

Abstract
Aim: This study aims to investigate the localization and expression of protein kinase C-beta I and beta II in kidney cortex of diabetic nephropathy mice and their roles in telmisartan treatment. Methods: 18 mice were randomly divided into three groups: normal group, diabetic nephropathy group and telmisartan-treated group. The localization and expression of protein kinase C-beta I and beta II were measured with confocal immunofluorescence laser scanning microscopy, immunohistochemistry and western blotting. The expression of transforming growth factor-beta 1 and vascular endothelial growth factor in glomeruli was detected by immunohistochemistry. Results: Compared to the normal mice, the expression and localization of protein kinase C-beta I and beta II are differed in diabetic nephropathy mice, with increased expression of protein kinase C-beta I but decreased level of protein kinase C-beta II. Meanwhile, the expression of transforming growth factor-beta 1 and vascular endothelial growth factor showed increase in the glomeruli of diabetic nephropathy, compared to the controls. Also, protein kinase C-beta I exhibited a positive correlation to transforming growth factor-beta 1 (r = 0.649, P = 0.030), but no correlation to vascular endothelial growth factor (r = 0.387, P = 0.079). Telmisartan treatment exercised significant beneficial role in diabetic nephropathy, which is associated with protein kinase C-beta I, but not beta II. Conclusions: The expression and localization of protein kinase C-beta I and beta II differ in the diabetic nephropathy, and such difference is associated with the pathogeneses of diabetic nephropathy.
Keywords: Diabetic nephropathy, protein kinase C, telmisartan, transforming growth factor-beta1, vascular endothelial growth factor
Introduction
Diabetic nephropathy (DN), characterized by the accumulation of extracellular matrix protein in the mesangial space with mesangial expansion, thickening of the glomerular and tubular basement membranes, and tubulointerstitial fibrosis [1,2], is the leading cause of end-stage kidney failure in many countries, like United States [3]. The exact pathogenesis of diabetic nephropathy is unknown; however, hyperglycemia has been regarded as a promoter to accelerate the development of clinical nephropathy [3,4]. Hyperglycemia drives the production or expression of many factors associated with the development of DN, including angiotensin II (Ang II), TGF-β, connective tissue growth factor (CTGF) and vascular endothelial growth factor (VEGF) [5]. Hyperglycemia alone, or with others like AngII [6], promotes the activation of protein kinase C (PKC), which phosphorylates serine or threonine residues of various intracellular proteins and thus is involved in a wide range of cellular functions that may be relevant to the pathophysiology of diabetic nephropathy [7,8]. PKCs have been classified into three groups: group 1 (the conventional PKCs), including α, βI, βII, as well as γ; group 2 (novel PKCs), containing δ, ε, η, θ, and μ; group 3 (atypical PKCs) composing ζ, ι, and λ [8-10]. Using single isoform-specific knockout mice, the functional role of distinct PKC isoforms in the development of diabetic nephropathy has been uncovered [7,11-14]. It is well regarded that PKC-α-dependent signaling pathway regulates perlecan and VEGF as well as nephrin expression, leading to diabetic albuminuria [7,11]. The activation of the PKC-β isoforms contributes to high-glucose-induced, TGF-β1-mediated renal hypertrophy and extracellular matrix expansion [7,15]. The inhibition of PKC-β signaling protects against diabetic nephropathy [7,16,17]. Although PKC-β isoforms, including PKC-β1 and PKC-β2, plays critical role in the pathogenesis of diabetic nephropathy [17-19], we are missing the comprehensive knowledge about the localization and expression of PKC-β isoforms in the kidney of diabetic nephropathy.
Thus, this study aims to investigate the expression and localization of PKC-β isoforms in diabetic nephropathy mouse. This study also explores the relation between PKC-β isoforms and TGF-β1, VEGF, as well as renin-angiotensin system.
Materials and methods
Animals
8-week-old male C57BL/6 mice were obtained from Huazhong University of Science and Technology (Wuhan, China). Mice were individually housed in cages and acclimated for two weeks in animal facility conditions (22±1°C and 50±1% humidity with a 12 h in the light/dark). Diabetes was induced with intraperitoneal (i.p.) injection of 100 mg/kg streptozotocin (STZ, Sigma-Aldrich Co., St Louis, MO, USA) in saline. As nondiabetic control, mice were injected with same dosages of saline in the same manner as the diabetic mice (group N, n = 6). One week later, the induction of diabetes was confirmed by measuring fasting blood glucose levels. Fasting blood glucose levels from the mouse tail vein were measured by using a one-touch blood glucose meter (LifeScan Inc., Milpitas, USA). Only those mice with fasting blood glucose above 16.7 mmol/L were considered as diabetic mice. Mice care and experiments were conducted in accordance with the guidelines of Huazhong University of Science and Technology.
Experimental design
The diabetic mice received telmisartan (9 mg·kg-1·d-1, Boehringer Ingelheim, group T, n = 6) or vehicle (group DN, n = 6) by gastric tube. During the experiment, the mice had free access to standard mouse chow and tap water and received no insulin and other treatments. One mouse from DN group died on day 21, while one from T group died on day 25 after the last administration of STZ. 24 h urine samples were collected from the mouse on day 41 in metabolic cages. Forty-two days after the application of STZ or vehicle, mice were sacrificed to collect clinical data with the following experiments were performed, such as blood glucose levels, body weight and the weight of right kidney. Meanwhile, the left kidneys were collected for further analysis.
Urine albumin analysis
Before sacrifice, each mouse was housed in individual metabolic cages (KAT-DXL-S; Shanghai TongYu Educational Instrument Manufacture CO., Ltd., China) with free access to water and rodent mash. The 24-h urine was collected, centrifuged at 8,000 g for 5 min (to remove debris), and flash-frozen for further analysis. An indirect competitive ELISA kit (Shanghai Shanghai YuBo Biotech Co., Ltd., China) was used according to the manufacturer’s instruction to measure urinary albumin concentration.
Histological studies
Kidneys from the normal and experimental animals were fixed in 10% buffered formalin and were processed for paraffin sectioning. Sections of about 5 μm thickness were stained with hematoxylin and eosin to evaluate the pathophysiological changes under light microscope. The image analysis software (Image-pro plus, Version 5.1.0.20, Media Cybernetics, Inc) was used to analyze the clinical data, like average diameter of glomeruli, in ten different visual fields. The double-blind experiment was carried out in the histological examination, and the person who evaluated the morphologic changes was blinded to the researcher who performed the treatment.
Immunofluorescent staining
To fix the kidney for immunofluorescent staining, the kidneys were first perfused with the PBS to flush out the blood and then freshly prepared 4% paraformaldyhide in PBS for 10 min. The kidneys were excised and immersed in the same fixative for 2 h at 4°C. The kidneys were then rinsed with PBS and cryoprotected in 30% sucrose in PBS at 4°C for 24 h. Sections of 8-μm thick were cut with a cryostat at -25°C, mounted on glass slides, and immediately processed for immunofluorescent localization of PKC-βI and PKC-βII expression. Briefly, sections were rinsed 5 min with PBS, incubated in washing buffer (PBS containing 50 mM NH4Cl) for 10 min, and in blocking buffer (washing buffer with 2% BSA and 0.05% Saponin) for 20 min. Sections were incubated with polyclonal antibodies developed in rabbits against PKC-βI (3 μg/ml, Santa Cruz Biotechnology, Dallas, Texas, U.S.A.; cat. no. sc-209), and PKC-βII (3 μg/ml, Santa Cruz Biotechnology, Dallas, Texas, U.S.A.; cat. no. sc-210), goats against Tamm-Horafall (2 μg/ml, Santa Cruz Biotechnology, Dallas, Texas, U.S.A.; cat. no. sc-19554), and goats against aquaporin-2 (2 μg/ml, Santa Cruz Biotechnology, Dallas, Texas, U.S.A.; cat. no. sc-28629), respectively. After 4 × 5 min washing, sections were incubated in Cy3 labeled goat anti-rabbit IgG (1:50; EarthOxLLC., U.S.A.; cat. no. E031610) and FITC-conjugated phalloidin (0.5 μg/ml, Alexis, Switzerland; cat. no. ALX-350-265), or FITC labeled donkey anti-rabbit IgG (1:50; EarthOxLLC., U.S.A.; cat. no. E031220) and Texas Red labeled donkey anti-goat IgG (1:50; EarthOxLLC., U.S.A.; cat. no.E031330) for 2 h at room temperature. Phalloidin is the maker of Glomerular podocytes and epithelial cells in the proximal tubule, while Tamm-Horsfall is the maker of the ascending thick limb of Henle’s loop, and aquaporin-2 is the maker of the collecting duct. Sections were imaged with confocal laser-scanning microscopy (Olympus FV500, Tokyo, Japan).
For expression of PKC-βI, PKC-βII, TGF-β1 and VEGF, sections were incubated with respective primary antibody or preimmune rabbit serum in the blocking buffer overnight at 4°C. Primary antibodies used in these experiments are polyclonal antibodies developed in rabbits against PKC-βI, PKC-βII, TGF-β1 (1:100, Boster, Wuhan, China; cat. no. BA0294), and VEGF (1:100, Peprotech, England ; cat. no. PeproTech 450-32), respectively. After 4 × 5 min washing, sections were incubated in Cy3 labeled goat anti-rabbit IgG for 2 h at room temperature. Sections were mounted with ProLong antifade medium and imaged with Olympus FV500 confocal microscope. Images were analyzed with the Image-Pro Plus version 5.0 System (Media Cybernetics Inc, Georgia, USA).
Immunoblotting
Western blot analysis was conducted according to a standard protocol [20]. Briefly, equal amounts of membrane proteins obtained from kidney cortex were separated by a reducing SDS-PAGE electrophoresis. The membrane proteins were transferred onto polyvinyli-dene difluoride membranes (Millipore) and blocked with 5% nonfat milk in Tris-Tween-buffered saline buffer (20 mM Tris, pH 7.5, 150 mM NaCl, and 0.1% Tween 20) for 3 h. The primary antibodies were incubated overnight at 4°C; the horseradish peroxidase-conjugated secondary antibodies were subsequently incubated for 1 h at 25°C before development of the blots using the Alpha Imager 2200 software (Alpha Innotech). We quantified the resultant signals and normalized the data to the abundance of actin.
Statistical methods
Data, expressed as means±SEMs, were analyzed statistically using SPSS 11.0 software (SPSS Inc Chicago, USA). Data were analyzed by One-Way ANOVA method. Correlation analysis was conducted using Pearson method. Probability values < 0.05 were taken to indicate statistical significance.
Results
Diabetic renal injury
Compared to the NG group, 24-hour urinary protein and blood glucose levels were significant (P < 0.05) higher in DN group (Table 1). Meanwhile, gross morphological assessment of the kidneys revealed the increased glomerular cell number, the renal weight as well as the glomerular size in DN group, compared those in NG group (P < 0.05) (Tables 1 and 2). However, telmisartan treatment significantly (P < 0.05) alleviated these clinical parameters, except the blood glucose levels (Tables 1 and 2).
Table 1.
Blood glucose, 24 hour urine protein and glomerular cells number
| Group | Blood glucose (mmol/L) | 24 hour urine protein (mg) | glomerular cells number (n/glomeruli) |
|---|---|---|---|
| NG | 7.10±0.68 | 0.32±0.03 | 32.11±3.11 |
| DN | 33.33±0** | 5.52±0.49** | 54.45±4.16** |
| T | 31.97±3.44** | 4.98±0.28**,▲▲ | 44.97±4.02**,▲▲ |
Values are Means±SEM, n = 6.
Different from NG, P < 0.01;
Different from DN P < 0.01.
Table 2.
Clinical parameters in different groups
| G | A (× 10-2) (mg) | B (× 10-1) (g) | C (× 102) | D (× 10-2) (μm2) | E (× 10-1) (μm) |
|---|---|---|---|---|---|
| NG | 1.23±0.11 | 2.46±0.28 | 0.51±0.057 | 96.05±5.34 | 34.70±0.97 |
| DN | 1.55±0.14** | 1.76±0.26** | 0.90±0.112** | 158.35±12.91** | 44.57±1.81** |
| T | 1.37±0.03*,▲ | 2.11±0.21*,▲ | 0.65±0.057*,▲▲ | 125.60±8.75**,▲▲ | 39.70±1.38**,▲▲ |
A: the weight of right kidney; B: the body weight; C: the right kidney index (A/B); D: glomerular area; E: glomerular perimeter. Values are Means±SEM, n = 6.
Different from NG, P < 0.01;
Different from NG, P < 0.05;
Different from DN P < 0.01;
Different from DN P < 0.05.
Microscopic analysis also shown higher (P < 0.05) glomerular size, glomerular mesangial region and glomerular cell number in DN group than those in the NG group, while mice in T group had lower (P < 0.05) microscopic lesion in the kidneys, compared to the DN group (Figure 1). Summarily, mice in DN group show significant diabetic renal injury, while telmisartan treatment alleviates the diabetic renal injury.
Figure 1.
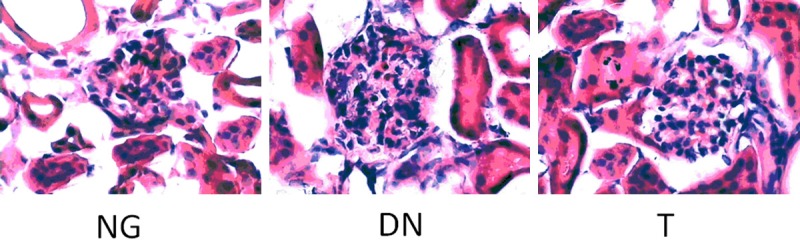
Microscopic observation (*400).
Localization of PKC-βI
PKC-βI in NG group expressed in podocytes, cortical thick ascending limbs of Henle’s loop, as well as in cortical collecting duct and interstitial cells. It also had a weaker expression in epithelial cells in the proximal tubule in NG group (Figure 2A). Like the NG group, PKC-βI also expressed in podocytes, cortical thick ascending limbs of Henle’s loop, as well as cortical collecting duct in DN and T group (Figure 2B). However, a mark expression of PKC-βI was found in epithelial cells in the proximal tubule in DN and T group (Figure 2C).
Figure 2.
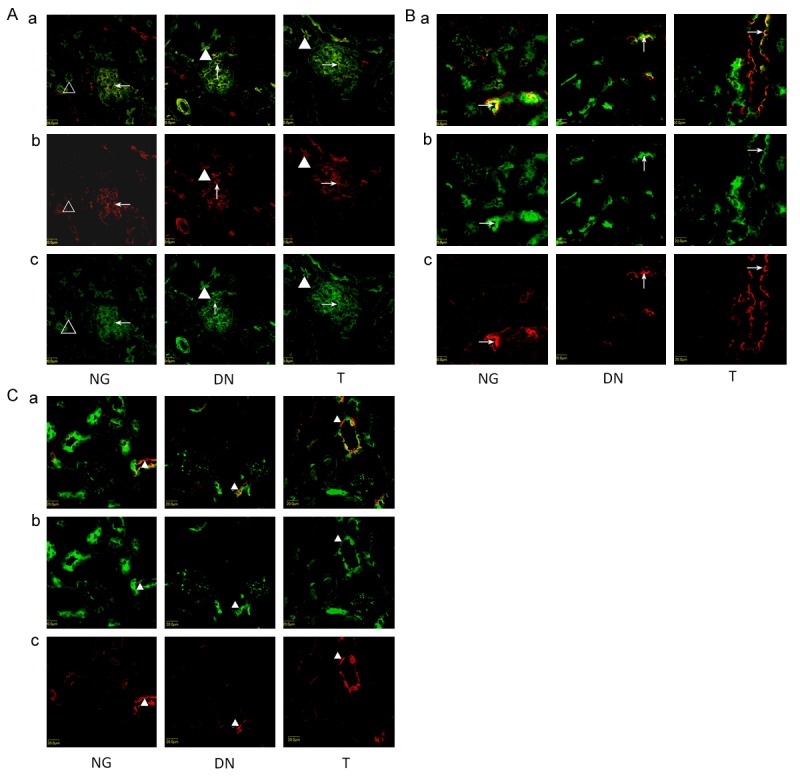
A. Localization of PKC-βI in glomerulus and proximal tubule. a shows the overlay of Phalloidin staining (green) and PKC-βI antibody staining (red). Kidneys were immunostained with PKC-βI antibody (red) in panel b showing the localization of PKC-βI. In panel c, kidneys were stained with the Phalloidin (green). Left column: PKC-βI was detected in podocytes (arrow) and suspicious expression in proximal tubule (Triangle ) in NG. Middle column: 6 weeks after STZ induction, enhanced accumulation of PKC-βI in podocytes (arrow)and proximal tubule brush border (Triangle) were observed. Right column: Telmisartan treatment attenuated enhanced accumulation of DN mice. B. Localization of PKC-βI in cortical thick ascending limbs of Henle’s loopon three groups (arrow). a shows the overlay of Tamm-Horsfall antibody staining (red) and PKC-βI antibody staining (green). Kidneys were immunostained with PKC-βI antibody (green) in panel b showing the localization of PKC-βI. In panel c, kidneys were stained with the Tamm-Horsfall antibody staining (red). C. Localization of PKC-βI in cortical collecting duct on three groups (Triangle). a shows the overlay of AQP2 antibody staining (red) and PKC-Βi antibody staining (green). Kidneys were immunostained with PKC-βI antibody (green) in panel b showing the localization of PKC-βI. In panel c, kidneys were stained with the aquaporin-2 antibody staining (red).
Localization of PKC-βII
In NG group, PKC-βII expressed in podocytes, thick ascending limbs, and principal cells in cortical collecting duct. No expression was found in epithelial cells in the proximal tubule in NG group (Figure 3A). In DN group, PKC-βII expressed in podocytes, thick ascending limbs, and epithelial cells in the proximal tubule, but not in the principal cells in cortical collecting duct (Figure 3B). In T group, PKC-βII expressed in podocytes, as well as thick ascending limbs, but not in the epithelial cells in the proximal tubule and the principal cells in cortical collecting (Figure 3C).
Figure 3.
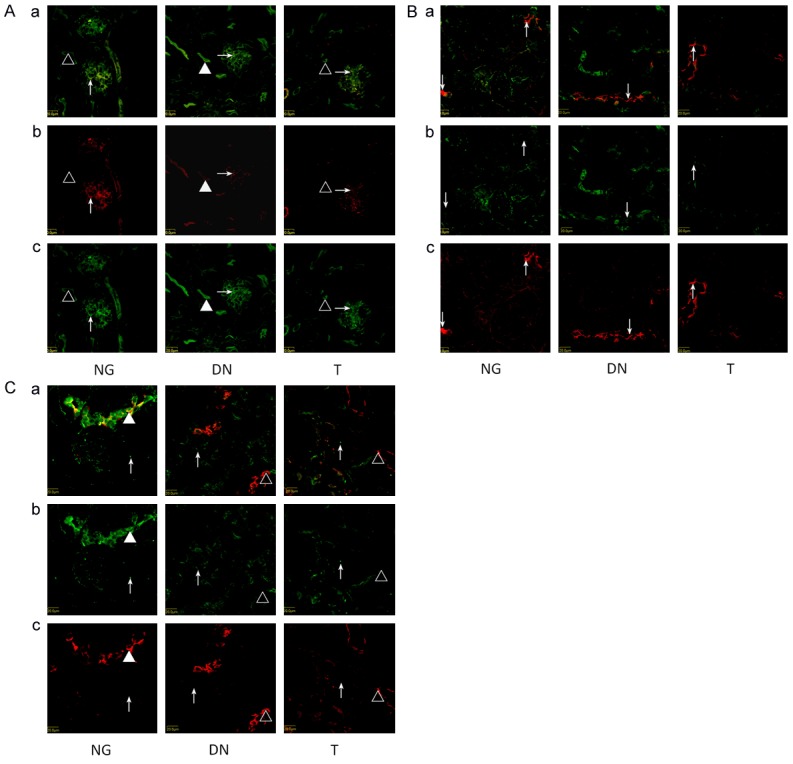
A. Localization of PKC-βII in glomerulus and proximal tubule. a shows the overlay of Phalloidin staining (green) and PKC-βII antibody staining (red). Kidneys were immunostained with PKC-βII antibody (red) in panel b showing the localization of PKC-βII. In panel c, kidneys were stained with the phalloidin (green). Left column: PKC-βII was detected in podocytes (arrow) and no expression in proximal tubule (Triangle ) in NG. Middle column: 6 weeks after STZ induction, enhanced accumulation of PKC-βII in podocytes (arrow) and expression on proximal tubule were observed (Triangle). Right column: Telmisartan treatment attenuated enhanced accumulation of DN mice. B. Localization of PKC-βII in cortical thick ascending limbs of Henle’s loop on three groups (arrow). a shows the overlay of Tamm-Horsfall antibody staining (red) and PKC-βII antibody staining (green). Kidneys were immunostained with PKC-βII antibody (green) in panel b showing the localization of PKC-βII. In panel c, kidneys were stained with the Tamm-Horsfall antibody staining (red). C. Localization of PKC-βII in cortical collecting duct (Triangle) and interstitial cells (arrow). a shows the overlay of AQP2 antibody staining (red) and PKC-βII antibody staining (green). Kidneys were immunostained with PKC-βII antibody (green) in panel b showing the localization of PKC-βII. In panel c, kidneys were stained with the aquaporin-2 antibody staining (red). Left column: PKC-βII was detected in cortical collecting duct (Triangle). Middle column: 6 weeks after STZ induction, the expression of PKC-βII disappear in DN. Right column: Telmisartan treatment can not restore the PKC-βII expression in cortical collecting duct of DN mice. PKC-βIIwas detected in interstitial cells of all three groups (arrow).
Expression of PKC-βI, PKC-βII, TGF-β1 and VEGF
Compared to the NG group, the expression of PKC-βI, TGF-β1 and VEGF significantly (P < 0.01) increased in DN group (Figure 4; Table 3), while the expression of PKC-βII significantly (P < 0.01) lowered in DN group (Figure 4; Table 3). Telmisartan treatment significantly decreased the expression of PKC-βI (P < 0.01), TGF-β1 (P < 0.05) and VEGF (P < 0.05), but had little effect the expression of PKC-βII, compared to the DN group (Figure 4; Table 3). Further analysis with the Pearson’s Correlation shown that PKC-βI exhibited a positive correlation to TGF-β1 (r = 0.649, P = 0.030), but no correlation to VEGF (r = 0.387, P = 0.079). No correlation was found between the expression of PKC-βII to TGF-β1 (r = 0.287, P = 0.085) and VEGF (r = 0.341, P = 0.082). Further evidence from Western blotting also showed that mice in DN groups had greater (P < 0.01) protein abundance of PKC-βI but lower (P < 0.01) protein abundance of PKC-βII in the cortex than those in the NG group (Figure 5; Table 4). Telmisartan treatment significantly (P < 0.01) decreased the expression of PKC-βI in the cortex, but increased the expression of PKC-βII in the cortex, compared to the DN group (Figure 5; Table 4).
Figure 4.
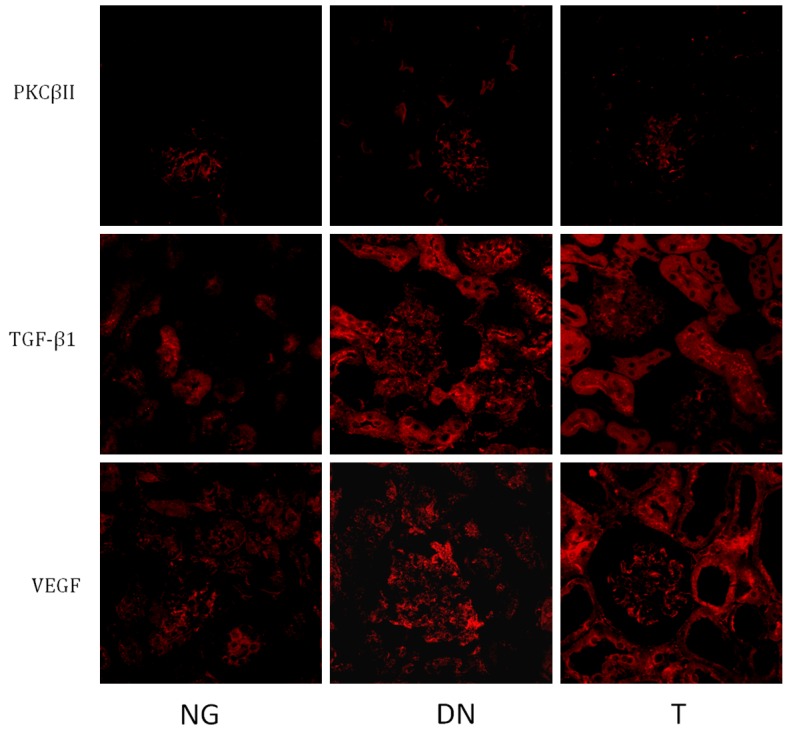
Expression of PKC-βI, PKC-βII, TGF-β1 and VEGF. Mice in DN group treated with STZ, while mice in T group treated with STZ and telmisartan, and mice in NG group didn’t receive any treatment.
Table 3.
Expression of PKC-βI, PKCβII, TGF-β1 and VEGF
| G | PKC-βI | PKCβII | TGF-β1 | VEGF |
|---|---|---|---|---|
| NG | 52.50±4.51 | 43.50±3.70 | 45.28±3.24 | 56.31±4.51 |
| DN | 72.67±4.73** | 31.68±1.58** | 54.27±5.79** | 69.94±6.48** |
| T | 66.15±3.87**,▲▲ | 33.67±3.01** | 49.38±5.48*,▲ | 63.58±5.14*,▲ |
Values are Means±SEM, n = 6. The data calculated from Figure 4.
Different from NG, P < 0.01;
Different from NG, P < 0.05;
Different from DN P < 0.01;
Different from DN P < 0.05.
Figure 5.
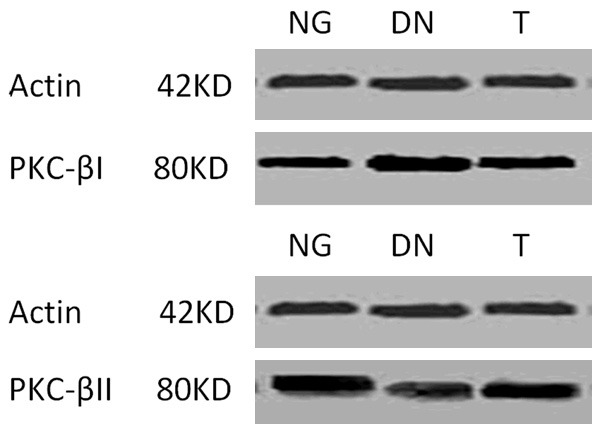
Protein of PKC-βI and PKC-βII on contex.
Table 4.
Expression of PKC-βI and PKCβII
| G | PKC-βI | PKC-βII |
|---|---|---|
| NG | 100.09±2.28 | 102.52±2.50 |
| DN | 121.82±1.06** | 84.15±2.48** |
| T | 118.24±2.43**,▲▲ | 91.37±1.93**,▲▲ |
Values are Means±SEM, n = 6. The data calculated from Figure 5.
Different from NG, P < 0.01;
Different from DN P < 0.01.
Discussion
The PKC family comprises more than ten isoforms categorized into three classes (classical, novel, and typical) based on their structure and cofactor regulation. PKC β-II is an essential isozyme among the PKC isoforms, which is activated in vascular tissues during hyperglycemia-induced situation, leading to the pathogenesis of diabetic complications, like nephropathy [18,21]. PKCβ1 also has an important role in the pathogenesis of diabetic nephropathy. For example, the activation of PKCβ1 and its downstream effects, including upregulation of TGF-β1, is necessary for RhoA activation induced by high glucose in mesangial cells, resulting in matrix up-regulation [19,22]. These compelling studies suggest that both PKC β-I and PKC β-II pays critical roles in the pathogenesis of diabetic nephropathy, but there are different in their functions in diabetic nephropathy. Indeed, our discovery of different expression and distribution of PKC β-I and PKC β-II in the diabetic nephropathy also supports this conclusion. Similarly, the expression and localization of PKC β-I and PKC β-II is also differently affected by telmisartan in the treatment of diabetic nephropathy. However, their exact function in pathogenesis of diabetic nephropathy needs further investigation. Selective inhibition of PKC β-I or PKC β-II will be one of the favorable approaches to uncover their different roles in diabetic nephropathy, however, selective inhibitors of these enzymes are under clinical trials but to date, success has not been achieved because of the high sequence similarities among PKC isoforms [18].
The expression of PKC β-I increases in diabetic nephropathy, while the expression of PKC β-II decreases in diabetic nephropathy. Not just PKC β-I, the expression of TGF-β1 and VEGF also increases in diabetic nephropathy. It is well known that diabetes mellitus leads to sustained hyperglycemia, which in turn results in over-expression of PKC, TGF-β and VEGF in the kidney, increasing synthesis and accumulation of extracellular matrix components to lead to diabetic nephropathy [3,8,23]. Different from our observation, some previous studies have indicated that production of PKC-βI increases in diabetic kidneys, and it may induce production of growth factors, such as TGF-β, CTGF, and VEGF [15,24,25]. However, similar to our reports, Yao also found that the expression of PKC-α VEGF and TGF-β1 increases significantly, but the expression of PKC-βII decreases markedly in glomeruli of kidney tissues from patients with diabetic nephropathy [26]. Such difference may relate to the discrepancy of samples and analysis methods. Telmisartan treatment excises significant beneficial role in diabetic nephropathy, associating with the regulation of PKC β. Indeed, numerous well-designed works have shown that we can manipulate the pathogeneses of diabetic nephropathy through regulation of PKC β [7,23,27]. For example, genistein might be a good protective substance for diabetic nephropathy, in particular, for diabetes patients with medium-high blood glucose levels with reducing the activation of PKC β [8].
Intriguingly, PKC-βI expresses in podocytes, cortical thick ascending limbs of Henle’s loop, principal cells in cortical collecting duct as well asepithelial cells in the proximal tubule. PKC-βII expresses in podocytes, thick ascending limbs of Henle’s loop, and principal cells in cortical collecting duct and interstitial cells, but not in proximal tubule. These compelling findings indicate the different functions of PKC-βI and PKC-βII in the pathogeneses of diabetic nephropathy. As few reports on the localization of PKC-βI and PKC-βII in diabetic nephropathy, thus it is difficult to state the underlying reason for this distribution, but it may associate with their normal function in kidney.
In conclusion, the expression and localization of PKC-βI and PKC-βII differ in the diabetic nephropathy, and such difference is associated with the pathogeneses of diabetic nephropathy. Telmisartan treatment excises significant beneficial role in diabetic nephropathy associating with the regulation of PKC β.
Acknowledgements
This study was supported by the Department of Education of Zhejiang Province (No. Y201431742). The authors thank Yan MAO for assistance.
Disclosure of conflict of interest
None.
References
- 1.Mauer SM, Steffes MW, Ellis EN, Sutherland DE, Brown DM, Goetz FC. Structural-functional relationships in diabetic nephropathy. J Clin Invest. 1984;74:1143–1155. doi: 10.1172/JCI111523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ibrahim HN, Hostetter TH. Diabetic nephropathy. J Am Soc Nephrol. 1997;8:487–493. doi: 10.1681/ASN.V83487. [DOI] [PubMed] [Google Scholar]
- 3.Heilig CW, Deb DK, Abdul A, Riaz H, James LR, Salameh J, Nahman NS Jr. GLUT1 regulation of the pro-sclerotic mediators of diabetic nephropathy. Am J Nephrol. 2013;38:39–49. doi: 10.1159/000351989. [DOI] [PubMed] [Google Scholar]
- 4.Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, Hadden D, Turner RC, Holman RR. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. 2000;321:405–412. doi: 10.1136/bmj.321.7258.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiarelli F, Gaspari S, Marcovecchio ML. Role of growth factors in diabetic kidney disease. Horm Metab Res. 2009;41:585–493. doi: 10.1055/s-0029-1220752. [DOI] [PubMed] [Google Scholar]
- 6.Osicka TM, Yu Y, Panagiotopoulos S, Clavant SP, Kiriazis Z, Pike RN, Pratt LM, Russo LM, Kemp BE, Comper WD, Jerums G. Prevention of albuminuria by aminoguanidine or ramipril in streptozotocin-induced diabetic rats is associated with the normalization of glomerular protein kinase C. Diabetes. 2000;49:87–93. doi: 10.2337/diabetes.49.1.87. [DOI] [PubMed] [Google Scholar]
- 7.Menne J, Shushakova N, Bartels J, Kiyan Y, Laudeley R, Haller H, Park JK, Meier M. Dual inhibition of classical protein kinase C-alpha and protein kinase C-beta isoforms protects against experimental murine diabetic nephropathy. Diabetes. 2013;62:1167–1174. doi: 10.2337/db12-0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thallas-Bonke V, Cooper ME. Tandem inhibition of PKC in Dialphabetaetic nephropathy: it takes two to tango? Diabetes. 2013;62:1010–1. doi: 10.2337/db12-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang N, Alexander G, Park JK, Maasch C, Buchwalow I, Luft FC, Haller H. Differential expression of protein kinase C isoforms in streptozotocin-induced diabetic rats. Kidney Int. 1999;56:1737–1750. doi: 10.1046/j.1523-1755.1999.00725.x. [DOI] [PubMed] [Google Scholar]
- 10.Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 11.Menne J, Park JK, Boehne M, Elger M, Lindschau C, Kirsch T, Meier M, Gueler F, Fiebeler A, Bahlmann FH, Leitges M, Haller H. Diminished loss of proteoglycans and lack of albuminuria in protein kinase C-alpha-deficient diabetic mice. Diabetes. 2004;53:2101–2109. doi: 10.2337/diabetes.53.8.2101. [DOI] [PubMed] [Google Scholar]
- 12.Meier M, Menne J, Park JK, Haller H. Nailing down PKC isoform specificity in diabetic nephropathy two’s company, three’s a crowd. Nephrol Dial Transplant. 2007;22:2421–2425. doi: 10.1093/ndt/gfm320. [DOI] [PubMed] [Google Scholar]
- 13.Meier M, Menne J, Haller H. Targeting the protein kinase C family in the diabetic kidney: lessons from analysis of mutant mice. Diabetologia. 2009;52:765–775. doi: 10.1007/s00125-009-1278-y. [DOI] [PubMed] [Google Scholar]
- 14.Meier M, Menne J, Park JK, Holtz M, Gueler F, Kirsch T, Schiffer M, Mengel M, Lindschau C, Leitges M, Haller H. Deletion of protein kinase C-epsilon signaling pathway induces glomerulosclerosis and tubulointerstitial fibrosis in vivo. J Am Soc Nephrol. 2007;18:1190–1198. doi: 10.1681/ASN.2005070694. [DOI] [PubMed] [Google Scholar]
- 15.Meier M, Park JK, Overheu D, Kirsch T, Lindschau C, Gueler F, Leitges M, Menne J, Haller H. Deletion of protein kinase C-beta isoform in vivo reduces renal hypertrophy but not albuminuria in the streptozotocin-induced diabetic mouse model. Diabetes. 2007;56:346–354. doi: 10.2337/db06-0891. [DOI] [PubMed] [Google Scholar]
- 16.Mima A, Hiraoka-Yamomoto J, Li Q, Kitada M, Li C, Geraldes P, Matsumoto M, Mizutani K, Park K, Cahill C, Nishikawa S, Rask-Madsen C, King GL. Protective effects of GLP-1 on glomerular endothelium and its inhibition by PKCbeta activation in diabetes. Diabetes. 2012;61:2967–2979. doi: 10.2337/db11-1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soetikno V, Watanabe K, Sari FR, Harima M, Thandavarayan RA, Veeraveedu PT, Arozal W, Sukumaran V, Lakshmanan AP, Arumugam S, Suzuki K. Curcumin attenuates diabetic nephropathy by inhibiting PKC-alpha and PKC-beta1 activity in streptozotocin-induced type I diabetic rats. Mol Nutr Food Res. 2011;55:1655–1665. doi: 10.1002/mnfr.201100080. [DOI] [PubMed] [Google Scholar]
- 18.Sobhia ME, Grewal BK, Bhat J, Rohit S, Punia V. Protein kinase C betaII in diabetic complications: survey of structural, biological and computational studies. Expert Opin Ther Targets. 2012;16:325–344. doi: 10.1517/14728222.2012.667804. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Peng F, Gao B, Ingram AJ, Krepinsky JC. High glucose-induced RhoA activation requires caveolae and PKCbeta1-mediated ROS generation. Am J Physiol Renal Physiol. 2012;302:F159–172. doi: 10.1152/ajprenal.00749.2010. [DOI] [PubMed] [Google Scholar]
- 20.Ren W, Chen S, Yin J, Duan J, Li T, Liu G, Feng Z, Tan B, Yin Y, Wu G. Dietary Arginine Supplementation of Mice Alters the Microbial Population and Activates Intestinal Innate Immunity. J Nutr. 2014;144:988–995. doi: 10.3945/jn.114.192120. [DOI] [PubMed] [Google Scholar]
- 21.Vijayakumar B, Velmurugan D. Designing of Protein Kinase C beta-II Inhibitors against Diabetic complications: Structure Based Drug Design, Induced Fit docking and analysis of active site conformational changes. Bioinformation. 2012;8:568–573. doi: 10.6026/97320630008568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng F, Wu D, Gao B, Ingram AJ, Zhang B, Chorneyko K, McKenzie R, Krepinsky JC. RhoA/Rho-kinase contribute to the pathogenesis of diabetic renal disease. Diabetes. 2008;57:1683–1692. doi: 10.2337/db07-1149. [DOI] [PubMed] [Google Scholar]
- 23.Jamuna JB, Nandini CD. Feeding of banana flower and pseudostem to diabetic rats results in modulation of renal GLUTs, TGFbeta, PKC and extracellular matrix components. Nutr Metab Cardiovasc Dis. 2014;24:623–631. doi: 10.1016/j.numecd.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 24.Kim MJ, Lim Y. Protective effect of short-term genistein supplementation on the early stage in diabetes-induced renal damage. Mediators Inflamm. 2013;2013:510212. doi: 10.1155/2013/510212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ikeda A, Matsushita S, Sakakibara Y. Inhibition of protein kinase C beta ameliorates impaired angiogenesis in type I diabetic mice complicating myocardial infarction. Circ J. 2012;76:943–949. doi: 10.1253/circj.cj-11-0881. [DOI] [PubMed] [Google Scholar]
- 26.Yao L, Wang J, Mao Y, Zhu H, Deng A, Zhu Z. Different expressions of protein kinase C-alpha, beta I and beta II in glomeruli of diabetic nephropathy patients. J Huazhong Univ Sci Technolog Med Sci. 2006;26:651–653. doi: 10.1007/s11596-006-0605-5. [DOI] [PubMed] [Google Scholar]
- 27.Ke JT, Li M, Xu SQ, Zhang WJ, Jiang YW, Cheng LY, Chen L, Lou JN, Wu W. Gliquidone decreases urinary protein by promoting tubular reabsorption in diabetic Goto-Kakizaki rats. J Endocrinol. 2014;220:129–141. doi: 10.1530/JOE-13-0199. [DOI] [PubMed] [Google Scholar]