

Abstract
Parkinson's disease (PD) is a major movement disorder characterized by the loss of dopamine neurons and formation of Lewy bodies. Clinical and pathological evidence indicates that multiple brain regions are affected in PD in a spatiotemporal manner and are associated with a variety of motor and nonmotor symptoms, including disturbances in mood, executive function, and memory. The common PD-associated gene for leucine-rich repeat kinase, leucine-rich repeat kinase 2 (LRRK2), is highly expressed in brain regions that are involved with nonmotor functions, including the neocortex and hippocampus, but whether mutant LRRK2 contributes to neuronal dysfunction in these regions is unknown. Here, we use bacterial artificial chromosome transgenic mouse models of LRRK2 to explore potential nonmotor mechanisms of PD. Through electrophysiological analysis of the Schaffer collateral–CA1 synapse in dorsal hippocampus, we find that overexpression of LRRK2-G2019S increases basal synaptic efficiency through a postsynaptic mechanism, and disrupts long-term depression. Furthermore, these effects of the G2019S mutation are age dependent and can be normalized by acute inhibition of LRRK2 kinase activity. In contrast, overexpression of wild-type LRRK2 has no effect under the same conditions, suggesting a specific phenotype for the G2019S mutation. These results identify a pathogenic function of LRRK2 in the hippocampus that may contribute to nonmotor symptoms of PD.
SIGNIFICANCE STATEMENT Parkinson's disease (PD) is among the most common neurological diseases and is best known for its adverse effects on brain regions that control motor function, resulting in tremor, rigidity, and gait abnormalities. Less well appreciated are the psychiatric symptoms experienced by many PD patients, including depression and memory loss, which do not respond well to currently available treatments for PD. Here, we describe functional effects of a common PD-linked mutation of leucine-rich repeat kinase 2 in the mouse hippocampus, an area of the brain that is responsible for encoding and retaining memories. By providing a potential mechanism for some of the cognitive symptoms produced by this mutation, our findings may lead to novel approaches for the treatment of nonmotor symptoms of PD.
Keywords: aged, electrophysiology, long-term depression, LRRK2, nonmotor deficits, transgenic
Introduction
Parkinson's disease (PD) is known for its characteristic motor function deficits, but patients also suffer from a wide variety of nonmotor symptoms that represent a significant quality-of-life issue (Hely et al., 2008; Leroi et al., 2012). These symptoms can include cognitive impairment and depression, and may result from dysfunction of interconnected systems, including the striatum, neocortex, and hippocampus (Williams-Gray et al., 2009; Kehagia et al., 2010). This concept is supported by the progression of PD through multiple brain regions as the severity of the disease increases (Braak et al., 2003). Notably, these symptoms are not improved by current dopaminergic therapies.
Given the presumed multisystem nature of the symptoms PD patients experience, it is important to explore the role of nonmotor brain regions. While the hippocampus is well known for its role in learning and memory, there is evidence for hippocampal involvement in PD (Pereira et al., 2013; Kandiah et al., 2014). Recent findings suggest that the hippocampus may play a compensatory role in some of the cognitive symptoms that PD patients experience (Nagano-Saito et al., 2014). A better understanding of the neuronal changes in the hippocampus that produce nonmotor symptoms may provide insights into the basic mechanisms of PD and lead to new therapeutic approaches.
Mutations in leucine-rich repeat kinase 2 (LRRK2) represent the most common genetic factors associated with Parkinson's disease, and the most frequent of these is G2019S, which increases LRRK2 kinase activity (Sheng et al., 2012). Unlike other genetic factors, LRRK2 mutations also are associated with idiopathic occurrences of the disease. The LRRK2-G2019S mutation is linked to nonmotor symptoms that are similar in severity to the idiopathic population, and non-PD carriers of the mutation have lower cognitive performance (Shanker et al., 2011; Alcalay et al., 2013). LRRK2 is highly expressed in brain areas receiving dopaminergic innervation, such as the striatum, hippocampus, cortex, and cerebellum (Taymans et al., 2006). LRRK2 has been shown to play a role in many aspects of neuronal function, including neurogenesis, axonal outgrowth, mitochondrial function, autophagy, and synaptic function (Shin et al., 2008; Winner et al., 2011; Matta et al., 2012; Wang et al., 2012; MacLeod et al., 2013; Sepulveda et al., 2013; Godena et al., 2014; Law et al., 2014; Parisiadou et al., 2014; Stafa et al., 2014). The protein expression pattern of LRRK2 in the brain and the prevalence of PD-associated mutations make it an attractive target in for understanding and possibly treating the nonmotor symptoms of PD.
In this study we report that the LRRK2-G2019S mutation affects plasticity in the hippocampus of aged bacterial artificial chromosome (BAC) transgenic mice. We observe that LRRK2-G2019S leads to an increase in basal synaptic efficiency and a profound reduction of long-term depression (LTD) at the Schaffer collateral–CA1 synapse in aged hippocampus, but no apparent change in presynaptic function. Further, we find that the effects of the mutation can be rescued by a LRRK2 kinase inhibitor. These results highlight the role of LRRK2 beyond the extrapyramidal system, and suggest the therapeutic potential of LRRK2 kinase inhibitors.
Materials and Methods
Mouse maintenance.
BAC transgenic heterozygous LRRK2-G2019S mice, heterozygous LRRK2 wild-type (LRRK2-Wt) mice, and nontransgenic littermates (Li et al., 2010) of either sex were maintained according to Icahn School of Medicine at Mount Sinai Institutional Animal Care and Use Committee guidelines. Except where noted, all mice used in this study were 8–12 months old.
Field electrophysiology.
Brains were removed and chilled in bubbled (95% O2, 5% CO2) ice-cold ACSF containing the following (in mm): 1.25 NaH2PO4, 3.5 KCl, 24 NaHCO3, 118 NaCl, 1.3 MgSO4, 2.5 CaCl2, 15 dextrose, pH 7.4. The hippocampus was removed and 400-μm-thick transverse slices of the dorsal hippocampus were cut on a McIlwain tissue chopper at 4°C. Slices were maintained at room temperature in a humidified, bubbled interface chamber containing ACSF for ≥2.5 h.
For recording, slices were submerged on a nylon mesh, superfused with bubbled ACSF and maintained at 31 ± 1°C. A bipolar stainless steel stimulating electrode was placed in stratum radiatum of area CA3, and field EPSPs were recorded in stratum radiatum of area CA1 using ACSF-filled pipettes (2–4 MΩ). Recordings were acquired with a Multiclamp 700B and Digidata 1440A (Molecular Devices). fEPSPs were evoked with a 100 μs stimulus delivered by IsoFlex stimulators (AMPI). All signals were low-pass filtered at 2 kHz and digitized at 10 kHz. Baseline recordings (stable for ≥20 min) were made every 30 s using stimuli giving a response equal to ∼30–40% of spike threshold. fEPSP slope was measured for all analyses. Long-term potentiation (LTP) was induced using two 100 Hz stimulus trains, each 1 s long, separated by 20 s, and delivered at an intensity giving a baseline response equal to 75% of spike threshold. LTD was induced using a 1 Hz train of 900 bursts, each burst containing three stimuli delivered at 20 Hz, at 120% of spike threshold. Paired-pulse facilitation was determined using pairs of stimuli delivered every 60 s. Each interstimulus interval was repeated three times and the resulting potentials were averaged. Post-tetanic potentiation (PTP) was induced by a single stimulus train delivered at 100 Hz, at baseline stimulation intensity. Response rundown was determined with a single 30 s, 10 Hz train, delivered at baseline stimulation intensity. The LRRK2 kinase inhibitor G1023 (Sheng et al., 2012) was resuspended in ACSF and applied in the recording bath at a final concentration of 1 μm for 30 min before recording.
Whole-cell patch electrophysiology.
With a vibratome (Leica VT1000), 200-μm-thick slices of dorsal hippocampus were prepared in ice-cold sucrose cutting solution containing the following (in mm): 1.25 Na2HPO4, 3 KCl, 25 NaHCO3, 254 sucrose, 10 dextrose, 2 MgSO4, 2 CaCl2, pH 7.4. The slices recovered while submerged at 30°C for 30 min in bubbled ACSF containing the following (in mm): 1 NaH2PO4, 2.5 KCl, 18 NaHCO3, 131 NaCl, 2 MgCl2, 2 CaCl2, 11 dextrose, pH 7.4). The slices were then kept for ≥30 min at room temperature. Recordings were obtained in submersion chambers with room temperature ACSF using a Multiclamp 700B and Digidata 1440A (Molecular Devices). Cells were visualized on an Olympus BX51W1 microscope with differential interference contrast-infrared optics, and whole-cell and perforated-patch recordings were made from pyramidal cells in the CA1 region. Signals were filtered using a 2 kHz low-pass filter and digitized at 20 kHz. All recordings were made using pipettes filled with the following (in mm): 130 Cs-methanesulfonate, 10 HEPES, 0.5 EGTA, 8 NaCl, 5 tetraethylammonium chloride, 4 Mg-ATP, 0.4 Na-GTP, 10 Na-phosphocreatine, 1 QX-314, 290–300 mOsM, pH 7.3. Perforated-patch recordings were performed by including 4 μm amphotericin in the pipette filling solution. Miniature EPSCs (mEPSCs) were recorded using whole-cell patch and events were blindly identified, using threshold-based automatic analysis followed by manual confirmation, and analyzed for amplitude and frequency in MiniAnalysis (Synaptosoft). The ratio of evoked AMPAR/NMDAR-mediated currents was determined using whole-cell patch and perforated-patch techniques in the presence of 50 μm picrotoxin. EPSCs were evoked by 100 μs stimuli delivered by a bipolar electrode in area CA3 stratum radiatum. The AMPAR current was measured at its peak (Vm = −70 mV), and the NMDAR current was measured 50 ms after the stimulus (Vm = +40 mV).
Statistics.
All comparisons were done using two-tailed, unpaired Mann–Whitney t tests or unpaired t test with Welch's correction for normal data using Prism 5.0 (Graphpad). Outlier analysis was performed using single Grubbs' test with α = 0.05.
Results
Basal synaptic efficiency is increased and LTD is reduced in aged LRRK2-G2019S BAC transgenic mice
We previously described BAC transgenic mice overexpressing LRRK2-G2019S under the control of the endogenous promoter, which displays reduced dopamine release in the striatum, but not neurodegeneration (Li et al., 2010). We used these mice to investigate the effect of the G2019S mutation on hippocampal function. Nontransgenic littermates and BAC transgenic mice overexpressing wild-type LRRK2 (LRRK2-Wt) served as controls to identify specific effects of the LRRK2-G2019S mutant. Basal efficiency was increased at Schaffer collateral–CA1 synapses in aged LRRK2-G2019S mice relative to their littermate controls (p = 0.0004), while the LRRK2-Wt mice did not differ significantly from their littermates (Fig. 1A). Strikingly, LRRK2-G2019S mice showed a profound deficit in synaptically induced long-term depression (LTD; p = 0.0140), while LRRK2-Wt mice had normal LTD (Fig. 1B). Despite the changes in basal synaptic efficiency and LTD, the G2019S mutation did not affect LTP (Fig. 1C). In slices from younger LRRK2-G2019S mice (3–6 months), basal efficiency and LTD did not differ from those of littermate controls (Fig. 1D). The previously characterized deficits in striatum of the LRRK2-G2019S mice were also age dependent, indicating a consistent role for the LRRK2 mutation in the aged mouse brain (Li et al., 2010). These results show that the LRRK2-G2019S mutation causes age-related dysfunction in basal synaptic efficiency and plasticity in the hippocampus.
Figure 1.

LRRK2-G2019S increases basal synaptic efficiency and prevents LTD in aged mice. A, LRRK2-G2910S mice, but not LRRK2-Wt mice, show increased basal synaptic efficiency. Input–output relationships and sample traces are shown for LRRK2-Wt mice (n = 14 slices, 7 mice) versus littermate controls (n = 15 slices, 8 mice; left), and LRRK2-G2019S mice (n = 19 slices, 8 mice) versus littermate controls (n = 20 slices, 8 mice; right; p = 0.0231). B, LRRK2-G2019S interferes with low-frequency stimulation induced LTD. LRRK2-Wt (n = 6 slices, 3 mice) versus control (n = 5 slices, 3 mice) mice, and LRRK2-G2019S (n = 7 slices, 4 mice) versus control (n = 8 slices, 6 mice) mice (p = 0.0140). C, LTP is not affected by LRRK2-G2019S. LRRK2-Wt (n = 6 slices, 3 mice) mice versus controls (n = 5 slices, 3 mice) and LRRK2-G2019S mice (n = 8 slices, 4 mice) versus controls (n = 6 slices, 3 mice). Mice that were analyzed in A–C were 8–12 months old. D, Basal synaptic efficiency (left) and LTD (right) are intact in animals aged 3–5 months. LRRK2-G2019S mice (n = 13 slices, 5 mice) versus controls (n = 13 slices, 4 mice). Scale bars: 0.5 mV, 10 ms.
The LRRK2-G2019S mutation does not affect presynaptic function
It has been suggested that LRRK2 plays a role in presynaptic function by regulating synaptic machinery and neurotransmitter release (Piccoli et al., 2011; Matta et al., 2012; Cirnaru et al., 2014; Beccano-Kelly et al., 2015). To determine whether the LRRK2-G2019S mutation alters presynaptic function in the adult hippocampus, several phenomena that depend on presynaptic mechanisms were examined. First, paired-pulse facilitation was measured, and no difference was seen between the transgenic (LRRK2-G2019S or LRRK2-Wt) and littermate control lines (Fig. 2A). To test for presynaptic effects that might be revealed only by more intense stimulation, PTP was examined. Here too, neither the amplitude of PTP nor its persistence was affected by overexpression of LRRK2-G2019S or LRRK2-Wt (Fig. 2B). Finally, we tested whether LRRK2-G2019S affects the rate of presynaptic vesicular depletion during a stimulus train (Rosahl et al., 1995). Neither LRRK2-G2019S nor LRRK2-Wt mice showed any difference in the rate of depletion when compared with their littermate controls (Fig. 2C). Together, these results indicate that presynaptic changes are unlikely to underlie the altered hippocampal synaptic function caused by the G2019S mutation.
Figure 2.
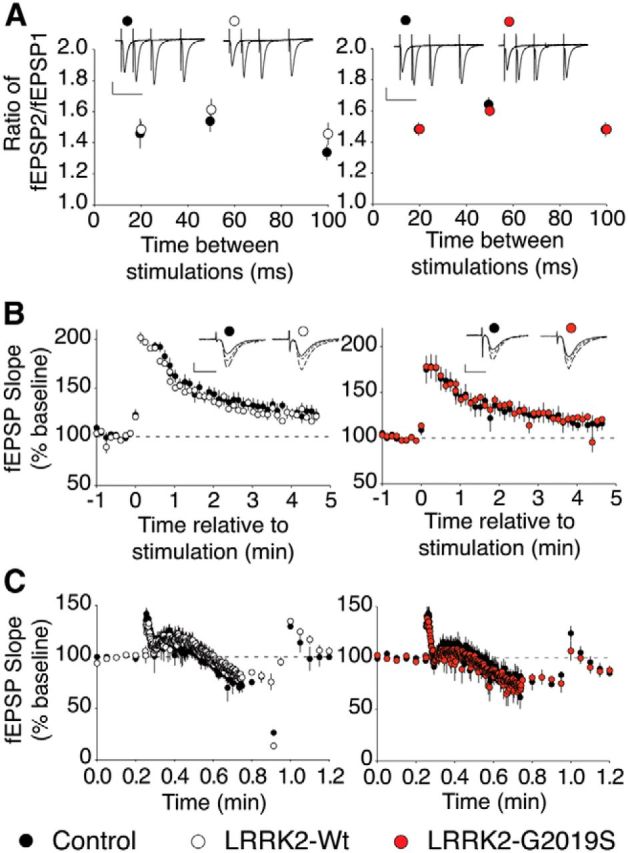
Presynaptic function is intact in aged LRRK2-G2019S mice. A, the paired-pulse ratio is not affected by LRRK2-G2019S or LRRK2-Wt. LRRK2-Wt mice (n = 6 slices, 3 mice) versus controls (n = 7 slices, 3 mice) and LRRK2-G2019S mice (n = 9 slices, 5 mice) versus controls (n = 11 slices, 6 mice). Scale bars: 0.5 mV, 50 ms. B, LRRK2-G2019S does not affect PTP. LRRK2-Wt (n = 4 slices, 3 mice) versus controls (n = 6 slices, 3 mice) and LRRK2-G2019S (n = 4 slices, 3 mice) versus controls (n = 6 slices, 3 mice). Scale bars: 0.5 mV, 10 ms. C, Synaptic rundown is not altered in LRRK2-Wt or LRRK2-G2019S animals. LRRK2-Wt mice (n = 6 slices, 4 mice) versus controls (n = 6 slices, 3 mice) and LRRK2-G2019S (n = 4 slices, 3 mice) mice versus controls (n = 6 slices, 3 mice).
The LRRK2-G2019S mutation enhances AMPAR-mediated synaptic transmission through a postsynaptic mechanism
One possible explanation for the effects of LRRK2-G2019S in field recordings is that postsynaptic endocytosis of AMPA-type glutamate receptors (AMPARs) is disrupted, leading to increased basal synaptic efficiency and a loss of LTD (Morishita et al., 2005). To test this possibility, we first measured EPSCs in whole-cell recordings of CA1 pyramidal cells. We found that LRRK2-G2019S did not alter the frequency of mEPSCs (Fig. 3B), in agreement with the evidence from field recordings that presynaptic function is intact. Surprisingly, mEPSC amplitude also was unaffected by G2019S, suggesting that the synaptic phenotype might be lost in whole-cell recordings (Fig. 3C). Indeed, the ratio of AMPAR-mediated to NMDAR-mediated evoked EPSCs, which provides a measure of postsynaptic changes in glutamatergic signaling, did not differ between neurons from LRRK2-G2019S mice and littermate controls (Fig. 3D). This indicated that the synaptic G2019S phenotype might depend on a diffusible intracellular factor that is lost during whole-cell patch recording. To reduce dialysis of the intracellular compartment, we performed additional recordings using the perforated-patch method. Under these conditions, the G2019S mutation increased the AMPAR/NMDAR ratio by ∼2-fold (Fig. 3E), consistent with the increase in synaptic efficiency observed in field recordings.
Figure 3.

LRRK2-G2019S enhances synaptic transmission through a postsynaptic mechanism. A, Sample mEPSC traces. The top trace shows currents from a control slice on an expanded time scale. Scale bars: top, 10 pA, 10 ms; middle and bottom, 10 pA, 5 s. B, C, mEPSC frequency (B) and amplitude (C) are unchanged in LRRK2-G2019S mice (n = 7 slices, 3 mice) compared with controls (n = 7 slices, 3 mice). D, The evoked AMPAR/NMDAR EPSC ratio in whole-cell patch is similar in LRRK2-G2019S (n = 11 slices, 7 mice) and control mice (n = 9 slices, 5 mice). Scale bars: top trace, 30 pA, 20 ms; bottom trace, 100 pA, 20 ms. E, The evoked AMPAR/NMDAR EPSC ratio using perforated patch is increased in LRRK2-G2019S (n = 11 slices, 6 mice) and control mice (n = 9 slices, 7 mice; p = 0.044). Scale bars: top trace, 5 pA, 20 ms; bottom trace, 5 pA, 20 ms.
Inhibition of LRRK2 kinase activity can rescue basal synaptic efficiency and LTD
Since the LRRK2-G2019S phenotype was age related, it is possible that the elevated LRRK2 kinase activity produced a gradual change in synaptic function. Alternatively, the phenotype could reflect an age-related susceptibility to high LRRK2 kinase activity, in which case acute inhibition of LRRK2 kinase might normalize synaptic function. To test this possibility, we examined synaptic basal efficiency and LTD in the presence of the LRRK2-kinase inhibitor G1023 (Genentech; Sheng et al., 2012). We found that acute treatment of the hippocampal slices with G1023 restored both synaptic efficiency and LTD to levels indistinguishable from littermate controls (Fig. 4).
Figure 4.

The LRRK2 kinase inhibitor G1023 rescues the normal synaptic phenotype in LRRK2-G2019S slices. A, Input–output curves in LRRK2-G2019S mice (n = 9 slices, 3 mice) are equivalent to control mice (n = 6 slices, 3 mice) after treatment with G1023. B, LFS-induced LTD in LRRK2-G2019S hippocampus (n = 9 slices, 3 mice) does not differ from control hippocampus (n = 5 slices, 3 mice) after treatment with G1023. Scale bars: 0.5 mV, 10 ms.
Discussion
LRRK2 regulates basal synaptic efficiency and long-term plasticity
We have shown that the LRRK2-G2019S mutation causes an increase in basal synaptic activity and a profound reduction of LTD in acute hippocampal slices from BAC transgenic mice (Fig. 1). This is, to our knowledge, the first description of an effect of LRRK2-G2019S on synaptic function in adult hippocampus. While the significance of the high expression levels of endogenous LRRK2 in hippocampal pyramidal neurons has been unclear, our data reveal a potential mechanism for the dysfunctional learning and memory observed in PD patients carrying LRRK2 mutations (Shanker et al., 2011)
Previous reports have suggested that the effects of PD-linked LRRK2 mutations may be due to changes in presynaptic neurotransmitter release through regulation of synaptic machinery, such as endophilin A and N-ethylmaleimide-sensitive factor (Li et al., 2010; Piccoli et al., 2011; Matta et al., 2012; Cirnaru et al., 2014; Beccano-Kelly et al., 2015). However, using several electrophysiological measures, we found no evidence for altered presynaptic function at the Schaffer collateral–CA1 synapse. Rather, LRRK2 was shown to play a role in postsynaptic signaling at excitatory synapses in the adult hippocampus. It is likely that specific brain regions and experimental paradigms contribute to the different synaptic phenotypes reported for LRRK2 mutations, and future detailed investigation is needed to resolve the apparent discrepancies and to gain a better understanding of LRRK2 pathophysiology. To date, much of the work on the consequences of LRRK2 mutations in adult brain has focused on the role of LRRK2 in medium spiny neurons in the striatum; in contrast, studies on other neuronal types have been mostly limited to primary neuronal cultures or invertebrate systems (Shin et al., 2008; Lee et al., 2010; Matta et al., 2012; Plowey et al., 2014). By exploring the role of LRRK2 in the adult hippocampus, we have provided new information on the physiological basis of PD-related cognitive symptoms. While the experiments described here relate specifically to the role of LRRK2-G2019S in PD, future studies of brain regions where LRRK2 is highly expressed should yield insights into the normal function of LRRK2 in the brain.
The effect of LRRK2-G2019S on basal synaptic efficiency was lost in whole-cell recordings, as indicated by an AMPAR/NMDAR EPSC ratio that did not differ from control cells. However, our data show that the mutation did increase the AMPAR/NMDAR ratio in perforated-patch recordings (Fig. 3). It is likely that the synaptic LRRK2-G2019S phenotype is mediated through a postsynaptic diffusible factor, which could be one of many signaling molecules upstream or downstream of LRRK2. Regardless of the identity of the signaling molecule, the acute loss of the phenotype upon whole-cell recording suggests that ongoing LRRK2-dependent signaling is required for the synaptic changes produced by the G2019S mutation. This conclusion agrees with our finding that acute treatment with a LRRK2 kinase inhibitor can rescue G2019S synaptic dysfunction.
The effects of LRRK2-G2019S that we observed in field and perforated-patch recordings are consistent with a scenario in which AMPAR endocytosis is reduced relative to exocytosis, resulting in aberrant accumulation of synaptic receptors (in agreement with enhanced basal synaptic efficiency; Fig. 1A) and deficient AMPAR removal, which is required for LTD (Fig. 1B). Among the mechanisms that could produce an imbalance between AMPAR insertion and removal are changes in the phosphorylation state of AMPARs, or in the function of proteins required for normal AMPAR cycling. Interestingly, LRRK2 has been implicated in AMPAR phosphorylation, and recently was reported to interact with proteins important for vesicle trafficking (Heo et al., 2010; MacLeod et al., 2013; Cirnaru et al., 2014). Another potential mechanism is suggested by the increases in spine density observed in cortical cultures overexpressing mutant LRRK2 (Plowey et al., 2014), but this effect apparently does not extend to the adult hippocampus since we found no increase in mEPSC frequency.
While it has been previously reported that LTD in the striatum is disturbed in a different line of LRRK2 transgenic mice (Chou et al., 2014), our study differs in several key ways in addition to the brain region studied. In the mice generated by Chou et al., the LRRK2 transgene is driven by a promoter that increases LRRK2 expression in the substantia nigra, among other areas, while the mice used in our work express LRRK2 under the endogenous promoter (Li et al., 2010). The two studies also differ in experimental technique: we used field recording to examine late forms of long-term plasticity in the hippocampus, while Chou et al. examined LTD deficits in whole-cell recordings.
LRRK2-G2019S effects on synaptic function are age related and acutely dependent on LRRK2 kinase activity
Our finding of normal basal efficiency and LTD in younger animals indicates that the synaptic consequences of LRRK2-G2019S are age-dependent. This result is in line with phenotypes reported previously in this BAC transgenic line and other LRRK2 transgenic animals models (Li et al., 2010), and suggests that the effects of LRRK2-G2019S interact with age-dependent processes to produce a phenotype. By rescuing the dysfunctional synaptic activity in aged G2019S mice with acute treatment by a LRRK2 kinase inhibitor, we show that such a drug may have the potential to ameliorate functional deficits induced by this mutation. The observation that hippocampal synaptic dysfunction in LRRK2-G2019S mice can be rapidly reversed pharmacologically, despite the age-dependent nature of the dysfunction, strongly suggests that aging makes these synapses more vulnerable to excessive LRRK2 kinase activity.
Together, our data indicate that LRRK2 plays a role in hippocampal synaptic physiology, and that an increase in kinase activity due to the LRRK2-G2019S mutation disrupts hippocampal function in aged mice, consistent with evidence for deficient hippocampus-mediated behavior in mice that overexpress LRRK2-G2019S (Beccano-Kelly et al., 2015). While more work must be done to determine the mechanisms involved, these findings are an important contribution to our understanding of LRRK2 function outside of the striatum, and help elucidate the role of LRRK2 in nonmotor symptoms that reduce the quality of life in patients suffering from PD.
Footnotes
This work was supported by the National Institutes of Health Grant 1R01NS072359 (R.B.D. and Z.Y.). We thank Nikos Tzavaras for his outstanding electrophysiological assistance, to the members of the Blitzer and Yue laboratories for valuable discussions, and to the reviewers for their insightful suggestions.
The authors declare no competing financial interests.
References
- Alcalay RN, Mirelman A, Saunders-Pullman R, Tang MX, Mejia Santana H, Raymond D, Roos E, Orbe-Reilly M, Gurevich T, Bar Shira A, Gana Weisz M, Yasinovsky K, Zalis M, Thaler A, Deik A, Barrett MJ, Cabassa J, Groves M, Hunt AL, Lubarr N, et al. Parkinson disease phenotype in Ashkenazi Jews with and without LRRK2 G2019S mutations. Mov Disord. 2013;28:1966–1971. doi: 10.1002/mds.25647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beccano-Kelly DA, Volta M, Munsie LN, Paschall SA, Tatarnikov I, Co K, Chou P, Cao LP, Bergeron S, Mitchell E, Han H, Melrose HL, Tapia L, Raymond LA, Farrer MJ, Milnerwood AJ. LRRK2 overexpression alters glutamatergic presynaptic plasticity, striatal dopamine tone, postsynaptic signal transduction, motor activity and memory. Hum Mol Genet. 2015;24:1336–1349. doi: 10.1093/hmg/ddu543. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/S0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Chou JS, Chen CY, Chen YL, Weng YH, Yeh TH, Lu CS, Chang YM, Wang HL. (G2019S) LRRK2 causes early-phase dysfunction of SNpc dopaminergic neurons and impairment of corticostriatal long-term depression in the PD transgenic mouse. Neurobiol Dis. 2014;68:190–199. doi: 10.1016/j.nbd.2014.04.021. [DOI] [PubMed] [Google Scholar]
- Cirnaru MD, Marte A, Belluzzi E, Russo I, Gabrielli M, Longo F, Arcuri L, Murru L, Bubacco L, Matteoli M, Fedele E, Sala C, Passafaro M, Morari M, Greggio E, Onofri F, Piccoli G. LRRK2 kinase activity regulates synaptic vesicle trafficking and neurotransmitter release through modulation of LRRK2 macro-molecular complex. Front Mol Neurosci. 2014;7:49. doi: 10.3389/fnmol.2014.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godena VK, Brookes-Hocking N, Moller A, Shaw G, Oswald M, Sancho RM, Miller CC, Whitworth AJ, De Vos KJ. Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat Commun. 2014;5:5245. doi: 10.1038/ncomms6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG. The Sydney multicenter study of Parkinson's disease: the inevitability of dementia at 20 years. Mov Disord. 2008;23:837–844. doi: 10.1002/mds.21956. [DOI] [PubMed] [Google Scholar]
- Heo HY, Kim KS, Seol W. Coordinate regulation of neurite outgrowth by LRRK2 and its interactor, Rab5. Exp Neurobiol. 2010;19:97–105. doi: 10.5607/en.2010.19.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandiah N, Zainal NH, Narasimhalu K, Chander RJ, Ng A, Mak E, Au WL, Sitoh YY, Nadkarni N, Tan LC. Hippocampal volume and white matter disease in the prediction of dementia in Parkinson's disease. Parkinsonism Relat Disord. 2014;20:1203–1208. doi: 10.1016/j.parkreldis.2014.08.024. [DOI] [PubMed] [Google Scholar]
- Kehagia AA, Barker RA, Robbins TW. Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson's disease. Lancet Neurol. 2010;9:1200–1213. doi: 10.1016/S1474-4422(10)70212-X. [DOI] [PubMed] [Google Scholar]
- Law BM, Spain VA, Leinster VH, Chia R, Beilina A, Cho HJ, Taymans JM, Urban MK, Sancho RM, Blanca Ramírez M, Biskup S, Baekelandt V, Cai H, Cookson MR, Berwick DC, Harvey K. A direct interaction between leucine-rich repeat kinase 2 and specific β-tubulin isoforms regulates tubulin acetylation. J Biol Chem. 2014;289:895–908. doi: 10.1074/jbc.M113.507913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Liu HP, Lin WY, Guo H, Lu B. LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J Neurosci. 2010;30:16959–16969. doi: 10.1523/JNEUROSCI.1807-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroi I, McDonald K, Pantula H, Harbishettar V. Cognitive impairment in Parkinson disease: impact on quality of life, disability, and caregiver burden. J Geriatr Psychiatry Neurol. 2012;25:208–214. doi: 10.1177/0891988712464823. [DOI] [PubMed] [Google Scholar]
- Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J Neurosci. 2010;30:1788–1797. doi: 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, MacCabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron. 2013;77:425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, De Bock PJ, Morais VA, Vilain S, Haddad D, Delbroek L, Swerts J, Chávez-Gutiérrez L, Esposito G, Daneels G, Karran E, Holt M, Gevaert K, Moechars DW, De Strooper B, et al. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron. 2012;75:1008–1021. doi: 10.1016/j.neuron.2012.08.022. [DOI] [PubMed] [Google Scholar]
- Morishita W, Marie H, Malenka RC. Distinct triggering and expression mechanisms underlie LTD of AMPA and NMDA synaptic responses. Nat Neurosci. 2005;8:1043–1050. doi: 10.1038/nn1506. [DOI] [PubMed] [Google Scholar]
- Nagano-Saito A, Habak C, Mejía-Constaín B, Degroot C, Monetta L, Jubault T, Bedetti C, Lafontaine AL, Chouinard S, Soland V, Ptito A, Strafella AP, Monchi O. Effect of mild cognitive impairment on the patterns of neural activity in early Parkinson's disease. Neurobiol Aging. 2014;35:223–231. doi: 10.1016/j.neurobiolaging.2013.06.025. [DOI] [PubMed] [Google Scholar]
- Parisiadou L, Yu J, Sgobio C, Xie C, Liu G, Sun L, Gu XL, Lin X, Crowley NA, Lovinger DM, Cai H. LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat Neurosci. 2014;17:367–376. doi: 10.1038/nn.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira JB, Junqué C, Bartrés-Faz D, Ramírez-Ruiz B, Marti MJ, Tolosa E. Regional vulnerability of hippocampal subfields and memory deficits in Parkinson's disease. Hippocampus. 2013;23:720–728. doi: 10.1002/hipo.22131. [DOI] [PubMed] [Google Scholar]
- Piccoli G, Condliffe SB, Bauer M, Giesert F, Boldt K, De Astis S, Meixner A, Sarioglu H, Vogt-Weisenhorn DM, Wurst W, Gloeckner CJ, Matteoli M, Sala C, Ueffing M. LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J Neurosci. 2011;31:2225–2237. doi: 10.1523/JNEUROSCI.3730-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowey ED, Johnson JW, Steer E, Zhu W, Eisenberg DA, Valentino NM, Liu YJ, Chu CT. Mutant LRRK2 enhances glutamatergic synapse activity and evokes excitotoxic dendrite degeneration. Biochim Biophys Acta. 2014;1842:1596–1603. doi: 10.1016/j.bbadis.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–493. doi: 10.1038/375488a0. [DOI] [PubMed] [Google Scholar]
- Sepulveda B, Mesias R, Li X, Yue Z, Benson DL. Short- and long-term effects of LRRK2 on axon and dendrite growth. PLoS One. 2013;8:e61986. doi: 10.1371/journal.pone.0061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanker V, Groves M, Heiman G, Palmese C, Saunders-Pullman R, Ozelius L, Raymond D, Bressman S. Mood and cognition in leucine-rich repeat kinase 2 G2019S Parkinson's disease. Mov Disord. 2011;26:1875–1880. doi: 10.1002/mds.23746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Z, Zhang S, Bustos D, Kleinheinz T, Le Pichon CE, Dominguez SL, Sollanoy HO, Drummond J, Zhang X, Ding X, Cai F, Song Q, Li X, Yue Z, van der Brug MP, Burdick DJ, Gunzer Toste J, Chen H, Liu X, Estrada AA, et al. Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med. 2012;4:164ra164. doi: 10.1126/scitranslmed.3004485. [DOI] [PubMed] [Google Scholar]
- Shin N, Jeong H, Kwon J, Heo HY, Kwon JJ, Yun HJ, Kim CH, Han BS, Tong Y, Shen J, Hatano T, Hattori N, Kim KS, Chang S, Seol W. LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res. 2008;314:2055–2065. doi: 10.1016/j.yexcr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Stafa K, Tsika E, Moser R, Musso A, Glauser L, Jones A, Biskup S, Xiong Y, Bandopadhyay R, Dawson VL, Dawson TM, Moore DJ. Functional interaction of Parkinson's disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum Mol Genet. 2014;23:2055–2077. doi: 10.1093/hmg/ddt600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taymans JM, Van den Haute C, Baekelandt V. Distribution of PINK1 and LRRK2 in rat and mouse brain. J Neurochem. 2006;98:951–961. doi: 10.1111/j.1471-4159.2006.03919.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, Perry G, Casadesus G, Zhu X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet. 2012;21:1931–1944. doi: 10.1093/hmg/dds003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Gray CH, Evans JR, Goris A, Foltynie T, Ban M, Robbins TW, Brayne C, Kolachana BS, Weinberger DR, Sawcer SJ, Barker RA. The distinct cognitive syndromes of Parkinson's disease: 5 year follow-up of the CamPaIGN cohort. Brain. 2009;132:2958–2969. doi: 10.1093/brain/awp245. [DOI] [PubMed] [Google Scholar]
- Winner B, Melrose HL, Zhao C, Hinkle KM, Yue M, Kent C, Braithwaite AT, Ogholikhan S, Aigner R, Winkler J, Farrer MJ, Gage FH. Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice. Neurobiol Dis. 2011;41:706–716. doi: 10.1016/j.nbd.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]