

Abstract
Emerging evidence has led to considerable interest in the role of Traf2- and Nck-interacting kinase (TNIK) in polycystic ovary syndrome (PCOS) development. However, the epigenetic mechanism regulating TNIK transcription remains largely unknown. Here, we show that (i) TNIK mRNA expression is significantly increased in PCOS ovarian tissues, compared to normal ovarian tissues; (ii) PCOS ovarian tissues exhibit a hypermethylation pattern at the cg10180092 site, (iii) and cg10180092 is the critical site for the transcriptional regulation of TNIK. Mechanistically, hypermethylated cg10180092 site-mediated loss of holocarboxylase synthetase (HLCS)-related H3K9me enrichment activated TNIK transcription in PCOS ovarian tissues. Notably, a substantial body of evidence indicates that DNA hypermethylation is an alternative mechanism for gene inactivation, and a new role for DNA hypermethylationmediated TNIK activating was observed in this study. This may improve our understanding of divergent transcriptional regulation in the initiation and progression of TNIK-related PCOS.
Keywords: TNIK, PCOS, epigenetic, DNA methylation, histone modifications
Introduction
Polycystic ovary syndrome (PCOS) is the most common endocrinopathy, affecting about 10% of the reproductive-age female population [1], with significant and diverse clinical symptoms including reproductive dysfunction, metabolic disorders, and psychological distress [1-4]. In addition to genetic predisposition, environmental and lifestyle factors contribute to the pathogenesis of PCOS [5]; an emerging body of evidence suggests that epigenetic events may in part be responsible for the development of PCOS [6,7]. It is well-known that DNA methylation at cytosines is a heritable epigenetic mark of cellular memory that modulates gene expression without affecting the DNA sequence, as an adaptation to environmental and lifestyle changes [8]. Recently, by the integrative analysis of differential DNA methylation and gene expression associated with PCOS using high-throughput microarray techniques, we found that PCOS ovarian tissues exhibit a specific methylation and expression pattern of the Traf2- and Nck-interacting kinase (TNIK) gene [9]. TNIK is a member of the germinal center kinase family, and is widely involved in cytoskeleton organization, neuronal dendrite extension, cell proliferation, and glutamate receptor regulation [10,11]. Interestingly, this is consistent with the suggestion that TNIK is a biomarker of embryo viability in PCOS patients, and may possibly be involved in the development of PCOS [12]. However, to date, little is known about the detailed expression patterns and transcriptional regulation of TNIK in PCOS. For this reason, the present study was first undertaken to investigate the methylation status and expression of TNIK in human ovary tissues and granulosa cells from PCOS patients and normal controls, and to provide novel insight into epigenetic-mediated dysfunction in the pathogenesis of PCOS.
Materials and methods
Ethical statements
This investigation was conducted in accordance with ethical standards according to the Helsinki Declaration of 1975.
Patients and tissue collection
This study was approved by the Institutional Review Board at China Medical University. Cervical cancer patients with normal ovaries and PCOS patients were enrolled between 2012 and 2014, and all patients gave informed consent. Fresh ovarian tissue was obtained from cervical cancer patients (n = 13) at the follicular phase, with regular menstrual cycles (25-35 days) and younger than 40 years of age, who underwent radical hysterectomy and pelvic lymphadenectomy for cervical cancer. Fresh ovarian tissue was obtained from PCOS patients (n = 13) at the follicular phase, with irregular menstrual cycle and younger than 40 years of age, who underwent laparoscopic wedge resection. Normal and PCOS ovarian granulosa cells were obtained from patients who underwent in vitro fertilization (n = 11). Follicle size was determined by determining the average follicular diameter, and follicular fluids from large follicles (17-22 mm) and small follicles (8-13 mm) were collected in separate tubes. Large and small follicles were examined separately to assess the differences between mature and immature dominant follicles. Follicular fluids from large and small follicles were pooled separately and centrifuged by Percoll gradient centrifugation. Luteinized granulosa cells were collected at the interface and washed with PBS. PCOS was defined according to the 2003 Rotterdam criteria [2].
DNA methylation analysis by pyrosequencing
Genomic DNA was bisulfite-modified using the EpiTect Plus DNA Bisulfite Kit (Qiagen) according to the manufacturer’s protocol. Bisulfite-converted DNA was amplified by EPIK™ Amplification Kit (Bioline) according to the manufacturer’s protocol. The specific primer sequences are listed in Table 1. The conditions were as follows: 95°C for 2 min, 40 cycles of 15 s at 95°C, 15 s at 56°C and 30 s at 72°C. Following amplification, the biotinylated PCR products were purified and incubated with the sequencing primer ATGTAGGAGGTTTATTAAGAA, which was designed to bind adjacent to the CpG site of interest. Pyrosequencing was conducted using PyroMark™ Gold Q96 reagents (Qiagen) with subsequent quantification of methylation levels determined with the PyroMark Q24 1.010 software. Relative peak height differences were used to calculate the percentage of methylated cytosines.
Table 1.
Primers used in this study
| Gene | Primers | Description |
|---|---|---|
| cg10180092-F | 5’-GTGTAATTTTGAGGGAAATAATAAAGGT | Methylation analysis for TNIK |
| cg10180092-R-biotin | 5’-AACTCCCCACATAATCAACTAA | |
| cg10180092-S | 5’-ATGTAGGAGGTTTATTAAGAA | |
| TNIK-RTP-F | 5’-CTTGTGGTCTTTGGGTATCAC | Real-time PCR for TNIK |
| TNIK-RTP-R | 5’-CCACTTCTTAGACTTCAGCC | |
| GAPDH-RTP-F | 5’-AGGTGAAGGTCGGAGTCA | Real-time PCR for GAPDH |
| GAPDH-RTP-R | 5’-GGTCATTGATGGCAACAA | |
| HLCS-SQP | Santa Cruz Biotech | Semi-quantitative PCR for HLCS |
| HLCS (h)-PR: sc-72198-PR | ||
| EHMT-1-SQP | Santa Cruz Biotech | Semi-quantitative PCR for EHMT-1 |
| EHMT-1 (h)-PR: sc-62261-PR | ||
| TNIK-F | 5’-AATAGAGCTCTCTCTGAGTTCTCCCCCTTA | Amplified TNIK |
| TNIK-R | 5’-AATAAAGCTTTCTCAACCAACTTCACACCC | |
| TNIK-ChIP-RTP-F | 5’-GGATTTCTTTGGGACTGGCT | ChIP for TNIK |
| TNIK-ChIP-RTP-R | 5’-TCTTGACTTCTCTCTCTGGC | |
| TNIK-Mutation-F | 5’-GTTTATTAAGAAATGCAATGATGGCAGGATTGGCCAG | A point mutation at the site of cg10180092 (C to T) |
| TNIK-Mutation-R | 5’-CTGGCCAATCCTGCC A TCATTGCATTTCTTAATAAAC |
Abbreviations: RTP, real-time PCR; SQP, semi-quantitative PCR; ChIP, chromatin immunoprecipitation; F, forward primer; R, reverse primer.
Real-time quantitative PCR
Total RNA was extracted using Trizol reagent (Invitrogen) according to the manufacturer’s protocol. DNA contamination was removed by adding DNase I (Invitrogen) according to the manufacturer’s protocol. Total RNA was then reverse-transcribed from 2 μg of RNA using the PrimeScript RT Master Mix kit (TaKaRa) and amplified by SYBR Premix Ex TaqTM II (TaKaRa) in a Roche LightCycler 2.0 instrument (Roche Diagnostics). The specific primer sequences for real-time PCR are listed in Table 1. GAPDH mRNA was amplified as an internal control for the normalization of each sample. All samples were analyzed in triplicate using the 2-∆∆CT method.
Chromatin immunoprecipitation assay (ChIP)
ChIP assays were performed using the EpiQuik™ Tissue Chromatin Immunoprecipitation kit (Epigentek Group Inc.), as previously reported [13-15]. Briefly, small pieces of normal and PCOS ovarian tissues (1-2 mm3) were crosslinked with 1% formaldehyde. Cross-linking was terminated using 1.25 M glycine. The tissues and cells were added to homogenizing buffer, triturated, disaggregated, and centrifuged at 1000 g for 5 min at 4°C. After the removal of supernatants, protease inhibitors were added and the disaggregated tissue pellet was resuspended. Chromatin was sheared by sonication. Immunoprecipitation was performed at room temperature for 90 min. The specific antibodies (H3K9ac, ab4441; H3K18ac, ab1191; H3K4me1, ab8895; H3K4me2, ab7766; H3K4me3, ab8580; H3K36me3, ab9050; H3K79me, ab2886; H3K9me, ab9045; H3K9me3, ab8898) and (H3K27ac, 07-360; H3K9me2, 07-441; H3K27me, 07-448; H3K27me2, 07-452) for ChIP were purchased from Abcam and Millipore, respectively. Crosslinking was reversed at 65°C for 90 min. Finally, genomic DNA was eluted for PCR analysis. The specific primer sequences for ChIP are listed in Table 1. The thermocycle was 94°C for 2 min, then 30 cycles of 45 s at 94°C, 45 s at 56°C and 45 s at 72°C.
Generation of TNIK luciferase constructs with cg10180092 site-directed mutagenesis
Genomic DNA from ovarian tissues was extracted using the TIANamp Genomic DNA kit (Tiangen Biotech). The sequence containing cg10180092 site was obtained by PCR amplification. The specific primer sequences are provided in Table 1. The fragments were sub-cloned into the pGL4 luciferase reporter vector (Promega). For site-directed mutagenesis, the recombinant vector was used as a template to insert a C to T base mutation (cg10180092 site) following the manufacturer’s protocol (Stratagene). Briefly, Pfu Turbo DNA polymerase was used for the PCR reaction. The conditions were as follows: 95°C for 5 min, 30 cycles of 40 s at 95°C, 30 s at 56°C and 2 min at 72°C, then 10 min at 72°C. PCR products were digested with DpnI to specifically eliminate the methylated and hemimethylated DNA containing the nonmutated sequence. Subsequently, the nicked vector containing the expected mutation was transformed into Epicurian Coli XL1-Blue Supercompetent Cells. After transformation, the XL1-Blue Supercompetent Cells repair the nicks in the mutated pGL4 vector. The specific primer sequences for site-directed mutagenesis are provided in Table 1, and the mutated cg10180092 site was verified by automated Sanger sequencing.
Cell culture, infection, and cell proliferation assay
Human 293T cells were maintained in DMEM with 10% fetal bovine serum (Invitrogen), and were tested and authenticated on June 1, 2012. The short hairpin RNAs (shRNAs), lentiviral particles of holocarboxylase synthetase (HLCS, sc-72198-V), and euchromatic histone-lysine N-methyltransferase 1 (EHMT-1, sc-62261-V) were purchased from Santa Cruz Biotechnology. Lentiviral infection was performed using polybrene and enhanced infection solution (Genechem) according to the manufacturer’s recommended protocol. Plasmid transfections were performed using Lipofectamine™ 2000 (Invitrogen) according to the manufacturer’s protocol. After 48 hours of transfection, cell proliferation was determined using the Cell-Light™ EdU Apollo®643 In Vitro Imaging Kit (Ribobio) following the manufacturer’s instructions.
Statistical analysis
The data are presented as mean ± standard deviation (SD). Statistical differences in the data were evaluated by Student’s t test or one-way analysis of variance (ANOVA) as appropriate, and were considered significant at P < 0.05.
Results
Hypermethylation of the cg10180092 site is accompanied by increased TNIK levels in ovarian tissues
The location of the methylated CpG site (cg10180092) in TNIK gene was as shown in Figure 1A. Generally, promoter methylation is an epigenetic modification that is involved in regulating gene expression in mammals [13]. Consistent with this idea, we showed that PCOS ovarian tissues with a hypermethylated cg10180092 site (Figure 1Bi and 1C) displayed increased expression of TNIK mRNA (Figure 1D) compared with the normal ovarian tissues. However, no significant TNIK expression and methylation differences were observed between normal and PCOS ovarian granulosa cells (Figure 1Bii, 1E and 1F for large follicles, Figure 1Biii, 1G and 1H for small follicles). These results indicate that TNIK hypermethylation may be responsible for the regulation of TNIK expression in ovarian tissues.
Figure 1.
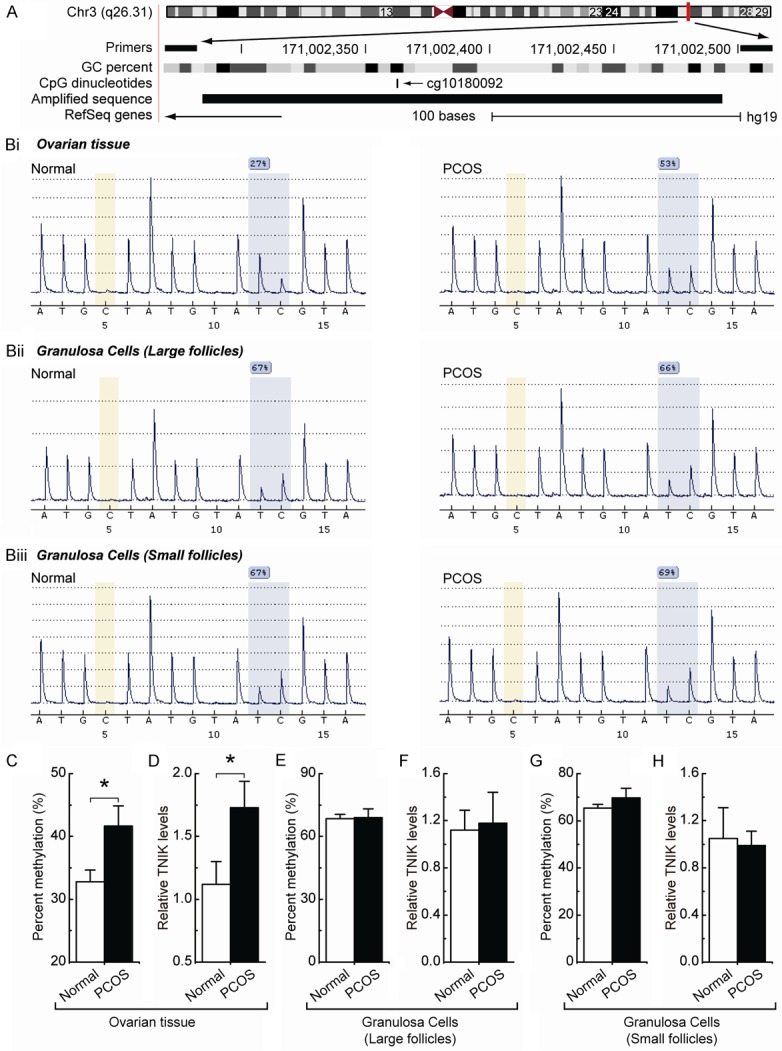
Expression and methylation pattern of TNIK in ovarian tissues and ovarian granulosa cells. A. Location of the methylated CpG site (cg10180092) in TNIK gene. Genomic coordinates are shown, along with the primer-amplified fragments, GC percentage, individual CpG dinucleotide, and TNIK RefSeq gene. The arrow indicates the direction of transcription. Bi-Biii. Comparative analysis of methylation patterns at the cg10180092 site of the TNIK gene in ovarian tissues and ovarian granulosa cells (large follicles and small follicles) in normal and PCOS samples. The yellow regions indicate control regions for automatic assessment of bisulfide conversion (unmethylated C should be fully converted to T), and the blue regions indicate the percentage of CpG methylation. C, E and G. Summary of the methylation levels of TNIK gene from the measurements shown in Bi, Bii and Biii, respectively. D, F and H. Relative TNIK mRNA levels were measured in ovarian tissues and ovarian granulosa cells (large follicles and small follicles) between normal and PCOS samples. Bar graphs show mean ± SD. *P < 0.05 vs. normal.
Loss of H3K9me enrichment around the cg10180092 site in PCOS ovarian tissues
Our previous study suggested that specific histone modification patterns play an important part in regulating gene transcription [14,15]. To obtain further understanding of the regulatory potential of histone modifications in controlling TNIK transcription, we examined the activating histone markers H3K9ac, H3K18ac, H3K27ac, H3K4me1, H3K4me2, H3K4me3, H3K36me3, and H3K79me, and the repressive histone markers H3K9me, H3K9me2, H3K9me3, H3K27me, and H3K27me2 around the cg10180092 site. Chromatin immunoprecipitation analysis indicated that the levels of H3K9me were only significantly decreased in PCOS ovarian tissues, compared with normal ovarian tissues (Figure 2A and 2B). Chromatin-modifying enzymes (HLCS and EHMT-1) that facilitate the creation of H3K9me were also analyzed. Although there was no significant change in the expression of EHMT-1, the expression levels of HLCS were reduced (Figure 2C). These results, together with Figure 1, suggest that TNIK transcription may be associated with decreased H3K9me enrichment in PCOS.
Figure 2.
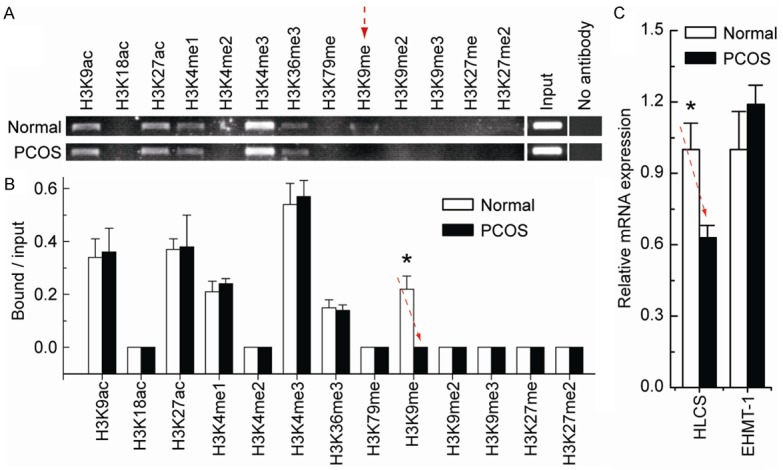
Comparative analysis of histone modification around the cg10180092 site between normal and PCOS ovarian tissues. A. Chromatin immunoprecipitation was performed using antibodies to H3K9ac, H3K18ac, H3K27ac, H3K4me1, H3K4me2, H3K4me3, H3K36me3, H3K79me, H3K9me, H3K9me2, H3K9me3, H3K27me, and H3K27me2. PCR was performed for regions around the cg10180092 site. A negative control without antibodies is included for comparison. B. Representative results of normal and PCOS ovarian tissues are shown. Bar graphs show mean ± SD. C. Expression levels of the chromatin-modifying enzymes HLCS and EHMT-1 in normal and PCOS ovarian tissues. Bar graphs show mean ± SD. *P < 0.05 vs. normal.
Decreased enrichment of H3K9me around the hypermethylated site in PCOS ovarian tissues
To further confirm the effect of hypermethylated cg10180092 site on H3K9me enrichment, the site was mutated and the vector was transfected into 293T cells. As shown in Figure 3, mutation of this site (AATGCAATGACGGCAGGATTGGC to AATGCAATGATGGCAGGATTGGC) in the TNIK gene induced the loss of H3K9me enrichment.
Figure 3.
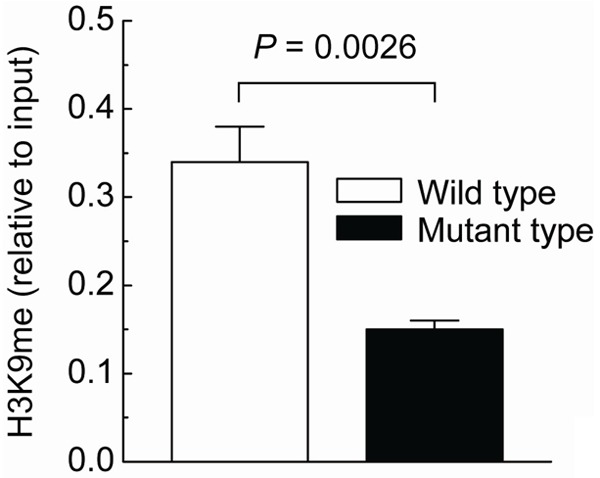
H3K9me pattern of the position cg10180092 mutation in 293T cells. 293T cells were transfected with constructs containing cg10180092 site-directed mutagenesis or not, respectively. At 24 hours after transfection, whole-cell extracts were analyzed for H3K9me enrichment. Each group, n = 12. Bar graphs show mean ± SD. *P < 0.05 vs. control.
H3K9me present around the cg10180092 site is responsible for transcriptional regulation of TNIK
We observed that the knockdown of HLCS and EHMT-1 (Figure 4A and 4B) had no detectable effect on cell morphology and proliferation (Figure 4C and 4D). Combined knockdown of HLCS and EHMT-1 specifically induced a decrease in H3K9me enrichment around the cg10180092 site (Figure 4E). Notably, after the deletion of H3K9me, the TNIK levels were significantly up-regulated (Figure 4F).
Figure 4.
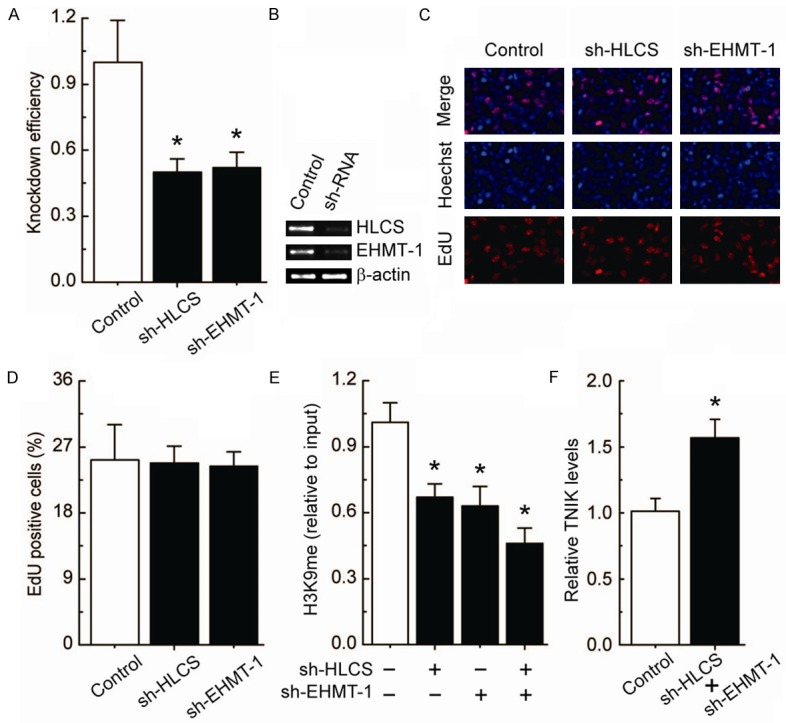
H3K9me-mediated transcriptional regulation of TNIK in 293T cells. A. RT-PCR showing HLCS and EHMT-1 levels before and after knockdown by shRNAs, and normalized to -actin expression (n = 3). B. Representative RT-PCR results are shown. C. EdU labeling showing the proliferation of HLCS and EHMT-1-silenced in 293T cells. Blue, Hoechst 33342 labeling of cell nuclei; red, EdU labeling of nuclei of proliferative cells. D. The EdU incorporation rate was expressed as the ratio of EdU-positive cells to total Hoechst 33342-positive cells. E. Analysis of histone modification H3K9me enrichment around the cg10180092 site after the deletion of HLCS and/or EHMT-1 in 293T cells. F. TNIK levels after the deletion of H3K9me around the cg10180092 site in 293T cells. Each group, n = 12. Bar graphs show mean ± SD. *P < 0.05 vs. control.
Discussion
Promoter methylation, with concurrent changes in histone modifications, is an epigenetic phenomenon that can affect the conformation of chromatin and tissue-specific gene expression [16,17]. In this study, we report that the TNIK transcript was up-regulated in PCOS ovarian tissues, compared with normal ovarian tissues, and that methylation of cg10180092 site is a key regulatory element for TNIK transcription. The molecular mechanism may involve hypermethylated cg10180092 site -mediated loss of the repressive histone marker H3K9me enrichment, which synergistically activates the transcription of TNIK.
Notably, an emerging body of evidence suggests that TNIK is a novel activator of Wnt signaling [18]. It is well-known that the Wnt signaling pathway and its key components play critical roles in the regulation of many cellular functions, including cellular proliferation, differentiation, migration, maintenance of pluripotency, and cancer development and progression [19]. Recent work has highlighted a potential role for Wnt signaling in metabolic homeostasis, and its misregulation has been implicated in obesity, insulin resistance, lipoprotein metabolism, and inflammation [20,21], which are often involved in PCOS pathophysiology [22]. However, to date, there are only a few studies that have correlated TNIK and PCOS, or Wnt signaling and PCOS, and no direct link has yet been established. For instance, TNIK plays essential roles in embryonic development, especially from zygote to blastocyst, and has been proposed as a biomarker of embryo viability in PCOS patients [12]. In addition, Wnt signal transduction is dysregulated in the PCOS theca cell [23]. Interestingly, a substantial body of evidence indicates that PCOS is characterised by proliferative disorders, often along with follicle growth arrest [24], abnormal granulosa cells [8,24,25], cumulus cells, [26] and endometrial proliferation [27]. It has been confirmed that TNIK is a potential proliferation regulator, and small interfering RNA-mediated silencing of TNIK results in significant inhibition of cell proliferation and induction of cell death [11,28,29]. Notably, a recent study suggested a possible role for TNIK in gastric cancer growth that was not dependent on its target pathway, Wnt signaling [11]. It should be noted that Wnt signaling itself also plays critical roles in cellular proliferation and differentiation [30]. Therefore, PCOS-related proliferative disorders may involve functional diversity of TNIK, and it seems beneficial for the understanding of PCOS-related biological features to know about the functional aspects of TNIK.
Taken together, our findings provide novel insights into the regulatory effects of promoter methylation and histone modifications on TNIK transcription, and further correlate the physiological properties of the TNIK gene with PCOS. In addition, accumulating evidence indicates that DNA hypermethylation is an alternative mechanism for gene inactivation, but a new role for DNA hypermethylation-mediated TNIK activation was observed. Based on these findings, there are some interesting issues that need to be considered in future studies; for example, (i) how DNA hypermethylation affects histone modification patterns; (ii) whether other factors could cooperate with these epigenetic events in controlling TNIK expression; (iii) how to evaluate the usefulness of TNIK as a novel therapeutic and diagnostic target for PCOS. All of this may improve our understanding of the basic molecular mechanisms underlying TNIK-related PCOS progression.
Acknowledgements
This work was supported by the Natural Science Foundation of China (No. 81402130) and Doctoral Start-up Foundation of Liaoning Province (No. 20141045).
Disclosure of conflict of interest
None.
References
- 1.Goodarzi MO, Dumesic DA, Chazenbalk G, Azziz R. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nat Rev Endocrinol. 2011;7:219–231. doi: 10.1038/nrendo.2010.217. [DOI] [PubMed] [Google Scholar]
- 2.Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81:19–25. doi: 10.1016/j.fertnstert.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Teede H, Deeks A, Moran L. Polycystic ovary syndrome: a complex condition with psychological, reproductive and metabolic manifestations that impacts on health across the lifespan. BMC Med. 2010;8:41. doi: 10.1186/1741-7015-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Witchel SF, Tena-Sempere M. The Kiss1 system and polycystic ovary syndrome: lessons from physiology and putative pathophysiologic implications. Fertil Steril. 2013;100:12–22. doi: 10.1016/j.fertnstert.2013.05.024. [DOI] [PubMed] [Google Scholar]
- 5.Insenser M, Montes-Nieto R, Murri M, Escobar-Morreale HF. Proteomic and metabolomic approaches to the study of polycystic ovary syndrome. Mol Cell Endocrinol. 2013;370:65–77. doi: 10.1016/j.mce.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 6.Qu F, Wang FF, Yin R, Ding GL, El-Prince M, Gao Q, Shi BW, Pan HH, Huang YT, Jin M, Leung PC, Sheng JZ, Huang HF. A molecular mechanism underlying ovarian dysfunction of polycystic ovary syndrome: hyperandrogenism induces epigenetic alterations in the granulosa cells. J Mol Med (Berl) 2012;90:911–923. doi: 10.1007/s00109-012-0881-4. [DOI] [PubMed] [Google Scholar]
- 7.Wang P, Zhao H, Li T, Zhang W, Wu K, Li M, Bian Y, Liu H, Ning Y, Li G, Chen ZJ. Hypomethylation of the LH/choriogonadotropin receptor promoter region is a potential mechanism underlying susceptibility to polycystic ovary syndrome. Endocrinology. 2014;155:1445–1452. doi: 10.1210/en.2013-1764. [DOI] [PubMed] [Google Scholar]
- 8.Das PM, Singal R. DNA methylation and cancer. J. Clin. Oncol. 2004;22:4632–4642. doi: 10.1200/JCO.2004.07.151. [DOI] [PubMed] [Google Scholar]
- 9.Wang XX, Wei JZ, Jiao J, Jiang SY, Yu DH, Li D. Genome-wide DNA methylation and gene expression patterns provide insight into polycystic ovary syndrome development. Oncotarget. 2014;5:6603–6610. doi: 10.18632/oncotarget.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coba MP, Komiyama NH, Nithianantharajah J, Kopanitsa MV, Indersmitten T, Skene NG, Tuck EJ, Fricker DG, Elsegood KA, Stanford LE, Afinowi NO, Saksida LM, Bussey TJ, O’Dell TJ, Grant SG. TNiK is required for postsynaptic and nuclear signaling pathways and cognitive function. J Neurosci. 2012;32:13987–13999. doi: 10.1523/JNEUROSCI.2433-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu DH, Zhang X, Wang H, Zhang L, Chen H, Hu M, Dong Z, Zhu G, Qian Z, Fan J, Su X, Xu Y, Zheng L, Dong H, Yin X, Ji Q, Ji J. The essential role of TNIK gene amplification in gastric cancer growth. Oncogenesis. 2014;2:e89. doi: 10.1038/oncsis.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang X, Hao C, Shen X, Liu X, Shan Y, Zhang Y, Chen L. Differences in the transcriptional profiles of human cumulus cells isolated from MI and MII oocytes of patients with polycystic ovary syndrome. Reproduction. 2013;145:597–608. doi: 10.1530/REP-13-0005. [DOI] [PubMed] [Google Scholar]
- 13.Li D, Bi FF, Cao JM, Cao C, Li CY, Liu B, Yang Q. Poly (ADP-ribose) polymerase 1 transcriptional regulation: a novel crosstalk between histone modification H3K9ac and ETS1 motif hypomethylation in BRCA1-mutated ovarian cancer. Oncotarget. 2014;5:291–297. doi: 10.18632/oncotarget.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li D, Bi FF, Cao JM, Cao C, Liu B, Yang Q. Regulation of DNA methyltransferase 1 transcription in BRCA1-mutated breast cancer: a novel crosstalk between E2F1 motif hypermethylation and loss of histone H3 lysine 9 acetylation. Mol Cancer. 2014;13:26. doi: 10.1186/1476-4598-13-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li D, Bi FF, Chen NN, Cao JM, Sun WP, Zhou YM, Cao C, Li CY, Yang Q. Epigenetic repression of phosphatidylethanolamine N-methyltransferase (PEMT) in BRCA1-mutated breast cancer. Oncotarget. 2014;5:1315–1325. doi: 10.18632/oncotarget.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang B, Zhou Y, Lin N, Lowdon RF, Hong C, Nagarajan RP, Cheng JB, Li D, Stevens M, Lee HJ, Xing X, Zhou J, Sundaram V, Elliott G, Gu J, Shi T, Gascard P, Sigaroudinia M, Tlsty TD, Kadlecek T, Weiss A, O’Geen H, Farnham PJ, Maire CL, Ligon KL, Madden PA, Tam A, Moore R, Hirst M, Marra MA, Zhang B, Costello JF, Wang T. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the M&M algorithm. Genome Res. 2013;23:1522–1540. doi: 10.1101/gr.156539.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim J, Moon SH, Kim BT, Chae CH, Lee JY, Kim SH. A novel aminothiazole KY-05009 with potential to inhibit Traf2- and Nck-interacting kinase (TNIK) attenuates TGF-beta1-mediated epithelial-to-mesenchymal transition in human lung adenocarcinoma A549 cells. PLoS One. 2014;9:e110180. doi: 10.1371/journal.pone.0110180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanaka SS, Kojima Y, Yamaguchi YL, Nishinakamura R, Tam PP. Impact of WNT signaling on tissue lineage differentiation in the early mouse embryo. Dev Growth Differ. 2011;53:843–856. doi: 10.1111/j.1440-169X.2011.01292.x. [DOI] [PubMed] [Google Scholar]
- 20.Almario RU, Karakas SE. Roles of Circulating WNT-Signaling Proteins and WNT-Inhibitors in Human Adiposity, Insulin Resistance, Insulin Secretion, and Inflammation. Horm Metab Res. 2015;47:152–157. doi: 10.1055/s-0034-1384521. [DOI] [PubMed] [Google Scholar]
- 21.Fuster JJ, Zuriaga MA, Thi-Minh Ngo D, Farb MG, Aprahamian T, Yamaguchi TP, Gokce N, Walsh K. Non-canonical Wnt signaling promotes obesity-induced adipose tissue inflammation and metabolic dysfunction independent of adipose tissue expansion. Diabetes. 2015;64:1235–1248. doi: 10.2337/db14-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moran LJ, Norman RJ, Teede HJ. Metabolic risk in PCOS: phenotype and adiposity impact. Trends Endocrinol Metab. 2015;26:136–143. doi: 10.1016/j.tem.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 23.Wood JR, Ho CK, Nelson-Degrave VL, McAllister JM, Strauss JF 3rd. The molecular signature of polycystic ovary syndrome (PCOS) theca cells defined by gene expression profiling. J Reprod Immunol. 2004;63:51–60. doi: 10.1016/j.jri.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 24.Wang Q, Kim JY, Xue K, Liu JY, Leader A, Tsang BK. Chemerin, a novel regulator of follicular steroidogenesis and its potential involvement in polycystic ovarian syndrome. Endocrinology. 2012;153:5600–5611. doi: 10.1210/en.2012-1424. [DOI] [PubMed] [Google Scholar]
- 25.Stubbs SA, Stark J, Dilworth SM, Franks S, Hardy K. Abnormal preantral folliculogenesis in polycystic ovaries is associated with increased granulosa cell division. J Clin Endocrinol Metab. 2007;92:4418–4426. doi: 10.1210/jc.2007-0729. [DOI] [PubMed] [Google Scholar]
- 26.Polzikov M, Yakovenko S, Voznesenskaya J, Troshina M, Zatsepina O. Overexpression of ribosomal RNA in cumulus cells of patients with polycystic ovary syndrome. J Assist Reprod Genet. 2012;29:1141–1145. doi: 10.1007/s10815-012-9827-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villavicencio A, Bacallao K, Gabler F, Fuentes A, Albornoz J, Casals A, Vega M. Deregulation of tissue homeostasis in endometria from patients with polycystic ovarian syndrome with and without endometrial hyperplasia. Gynecol Oncol. 2007;104:290–295. doi: 10.1016/j.ygyno.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 28.Jiao X, Hooper SD, Djureinovic T, Larsson C, Wärnberg F, Tellgren-Roth C, Botling J, Sjöblom T. Gene rearrangements in hormone receptor negative breast cancers revealed by mate pair sequencing. BMC Genomics. 2013;14:165. doi: 10.1186/1471-2164-14-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shitashige M, Satow R, Jigami T, Aoki K, Honda K, Shibata T, Ono M, Hirohashi S, Yamada T. Traf2- and Nck-interacting kinase is essential for Wnt signaling and colorectal cancer growth. Cancer Res. 2010;70:5024–5033. doi: 10.1158/0008-5472.CAN-10-0306. [DOI] [PubMed] [Google Scholar]
- 30.Tan SH, Senarath-Yapa K, Chung MT, Longaker MT, Wu JY, Nusse R. Wnts produced by Osterix-expressing osteolineage cells regulate their proliferation and differentiation. Proc Natl Acad Sci U S A. 2014;111:E5262–E5271. doi: 10.1073/pnas.1420463111. [DOI] [PMC free article] [PubMed] [Google Scholar]