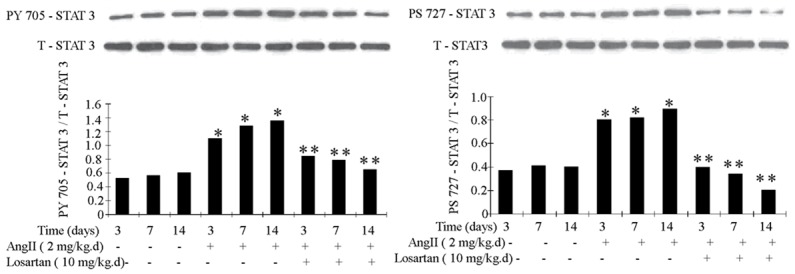
Phosphorylation of STAT3 in rat atria was measured by Western blot. Ang II infusion promoted the rat atrial phosphorylation of STAT3. The effect was attenuated by losartan infusion. n = 3 animals per group; data are mean ± SD. *P < 0.05 vs control group, **P < 0.05 vs Ang II infusion group.
Human atrial samples
Results from ELISA analysis indicated that Ang II concentration in atrial tissue is higher in AF patients than that in the sinus rhythmpatients. TUNEL staining revealed that the percentage of apoptotic cells in the atrial tissues of AF patients is higher than in the sinus rhythmpatients (Figure 6).
Figure 6.
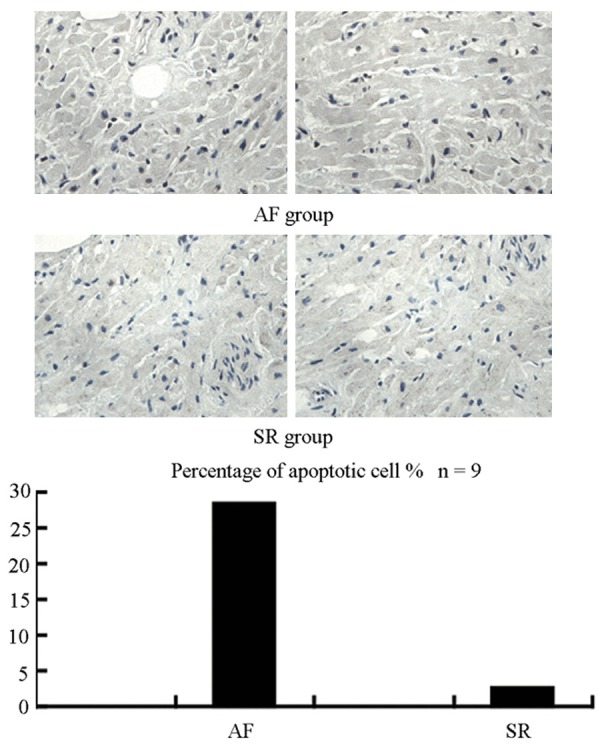
TUNEL staining showed level of apoptosis in the atria of AF patients was much higher than in SR patients. Upper panel shows representative photograph of TUNEL. Nucleusof normal cell was stained blue, while nucleus of apoptotic cell was stained brownish black. Bottom panel shows quantification by percentage.
Picric acid Sirius red staining results show that there is more collagen synthesis in the atrial tissues of AF patients compared to the sinus rhythmpatients (Figure 7).
Figure 7.
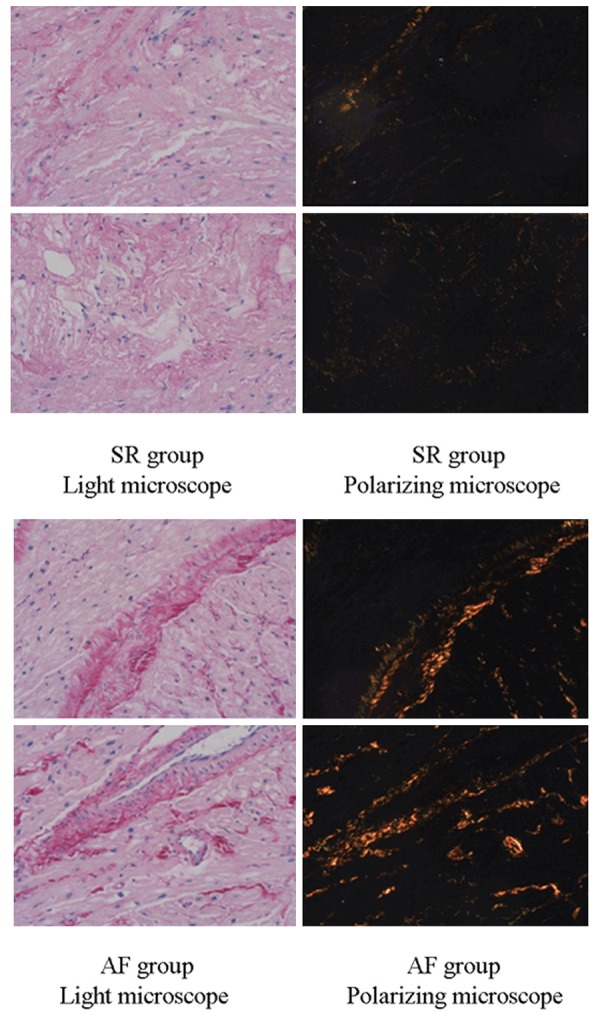
Synthesis of collagen in human atria was evaluated by Picric acid Sirius red staining. Composition of collagen in the atria of AF patients was higher than that in SR patients.
Expressions of collagen I, collagen III, MMP1 and MMP2 were higher in the atrial tissues of AF patients than those of the sinus rhythmpatients (Figure 8).
Figure 8.
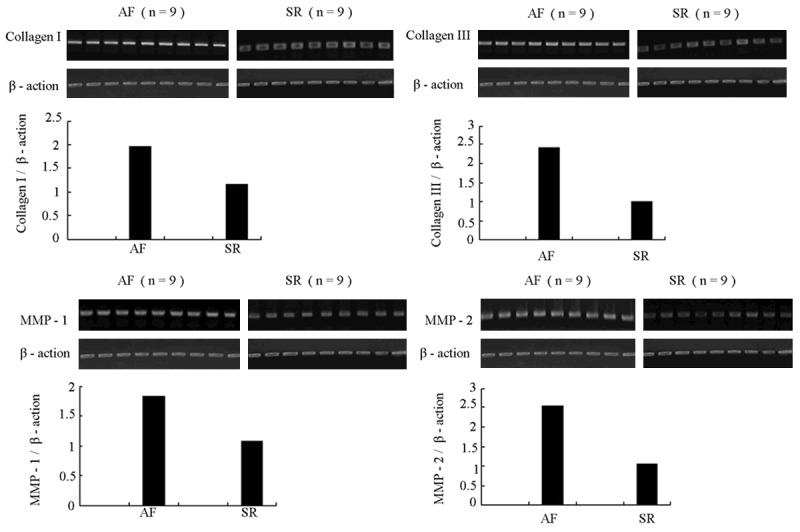
Results of RT-PCR showed that the expression levelsof collagen I, collagen III, MMP1 and MMP2 in atria of AF patients were higher than those in SR patients. Upper panel: representative RT-PCR. Bottom panel: quantification by densitometry.
Using immunohistochemistry staining, we found that the level of phosphorylated STAT3 protein in patients with AF is much higher than in those without AF (Figure 9).
Figure 9.
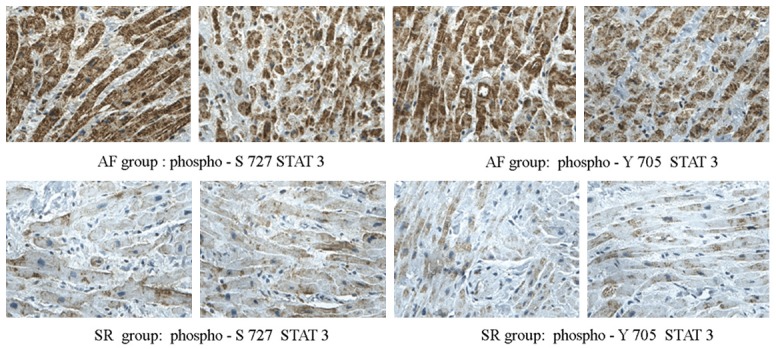
Expression of phosphorylated STAT3 was assayed by immunohistochemistry staining. There was more phosphorylated STAT3 in atrial sample of AF patients than SR patients.
Discussion
In the present study, by using in vivo rat models and human samples we have demonstrated that STAT3 played an important role in the regulation of Ang II-induced atrial structural changes, which were attenuated by losartan. These findings correspond well with previous observation and provide further insight into the influence of Ang II on particular cellular processes during atrial remodeling [15].
Fibrosis and apoptosis are two important features of structural remodeling [17-19], which occur with alterations in connexin expression. Structural remodeling delays conduction velocity (CV) by disrupting intermyocyte coupling [1,20]. In the human sample assay, we observed more significant structural changes in the AF patients compared to SR patients. We found that interstitial fibrosis (or collagen deposition) increased to different extents in AF patients. This result was consistent with previous studies [7,15]. Meanwhile, TUNEL assays of human specimens also revealed more apoptotic alterations in the atria of AF patients. This apoptosis-related loss of cells and fibrotic replacement increased the space between cardiomyocytes, caused conduction delays between the cells and allowed alternate pathways of conduction. In addition to the increase of apoptosis and fibrosis in atria, a significant expression of collagen I, collagen III, MMP1 and MMP2 were also observed in the human atria samples in our study. Production of ECM proteins is mainly carried out by fibroblasts, while their degradation occurs primarily under the action of matrix metalloproteinases (MMPs). Specific patterns of MMP induction in patients with AF have previously been reported [21-23]. Generally, MMPs contribute to matrix turnover and structural remodeling by breaking down the physiological collagens that are eventually replaced by fibrous interstitial deposits of various unorganized ECM proteins [24]. Enhanced synthesis and deposition of ECM proteins can occur either as the replacement fibrosis relating to the cell apoptosis, or as the reactive fibrosis relating to the degradation of physiological collagens. In particular, interstitial reactive fibrosis has been identified to be highly pro-arrhythmic in the atrial myocardium [25]. It also alters the biophysical properties of the atrial tissue which allows the initiation and perpetuation of AF. Specific subtypes of collagen are linked to the development and recurrence of AF and were recently suggested as prognostic biomarkers in the management of this condition [26,27].
In the present study, we observed a much higher level of Ang II in the atrial samples of AF patients than in the patients without AF. In rat model study, we demonstrated that Ang II infusion induced profound increases of collagen synthesis and apoptosis in the atria, strongly suggesting that Ang II advances the progress of atrial fibrosis and apoptosis.
In our study, Ang II infusion in vivo significantly improved the phosphorylation of STAT3, which demonstrated the activation of STAT3 by Ang II. Ang II also increased the level of atrial apoptosis in in vivo trials, which was inhibited by AT1 receptor antagonist losartan. Our data demonstrate that observed changes in the level of apoptosis, expression of collagen subtypes I and III, caspase-3, caspase-8, MMP1, MMP2 and redistribution of cytochrome C are specifically triggered by the infusion of Ang II, thus confirming the direct link between these AF-associated changes in atria and Ang II level. These data once again confirm the key role of Ang II in the development of AF and highlight the specific mechanisms and changes associated with this process.
Acknowledgements
This work was supported by the grants No. 2011BHKZ005 from the key support projects of Tianjin Binhai New Area of Medical Science and Technology projects.
Disclosure of conflict of interest
None.
References
- 1.Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. 2008;51:802–809. doi: 10.1016/j.jacc.2007.09.064. [DOI] [PubMed] [Google Scholar]
- 2.Muntean DM, Kohajda Z, Fazekas T, Jost N. Atrial Remodeling in Permanent Atrial Fibrillation: Mechanisms and Pharmacological Implications. J Clin Exp Cardiolog. 2013;4:11. [Google Scholar]
- 3.Pandit SV, Jalife J. Rotors and the dynamics of cardiac fibrillation. Circ Res. 2013;112:849–862. doi: 10.1161/CIRCRESAHA.111.300158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schotten U, Neuberger HR, Allessie MA. The role of atrial dilatation in the domestication of atrial fibrillation. Prog Biophys Mol Biol. 2003;82:151–162. doi: 10.1016/s0079-6107(03)00012-9. [DOI] [PubMed] [Google Scholar]
- 5.Frustaci A, Chimenti C, Bellocci F, Morgante E, Russo MA, Maseri A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation. 1997;96:1180–1184. doi: 10.1161/01.cir.96.4.1180. [DOI] [PubMed] [Google Scholar]
- 6.Tsai CT, Tseng CD, Hwang JJ, Wu CK, Yu CC, Wang YC, Chen WP, Lai LP, Chiang FT, Lin JL. Tachycardia of atrial myocytes induces collagen expression in atrial fibroblasts through transforming growth factor β1. Cardiovasc Res. 2011;89:805–815. doi: 10.1093/cvr/cvq322. [DOI] [PubMed] [Google Scholar]
- 7.Xu J, Cui G, Esmailian F, Plun-kett M, Marelli D, Ardehali A, Odim J, Laks H, Sen L. Atrial extracellular matrix remodeling and the maintenance of atrial fibrillation. Circulation. 2004;109:363–368. doi: 10.1161/01.CIR.0000109495.02213.52. [DOI] [PubMed] [Google Scholar]
- 8.Grammer JB, Bohm J, Dufour A, Benz M, Lange R, Bauernschmitt R. Atrial fibrosis in heart surgery patients decreased collagen III/I ratio in postoperative atrial fibrillation. Basic Res Cardiol. 2005;100:288–294. doi: 10.1007/s00395-005-0515-x. [DOI] [PubMed] [Google Scholar]
- 9.Rossi MA. Pathologic fibrosis and connective tissue matrix in left ventricular hypertrophy due to chronic arterial hypertension in humans. J Hypertens. 1998;16:1031–1041. doi: 10.1097/00004872-199816070-00018. [DOI] [PubMed] [Google Scholar]
- 10.Tan AY, Zimetbaum P. Atrial fibrillation and atrial fibrosis. J Cardiovasc Pharmacol. 2011;57:625–629. doi: 10.1097/FJC.0b013e3182073c78. [DOI] [PubMed] [Google Scholar]
- 11.Evans RA, Tian YC, Steadman R, Phillips AO. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of SMAD proteins. Exp Cell Res. 2003;282:90–100. doi: 10.1016/s0014-4827(02)00015-0. [DOI] [PubMed] [Google Scholar]
- 12.Chen K, Mehta JL, Li D, Joseph L, Joseph J. Transforming growth factor beta receptor endoglin is expressed in cardiac fibroblasts and modulates profibrogenic actions of angiotensin II. Circ Res. 2004;95:1167–1173. doi: 10.1161/01.RES.0000150369.68826.2f. [DOI] [PubMed] [Google Scholar]
- 13.Tokuda K, Kai H, Kuwahara F, Yasukawa H, Tahara N, Kudo H, Takemiya K, Koga M, Yamamoto T, Imaizumi T. Pressure-independent effects of angiotensin II on hypertensive myocardial fibrosis. Hypertension. 2004;43:499–503. doi: 10.1161/01.HYP.0000111831.50834.93. [DOI] [PubMed] [Google Scholar]
- 14.Aharinejad S, Krenn K, Paulus P, Schäfer R, Zuckermann A, Grimm M, Abraham D. Differential role of TGFbeta1/bFGF and ET-1 in graft fibrosis in heart failure patients. Am J Transplant. 2005;5:2185–2192. doi: 10.1111/j.1600-6143.2005.01006.x. [DOI] [PubMed] [Google Scholar]
- 15.Tsai CT, Lai LP, Kuo KT, Hwang JJ, Hsieh CS, Hsu KL, Tseng CD, Tseng YZ, Chiang FT, Lin JL. Angiotensin II activates signal transducer and activators of transcription 3 via Rac1 in atrial myocytes and fibroblasts: implication for the therapeutic effect of statin in atrial structural remodeling. Circulation. 2008;117:344–355. doi: 10.1161/CIRCULATIONAHA.107.695346. [DOI] [PubMed] [Google Scholar]
- 16.Disertori M, Barlera S, Staszewsky L, Latini R, Quintarelli S, Franzosi MG. Systematic review and meta-analysis: renin-Angiotensin system inhibitors in the prevention of atrial fibrillation recurrences: an unfulfilled hope. Cardiovasc Drugs Ther. 2012;26:47–54. doi: 10.1007/s10557-011-6346-0. [DOI] [PubMed] [Google Scholar]
- 17.Aimé-Sempé C, Folliguet T, Rücker-Martin C, Krajewska M, Krajewska S, Heimburger M, Aubier M, Mercadier JJ, Reed JC, Hatem SN. Myocardial cell death in fibrillation and dilated human right atria. J Am Coll Cardiol. 1999;34:1577–86. doi: 10.1016/s0735-1097(99)00382-4. [DOI] [PubMed] [Google Scholar]
- 18.Goette A, Juenemann G, Peters B, Klein HU, Roessner A, Huth C, Röcken C. Determinants and consequences of atrial fibrosis in patients undergoing open heart surgery. Cardiovasc Res. 2002;54:390–396. doi: 10.1016/s0008-6363(02)00251-1. [DOI] [PubMed] [Google Scholar]
- 19.Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ Arrhythm Electrophysiol. 2008;1:62–73. doi: 10.1161/CIRCEP.107.754564. [DOI] [PubMed] [Google Scholar]
- 20.Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87:425–456. doi: 10.1152/physrev.00014.2006. [DOI] [PubMed] [Google Scholar]
- 21.Nakano Y, Niida S, Dote K, Takenaka S, Hirao H, Miura F, Ishida M, Shingu T, Sueda T, Yoshizumi M, Chayama K. Matrix metalloproteinase-9 contributes to human atrial remodeling during atrial fibrillation. J Am Coll Cardiol. 2004;43:818–825. doi: 10.1016/j.jacc.2003.08.060. [DOI] [PubMed] [Google Scholar]
- 22.Anné W, Willems R, Roskams T, Sergeant P, Herijgers P, Holemans P, Ector H, Heidbüchel H. Matrix metalloproteinases and atrial remodeling in patients with mitral valve disease and atrial fibrillation. Cardiovasc Res. 2005;67:655–666. doi: 10.1016/j.cardiores.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 23.Mukherjee R, Herron AR, Lowry AS, Stroud RE, Stroud MR, Wharton JM, Ikonomidis JS, Crumbley AJ 3rd, Spinale FG, Gold MR. Selective induction of matrix metalloproteinases and tissue inhibitor of metalloproteinases in atrial and ventricular myocardium in patients with atrial fibrillation. Am J Cardiol. 2006;97:532–537. doi: 10.1016/j.amjcard.2005.08.073. [DOI] [PubMed] [Google Scholar]
- 24.Spinale FG. Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ Res. 2002;90:520–530. doi: 10.1161/01.res.0000013290.12884.a3. [DOI] [PubMed] [Google Scholar]
- 25.de Jong S, van Veen TA, van Rijen HV, de Bakker JM. Fibrosis and cardiac arrhythmias. J Cardiovasc Pharmacol. 2011;57:630–638. doi: 10.1097/FJC.0b013e318207a35f. [DOI] [PubMed] [Google Scholar]
- 26.Kallergis EM, Goudis CA, Kanoupakis EM, Mavrakis HE, Maliaraki NE, Tzanakis N, Vardas PE. Sinus rhythm restoration affects collagen turnover in patients with persistent atrial fibrillation. Europace. 2014;16:1726–1730. doi: 10.1093/europace/eut401. [DOI] [PubMed] [Google Scholar]
- 27.Löfsjögård J, Persson H, Díez J, López B, González A, Edner M, Mejhert M, Kahan T. Atrial fibrillation and biomarkers of myocardial fibrosis in heart failure. Scand Cardiovasc J. 2014;48:299–303. doi: 10.3109/14017431.2014.940063. [DOI] [PubMed] [Google Scholar]