

Abstract
Neuronal atrophy in neurodegenerative diseases is commonly viewed as an early event in a continuum that ultimately results in neuronal loss. In a mouse model of the polyglutamine disorder spinocerebellar ataxia type 1 (SCA1), we tested the hypothesis that cerebellar Purkinje neuron atrophy serves an adaptive role rather than being simply a nonspecific response to injury. In acute cerebellar slices from SCA1 mice, we find that Purkinje neuron pacemaker firing is initially normal but, with the onset of motor dysfunction, becomes disrupted, accompanied by abnormal depolarization. Remarkably, subsequent Purkinje cell atrophy is associated with a restoration of pacemaker firing. The early inability of Purkinje neurons to support repetitive spiking is due to unopposed calcium currents resulting from a reduction in large-conductance calcium-activated potassium (BK) and subthreshold-activated potassium channels. The subsequent restoration of SCA1 Purkinje neuron firing correlates with the recovery of the density of these potassium channels that accompanies cell atrophy. Supporting a critical role for BK channels, viral-mediated increases in BK channel expression in SCA1 Purkinje neurons improves motor dysfunction and partially restores Purkinje neuron morphology. Cerebellar perfusion of flufenamic acid, an agent that restores the depolarized membrane potential of SCA1 Purkinje neurons by activating potassium channels, prevents Purkinje neuron dendritic atrophy. These results suggest that Purkinje neuron dendritic remodeling in ataxia is an adaptive response to increases in intrinsic membrane excitability. Similar adaptive remodeling could apply to other vulnerable neuronal populations in neurodegenerative disease.
SIGNIFICANCE STATEMENT In neurodegenerative disease, neuronal atrophy has long been assumed to be an early nonspecific event preceding neuronal loss. However, in a mouse model of spinocerebellar ataxia type 1 (SCA1), we identify a previously unappreciated compensatory role for neuronal shrinkage. Purkinje neuron firing in these mice is initially normal, but is followed by abnormal membrane depolarization resulting from a reduction in potassium channels. Subsequently, these electrophysiological effects are counteracted by cell atrophy, which by restoring normal potassium channel membrane density, re-establishes pacemaker firing. Reversing the initial membrane depolarization improved motor function and Purkinje neuron morphology in the SCA1 mice. These results suggest that Purkinje neuron remodeling in ataxia is an active compensatory response that serves to normalize intrinsic membrane excitability.
Keywords: ataxia, atrophy, cerebellum, channel, purkinje neuron, SCA1
Introduction
Early events in neurodegeneration are poorly understood. Although neurodegenerative disorders ultimately end in neuronal loss, neurons show changes in morphology, including a reduction in dendritic spines, and a significant attenuation of the dendritic tree preceding cell death (Patt et al., 1991; Scott, 1993; Mavroudis et al., 2011). These early changes in neuronal morphology are viewed as an intermediate step in the ultimate loss of neurons. Neuronal remodeling is, however, a normal mechanism for neurons to regulate their plasticity. Dendrites of hippocampal neurons increase the number of spines when there is a loss of synaptic activity, as a homeostatic mechanism for a reduction in synaptic input (Kirov and Harris, 1999). Conversely, the normal pruning of excessive dendritic branches in Drosophila neurons occurs in response to segmental increases in intrinsic dendritic excitability (Kanamori et al., 2013). CNS neurons typically die rapidly after injury (Morimoto, 2012). Yet, in neurodegenerative disorders, despite presumed toxicity from proteotoxic or other insults, neurons survive for extended periods of time. In inherited cerebellar ataxia, as in other neurodegenerative disorders, cerebellar Purkinje neurons display a reduced dendritic arborization and decreased numbers of spiny branchlets before their death (Ferrer et al., 1994). It is unclear what purpose is served by these alterations in neuronal morphology. We considered the idea that Purkinje neuron atrophy in ataxia represents a compensatory, homeostatic response to increases in intrinsic membrane excitability.
The autosomal dominant spinocerebellar ataxias are a group of inherited disorders characterized by degeneration of the cerebellum and its associated pathways. Spinocerebellar ataxia type 1 (SCA1), one of the best characterized dominantly inherited ataxias (Orr and Zoghbi, 2001; Zoghbi and Orr, 2009), results from an expanded glutamine-encoding CAG repeat in the respective disease gene. As in many of the inherited ataxias, the disease protein in SCA1, ATXN1 (ataxin-1), is widely expressed in neuronal and non-neural tissues (Servadio et al., 1995), yet SCA1 causes preferential degeneration of cerebellar and brainstem neurons (Dürr et al., 1996). Is there something unique about these neurons that make them preferentially vulnerable? Examining the genetic causes of several other cerebellar ataxias suggests that this may be the case. A subset of inherited ataxias results from mutations in ion channel genes, important regulators of neuronal excitability. Mutations in the potassium channels cause SCA13 (Waters et al., 2006) and SCA19/22 (Duarri et al., 2012; Lee et al., 2012), mutations in calcium channel genes cause both SCA6 and episodic ataxia type 2 (Ophoff et al., 1996), and result in late onset progressive ataxia with cerebellar atrophy. These studies establish that ion-channel dysfunction can result in ataxia associated with cerebellar degeneration. These data also suggest a corollary: that in other degenerative ataxias not directly due to ion-channel mutations, ion channel dysfunction may play a role in disease pathogenesis. Here we investigate the electrophysiological underpinnings of neuronal dysfunction in a mouse model of the inherited ataxia, SCA1. We suggest that aberrant changes in potassium channel expression are a major driver of pathology and that these changes trigger a homeostatic response that results in neuronal atrophy.
Materials and Methods
Mice.
All animal procedures were approved by the University of Michigan Committee on the Use and Care of Animals. Mice used in the study include homozygous SCA1[82Q] transgenic mice that overexpress mutant human ATXN1 in cerebellar Purkinje neurons under the Purkinje neuron-specific murine Pcp2 (L7) promoter, maintained on an FVB background (Burright et al., 1995; Oz et al., 2010), wild-type FVB mice, or SCA1[30Q] transgenic mice that overexpress normal human ATXN1 under the Pcp2 (L7) promoter as described previously (Burright et al., 1995). Mice of either sex were used in these studies.
Western blotting.
Membrane enriched fractions of cerebellum were prepared following rapid dissection of cerebella in ice-cold PBS. Cerebella were homogenized in a detergent-free buffer containing 50 mm Tris-HCl, 10 mm EGTA and protease inhibitors (Roche). Following low speed centrifugation at 1500 × g, samples were centrifuged at 178,000 × g (Beckman Coulter). The supernatant was discarded and the membrane pellet was resuspended in fresh buffer. For immunoblotting, samples were resuspended in Laemmli buffer, electrophoresed, blotted, and probed with the BK channel antibody (1:200, mouse monoclonal antibody), followed by HRP-labeled goat anti-mouse IgG (Jackson ImmunoResearch Laboratories) and chemiluminescent detection (Thermo Scientific, Pierce Protein Biology). BK channel monoclonal antibodies were obtained from the UC Davis/NIH NeuroMab Facility, supported by NIH Grant U24NS050606 and maintained by the Department of Neurobiology, Physiology and Behavior, College of Biological Sciences, University of California, Davis.
Mass spectrometry.
The flufenamic acid (FFA) concentration in brain samples were analyzed by LC/MS. Following sample preparation by solid phase extraction as reported previously (Bakkali et al., 1999), ultra-high performance liquid chromatography (UPLC)/electrospray ionization-mass spectrometry analysis was performed with a Waters Acquity UPLC coupled to a ThermoFisher TSQ Quantum Access MAX mass spectrometer. The mobile phase consisted of methanol:water (80:20 v/v) with 0.2% formic acid. With the column temperature maintained at 24°C, FFA eluted at 1.5 min from the C18 column (Acquity UPLC BEH C18 1.7 μm; 2.1 × 50 mm) and was then detected and quantified by electrospray ionization MS/MS (capillary temperature 203°C, vaporizer temperature 300°C, spray voltage 3000 mV, collision energy 19 V, parent mass 282.066 m/z; Product Mass: 264.008). The limit of detection was 1 nm.
RNA isolation and quantitative real-time PCR.
Mice were killed following anesthesia with isoflurane, and cerebella were removed and flash-frozen in liquid nitrogen. Tissue was stored at −80°C until the time of processing. Total RNA from each mouse cerebellum was extracted using Trizol Reagent (Invitrogen) and subsequently purified using the RNeasy mini kit (Qiagen) following the manufacturer's instructions. cDNA was synthesized from 1.5 μg of purified RNA using the iScript cDNA synthesis kit (Bio-Rad). Quantitative real-time PCR assays were performed using the iQ SYBR Green Supermix (Bio-Rad) in a MyiQ Single Color Real-Time PCR Detection System (Bio-Rad), with each reaction performed at a 20 μl sample volume in an iCycler iQ PCR 96-well Plate (Bio-Rad) sealed with Microseal optical sealing tape (Bio-Rad). The relative amount of transcript mRNA was determined using the comparative Ct method for quantitation (Livak and Schmittgen, 2001), with gapdh or actb mRNA serving as the reference gene. Ct values for each sample were obtained in triplicate and averaged for statistical comparisons. The primers used for qRT-PCR are listed as follows: (1) kcnma1 N-terminus, (Forward: AGC CAA CGA TAA GCT GTG GT), (Reverse: AAT CTC AAG CCA AGC CAA CT) kcnma1 C-terminus, (Forward: GGG CCA AGA AAA GAA ATG GT), (Reverse: 5′–GAT CAG GCT GCT TGT GGA TT); (2) actb (Forward: CGG TTC CGA TGC CCT GAG GCT CTT), (Reverse: CGT CAC ACT TCA TGA TGG AAT TGA), and (3) Gapdh (Forward: TCT GAC GTG CCG CCT GGA GA), (Reverse: GAG AGC AAT GCC AGC CCC GG).
RNA sequencing.
RNA-seq studies used whole-cerebellar RNA from three biological replicates for each genotype at 5 and 12 weeks. RNA was extracted from the cerebellum. Library creation was completed using oligo-dT purification of polyadenylated RNA, and reverse-transcribed to create cDNA. The library was size selected (220 bp), normalized, pooled, and sequenced on an Illumina GAIIX using a 76 nt paired-end read strategy. Gene expression analysis with the Tuxedo pipeline was performed using the Galaxy platform hosted by the University of Minnesota Supercomputing Institute.
Immunofluorescence and Image analysis.
Mice were anesthetized with isoflurane and brains were removed, fixed in 1% paraformaldehyde for 1 h, immersed in 30% sucrose in PBS and sectioned on a cryostat. 14 μm parasagittal sections were processed for immunohistochemistry. Purkinje cells were labeled with anti-calbindin antibody (Swant 1:1000, or Cell Signaling Technologies 1:200) and an appropriate secondary goat anti-mouse AlexaFluor antibody (Life Technologies Invitrogen). Sections were imaged using a FV500 Olympus Confocal Microscope and single-plane images were obtained. Measurements of molecular layer thickness were made for each section, 100 μm from the depth of the primary fissure as previously described (Duvick et al., 2010). Purkinje neuron cell counts were obtained as the total number of neurons in each lobule in two sections from each mouse. Images were obtained using an Olympus FV500 Confocal Microscope with 60× magnification and were analyzed using ImageJ. Dual calbindin (rabbit, 1: 200, Cell Signaling Technology) and BK channel (1:500, mouse monoclonal antibodies were obtained from the UC Davis/NIH NeuroMab facility) immunostaining was performed. Measurement of staining intensity was performed in the cerebellar molecular layer. To eliminate selection bias, an area of strong Calbindin staining was identified for each image, and the corresponding region was analyzed for BK staining. Average staining intensity was measured in a circle of uniform area in each image. Approximately 30 images were analyzed for each genotype at each time point. The intensity values and the ratios were averaged by age and genotype, statistical analysis was performed using a one-way ANOVA, with a Bonferroni post hoc test. To quantify GIRK1 (Alomone Labs) staining, Purkinje cells that had a rim of GIRK1 staining were counted. Approximately 25 images were analyzed for each genotype at each time point.
Phenotype analysis.
Motor coordination was evaluated using a rotarod. The study was powered to detect a 25% improvement in motor performance, and estimated to require at least eight mice in each group. The groups were balanced with respect to gender, age, and number at the outset of the study. Approximately 1 week after virus injection, all mice were handled for four consecutive days. Once handling was complete, mice were trained for four consecutive days on the rotarod. The first 3 d were done on an accelerated protocol (4 to 40 rpm, at a rate of 0.12 rpm/s) and the last day was done at a constant speed of 24 rpm. Following the training period, mice were tested on the rotarod at a constant speed of 24 rpm on 4 consecutive days, with four trials per day. Latency score was recorded as the time taken before the animal either fell off the bar or if an animal made three full rotations on the rotating rod, to a maximum time of 300 s. The tester remained blinded to treatment condition. Performance was analyzed with a two-way repeated-measures ANOVA.
Viral vectors.
pBScMXT containing mSlo1 MBr5/3 (BK channel) was a gift from Larry Salkoff (Washington University in St. Louis, St. Louis, MO) (Addgene plasmid no. 16149; Butler et al., 1993) and cloned into a pAAVmcsCMV plasmid under a cytomegalovirus promoter. The pAAVmcsCMV plasmid was obtained from the University of Iowa vector core. Recombinant AAV serotype 2/5 vectors (AAV.mSlo1 and AAV.GFP) were generated by the University of Iowa Vector Core facility (http://www.medicine.uiowa.edu/vectorcore/) as previously described (Urabe et al., 2002). AAV vectors were resuspended in buffer, and titers (viral genomes/ml) were determined by qPCR.
Stereotaxic cerebellar viral delivery.
Stereotaxic administration of AAV2/5 was performed on 3.5- to 4-week-old ATXN1[82Q] transgenic mice placed under anesthesia using a mixture of O2 and isoflurane (dosage 4% for induction, 1.5% maintenance). Mice received bilateral intracerebellar injections (2 sites/hemisphere) of virus. For each injection, ∼5.0 × 1012 vg/ml of virus (1–3 μl) was delivered to the medial or lateral cerebellar nucleus at an infusion rate of 0.5 μl/min using a 10 μl Hamilton syringe (BD Biosciences). One minute after the infusion was completed, the micropipette was retracted 0.3 mm and allowed to remain in place for 4 min before complete removal from the mouse brain. Anterior–posterior coordinates were calculated separately for medial and lateral injection into each cerebellar hemisphere. The coordinates for the medial injection were −6.4 mm anterior–posterior, ±1.3 mm medial-lateral, and 1.9 mm dorsal-ventral as measured from bregma. The coordinates for the lateral injection were −6.0 mm anterior–posterior, ± 2.0 mm medial-lateral, and 2.2 mm dorsal-ventral as measured from bregma.
Cannula implantation.
Alzet osmotic pumps were primed per the manufacturer's protocol. The osmotic pump and cannula system were placed using a stereotaxic frame. The pump was inserted beneath the skin of the mouse and the cannula was directed to the midline cerebellum. Pumps were filled with 55 mm 2-hydroxypropyl β-cyclodextrin, and 0.1% fast green (vehicle) with or without 100 mm FFA. All FFA and vehicle tissue was immunostained with Calbindin antibodies. Measurements were made on the anterior side of lobule II and the posterior side of lobule III. The observer making measurements was blinded to the treatment. Measurements were made at the midpoint between the base and tip of each lobule.
Chemicals.
Reagents and chemical were obtained from Sigma-Aldrich unless otherwise specified. Iberiotoxin was obtained from Alomone Labs, apamin and tetrodotoxin (TTX) were obtained from Tocris Bioscience.
Electrophysiology
Preparation of brain slices for electrophysiological recordings.
Mice were anesthetized by isofluorane inhalation, decapitated, and the brains were chilled in ice-cold cutting solution containing the following (in mm): 87 NaCl, 2.5 KCl, 25 NaHCO3, 1 NaH2PO4, 0.5 CaCl2, 7 MgCl2, 75 sucrose, and 10 glucose, bubbled with 5% CO2/95% O2. Parasagittal cerebellar slices (300 μm) were cut using a vibratome. Slices were incubated at 33°C in artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 3.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, and 10 glucose, bubbled with 5% CO2 + 95% O2 (carbogen) for 45 min. Slices were stored at room temperature until use. Slices were then placed in a recording chamber and continuously perfused with carbogen-bubbled ACSF at 33°C with a flow rate of 2–3 mls/min.
Whole-cell recordings.
Purkinje neurons were identified for patch-clamp recordings in parasagittal cerebellar slices. Borosilicate glass patch pipettes (with resistances of 3–5 MΩ) were filled with internal recording solution containing the following (in mm): 119 K Gluconate, 2 Na gluconate, 6 NaCl, 2 MgCl2, 0.9 EGTA, 10 HEPES, 14 Tris-phosphocreatine, 4 MgATP, 0.3 tris-GTP, pH 7.3. Whole-cell recordings at 33°C were made in ACSF 1–5 h after slice preparation using an Axopatch 200B amplifier, Digidata 1440A interface, and pClamp-10 software (MDS Analytical Technologies) as previously described (Shakkottai et al., 2011). Voltage data were acquired in the fast current-clamp mode of the amplifier and filtered at 2 kHz. The fast current-clamp mode is necessary to reduce distortion of action potentials observed when patch-clamp amplifiers are used in current-clamp mode (Magistretti et al., 1996; Swensen and Bean, 2003). Series resistance was compensated up to 60% when possible. Cells were rejected if the series resistance changed by >20% in the course of the recording or if it exceeded 15 MΩ. In the majority of recordings, series resistance was <10 MΩ. Total cell capacitance was calculated from measurement of the area under current transients evoked from a 10 mV depolarizing step from −80 mV, following correction for input resistance. Data were digitized at 100 kHz. Voltage traces were corrected for a 10 mV liquid junction potential. To determine the effect of pharmacologic agents on firing, agents were perfused on slices for at least 8 min or until changes in firing reached equilibrium. Predominant firing frequency was calculated from the shortest interspike interval. The coefficient-of-variation was calculated as the ratio of the SD of the interspike interval divided by the mean interspike interval. To determine the amplitude of barium and FFA-sensitive currents, traces obtained following application of the agent, were subtracted from baseline current traces.
Statistical analysis.
Statistical significance for electrophysiology was assessed by either an unpaired Student's t test, Fisher's exact test, or one-way ANOVA. A paired Student's t test was used to determine the effect of pharmacologic agents on firing properties. Data were considered significant for p < 0.05. Data are expressed as mean ± SEM unless otherwise specified. Data were analyzed using SigmaPlot (Systat Software), GraphPad Prism, and Excel (Microsoft).
Results
Initial loss of repetitive spiking is subsequently followed by restored spiking in SCA1 Purkinje neurons
In the B05 ATXN1[82Q] mouse model of SCA1, mild motor impairment is evident by 5 weeks of age (Clark et al., 1997; Hourez et al., 2011), an age at which no gross changes in cerebellar morphology are present (Clark et al., 1997; Hourez et al., 2011). By 12–16 weeks of age, however, dendritic arborization of Purkinje cells is markedly reduced with corresponding atrophy of the molecular layer, without significant Purkinje cell loss (Clark et al., 1997; Orr and Zoghbi, 2001). We sought to determine Purkinje neuron function at these time points with their well defined behavioral and histopathological abnormalities. Purkinje neurons in wild-type mice normally exhibit spontaneous repetitive firing by 2 weeks of age (Raman and Bean, 1999). We first determined whether repetitive firing is normally established in ATXN1[82Q] Purkinje neurons. In cerebellar slices, using extracellular recordings to prevent disruption of cellular contents, we determined that Purkinje neurons from ATXN1[82Q] mice exhibit normal repetitive firing (Fig. 1A–E). At 5 weeks, however, when wild-type neurons continue to exhibit repetitive firing (Fig. 1F,H–J), the majority of ATXN1[82Q] Purkinje neurons no longer display spontaneous firing (Fig. 1G–I), with the remaining Purkinje neurons at this age displaying normal firing (Fig. 1J). Remarkably, in 15-week-old ATXN1[82Q] mice, the majority of Purkinje neurons again exhibit spontaneous spiking (Fig. 1L–N), though at a markedly reduced frequency compared with wild-type neurons (Fig. 1K,O). Between 5 and 15 weeks, ATXN1[82Q] Purkinje neurons undergo marked atrophy (Clark et al., 1997), as evidenced by thinning of the cerebellar molecular layer, without loss of Purkinje neurons (Clark et al., 1997). These data suggest that 5-week-old ATXN1[82Q] Purkinje neurons that lack spiking have a restoration of firing at 15 weeks.
Figure 1.

Initial cessation of spiking at 5 weeks is followed by restoration of repetitive spiking at 15 weeks in ATXN1[82Q] Purkinje neurons. A, Two-week-old wild-type and (B) ATXN1[82Q] Purkinje neurons exhibit repetitive firing, summarized in C. “Silent” refers to neurons in which no spike activity was detectable in cell attached recordings. D, The majority of Purkinje neurons in individual mice exhibit repetitive firing. The firing frequency in wild-type and SCA1 Purkinje neurons (E) is similar at 2 weeks. F, Wild-type Purkinje neurons continue to exhibit repetitive firing at 5 weeks. G, The majority of ATXN1[82Q] Purkinje neurons lack spiking at 5 weeks, summarized in H. I, Only ∼35% of Purkinje neurons in individual ATXN1[82Q] mice at 5 weeks exhibit repetitive spiking. ATXN1[82Q] Purkinje neurons (J) that do exhibit repetitive spiking have a firing frequency similar to wild-type neurons. K, Wild-type Purkinje neurons at 15 weeks exhibit repetitive firing similar to that seen at 2 and 5 weeks. L, The majority of ATXN1[82Q] Purkinje neurons at 15 weeks exhibit repetitive spiking, summarized in M. N, Even when these data are analyzed as a percentage of Purkinje neurons that exhibit repetitive spiking in individual mice, the majority of neurons in individual ATXN1[82Q] mice at 15 weeks exhibit repetitive spiking. The firing frequency of ATXN1[82Q] Purkinje neurons (O) is significantly lower than wild-type neurons. Data are expressed as mean ± SEM, and were analyzed with either a Student's t test for noncategorical data or Fisher's exact test for categorical data. Numbers above bars represent numbers of cells tested (C, E, H, J, M, O). Numbers within bars represent numbers of animals tested (D, I, N).
The loss of spiking in ATXN1[82Q] Purkinje neurons is associated with a reduction in function of BK channels and other subthreshold-activated potassium channels
To determine the membrane potential of nonfiring 5-week-old ATXN1[82Q] Purkinje neurons, we used the whole-cell configuration of the patch-clamp technique. Wild-type Purkinje neurons continued to exhibit tonic repetitive firing (Fig. 2A; n = 45), whereas nonfiring ATXN1[82Q] Purkinje neurons had a depolarized membrane potential of −42 ± 0.6 mV (Fig. 2C; n = 66). This was not due to altered synaptic activity, as the membrane potential remained depolarized in the presence of synaptic inhibitors [GABAA receptor antagonist, picrotoxin, 50 μm; AMPA and kainite receptor antagonist, DNQX (6,7-dinitrouinoxlaine–2,3-dione, 10 μm); and NMDA receptor antagonist CPP (3-(2-Carboxypiperazin-4-yl)propyl-1-phosphonic acid, 5 μm; n = 10, data not shown]. Because the major depolarizing conductance in Purkinje neurons is a TTX-sensitive voltage-gated sodium current, we determined whether blocking these channels would normalize the membrane potential. Application of TTX to wild-type neurons caused cessation of firing, revealing the resting membrane potential to be −58 ± 0.7 mV (Fig. 2B,F; n = 8). In contrast, TTX had little effect on the membrane potential of depolarized ATXN1[82Q] Purkinje neurons (Fig. 2D,F; −42 ± 0.8 mV pre-TTX, to −46 ± 1.2 mV post-TTX, n = 26). A combination of TTX and cadmium, an inhibitor of voltage-gated calcium channels was able to repolarize the membrane to the normal resting membrane potential (Fig. 2E, summarized in F). To investigate whether the depolarized membrane potential of ATXN1[82Q] Purkinje neurons represents depolarization block, we assessed the effect of depolarizing current injection from a negative holding potential. Whereas, wild-type neurons could sustain long trains of action potentials in response to depolarizing current (Fig. 2G; ATXN1[82Q] neurons could only sustain short spike trains before undergoing a plateau potential (Fig. 2H). ATXN1[82Q] Purkinje neurons were also unable to sustain high firing frequencies (Fig. 2I). These data suggest that a depolarization mediated by voltage-gated calcium channels, in addition to the normal depolarization mediated by voltage-gated sodium channels, results in a depolarized membrane potential in ATXN1[82Q] Purkinje neurons. The normal role of somatic calcium currents in Purkinje neurons is to repolarize the membrane by activating calcium-activated potassium channels (Raman and Bean, 1999). We therefore determined whether the after hyperpolarization (AHP; Fig. 2J), a measure of calcium-activated potassium channels (Raman and Bean, 1999; Womack and Khodakhah, 2003) was reduced in ATXN1[82Q] Purkinje neurons. There was a significant reduction in AHP amplitude in depolarized ATXN1[82Q] Purkinje neurons (Fig. 2K,L). This reduction in AHP in ATXN1[82Q] Purkinje neurons is similar to that produced in wild-type neurons by iberiotoxin (IbTX), a selective inhibitor of large conductance calcium-activated (BK) channels (Fig. 2M). Depolarized ATXN1[82Q] Purkinje neurons had a significantly smaller IbTX-sensitive AHP, consistent with a reduction of BK channel currents (Fig. 2N). Iberiotoxin applied to wild-type Purkinje neurons, however, induced bursting, but not persistent membrane depolarization (Fig. 2O), suggesting that BK channels alone are not responsible for the membrane depolarization in ATXN1[82Q] Purkinje neurons. We therefore determined whether abnormalities in other subthreshold-activated potassium channels, that can potentially play an important role in pacemaking (J. Li et al., 2013), were also involved. Because no specific blockers for these channels are available, we used barium, a nonselective inhibitor of potassium channels (Coetzee et al., 1999; J. Li et al., 2013). We determined the amplitude of barium-sensitive currents at subthreshold voltages, where adequate voltage-clamp of Purkinje neurons was also possible. A substantial barium-sensitive subthreshold-activated potassium current was present in wild-type neurons (Fig. 2P). The barium-sensitive current was significantly reduced in depolarized ATXN1[82Q] Purkinje neurons (Fig. 2Q, summarized in R). These data suggest that the depolarized membrane potential of ATXN1[82Q] Purkinje neurons is associated with a significant reduction in function of both BK channels and other subthreshold-activated potassium channels.
Figure 2.

The depolarized membrane potential in ATXN1[82Q] Purkinje neurons at 5 weeks is associated with the reduction in function of BK and other subthreshold-activated potassium channels. A, In whole-cell patch-clamp recordings, wild-type Purkinje neurons exhibit tonic repetitive spiking. B, Application of 1 μm TTX results in a cessation of spiking to reveal the resting membrane potential to be −60 mV. C, ATXN1[82Q] Purkinje neurons that lack spontaneous spiking in the cell attached configuration have a depolarized membrane potential in the whole-cell configuration. D, TTX is unable to repolarize the membrane of depolarized ATXN1[82Q] Purkinje neurons. E, A combination of TTX to block voltage-gated sodium channels and 100 μm cadmium chloride to block voltage-gated calcium channels is able to repolarize the membrane of ATXN1[82Q] Purkinje neurons, summarized in F. In response to injected current from a holding potential of −90 mV (G), wild-type Purkinje neurons can maintain high rates of repetitive spiking. H, ATXN1[82Q] Purkinje neurons can sustain only brief spike trains in response to injected current and undergo depolarization block with invasion of low amplitude calcium spikes into the plateau potential, summarized in I (n = 16 cells of each genotype). Schematic action potential (J) illustrating the AHP. K, Evoked spikes of depolarized ATXN1[82Q] Purkinje neurons have a reduction in the AHP amplitude, summarized in L. The reduction in AHP in ATXN1[82Q] Purkinje neurons is similar to that seen by inhibiting BK channels using 200 nm Iberiotoxin in wild-type Purkinje neurons (M). N, IbTX has a significantly smaller effect on the AHP in depolarized ATXN1[82Q] Purkinje neurons. O, IbTX causes burst firing in wild-type Purkinje neurons (n = 5). P, Five-week-old wild-type Purkinje neurons exhibit a barium-sensitive current at −80 mV. Q, The barium-sensitive current at these subthreshold voltages is significantly reduced in 5-week-old ATXN1[82Q] Purkinje neurons, summarized in R. Numbers within the bars represent numbers of cells tested. Data are expressed as mean ± SEM. Student's t tests were used for statistical analysis.
Expression of BK and GIRK1 channels is reduced in ATXN1[82Q] Purkinje neurons
We sought to determine the basis for reduced BK channel and subthreshold-activated potassium channel activity in ATXN1[82Q] Purkinje neurons. By immunofluorescence, BK channel expression in the molecular layer was reduced in 5-week-old ATXN1[82Q] cerebella (Fig. 3A,B), even after normalization for the mildly reduced calbindin staining(Fig. 3C–G). BK channel protein levels were also reduced in whole cerebellar membrane lysates from 5-week-old ATXN1[82Q] mice (Fig. 3H,I). This reduction reflected decreased BK channel gene expression as assayed by quantitative RT-PCR (Fig. 3J). To determine the identity of the subthreshold-activated potassium channels associated with membrane depolarization in ATXN1[82Q] Purkinje neurons, we examined the gene expression of the known subthreshold-activated potassium channels by RNA sequencing. In addition to the reduction in BK channels, (Kcnma1), the expression of Kcnj4 was significantly reduced at 5 weeks and Kcnj3 expression was reduced in 12-week-old ATXN1[82Q] cerebella (Table 1). Immunostaining for G-protein coupled inwardly rectifying potassium channel (GIRK1 encoded by Kcnj3) was therefore performed. Unlike BK channels, that are highly expressed in the entire Purkinje neuron dendritic tree in the molecular layer, and not in cerebellar granule cell neurites (Chen et al., 2010), GIRK1 is expressed only in Purkinje neuron somata and proximal dendrites. Also, cerebellar granule cell neurites express GIRK1 (Miyashita and Kubo, 1997). To detect Purkinje neuron-specific changes in GIRK1 in ATXN1[82Q] mice, Purkinje neuron somatic immunostaining for GIRK1 was examined. GIRK1 immunostaining was reduced in Purkinje neurons from ATXN1[82Q] mice (Fig. 3K–Q). These data suggest that reduced expression of BK channels and other subthreshold-activated potassium channels explains the loss of potassium channel function in ATXN1[82Q] mice.
Figure 3.
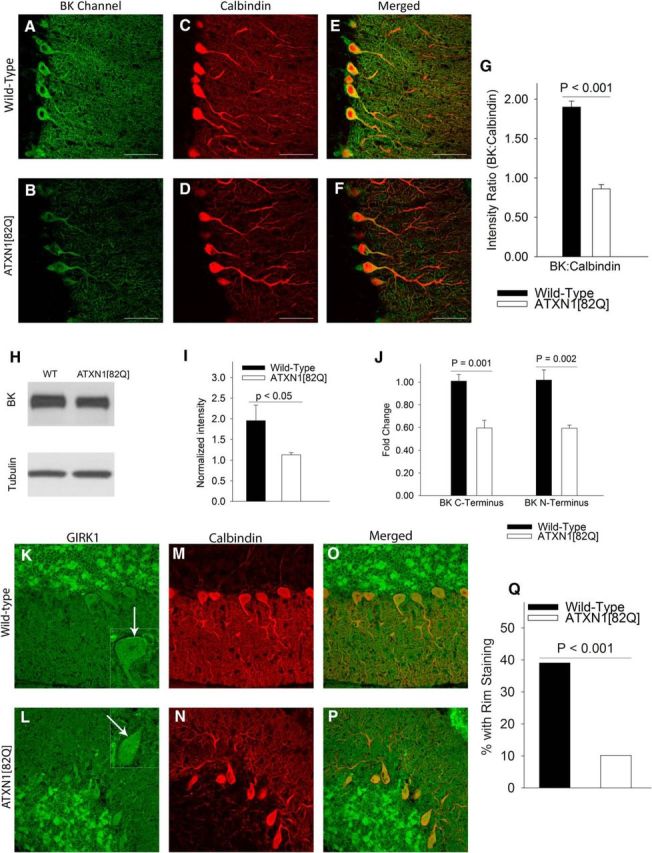
BK and GIRK1 channel expression is reduced in ATXN1[82Q] Purkinje neurons. A, Immunostaining for BK channels in wild-type cerebella at 5 weeks shows prominent staining of Purkinje neurons. B, There is a marked reduction in BK channel staining in ATXN1[82Q] Purkinje neurons. C, Calbindin immunostaining of wild-type cerebella. D, Calbindin immunostaining of ATXN1[82Q] cerebella. E, Merged images of calbindin and BK immunostaining in wild-type cerebella and ATXN1[82Q] cerebella (F), confirming that the BK channel staining corresponds to calbindin-positive Purkinje neurons. G, There is a significant reduction in BK channel immunostaining in the molecular layer of ATXN1[82Q] cerebella when normalized for the mild reduction in calbindin immunostaining (N = 3 animals of each genotype. Data were analyzed by a one-way ANOVA with a Bonferroni post hoc test). H, Western blots from the membrane fraction of cerebella from wild-type and ATXN1[82Q] mice show a reduction in BK channels (N = 3 animals of each genotype. Data were analyzed with a Student's t test) summarized in I. BK channel transcripts (J) are reduced as assayed by quantitative RT-PCR in ATXN1[82Q] cerebella (n = 5–6 animals of each genotype. Data were analyzed by a one-way ANOVA with a Bonferroni post hoc test). K, GIRK1 immunostaining in wild-type cerebella at 5 weeks. Note rim of staining of Purkinje neuron somata (inset, arrow). L, GIRK1 does not display the rim staining of Purkinje neurons in ATXN1[82Q] cerebella. M, Corresponding calbindin stained images from wild-type and (N) ATXN1[82Q] cerebella. O, Merged images suggesting that the rim of GIRK1 staining likely corresponds to the location of the plasma membrane of Purkinje neurons in wild-type and (P). ATXN1[82Q] cerebella, summarized in Q. Data are expressed as mean ± SEM.
Table 1.
Ion channel transcript levels derived by RNA sequencing from cerebella from ATXN1[82Q] and wild-type mice at 5 and 12 weeks
| Gene (protein) | 5 weeks |
12 weeks |
||||
|---|---|---|---|---|---|---|
| Wild-type | ATXN1 [82Q] | q value | Wild-type | ATXN1 [82Q] | q value | |
| Kcnma1 (BK) | 24.4879 | 17.2747 | 0.00235 | 29.3762 | 13.2709 | 0 |
| Kcnj3 (Kir3.1, GIRK1) | 42.2346 | 39.9454 | 0.99962 | 46.2715 | 26.4705 | 5.28E-09 |
| Kcnj4 (Kir2.3) | 0.414573 | 0.177637 | 0.03536 | 0.440548 | 0.539496 | 1 |
Restoration of spiking in atrophic ATXN1[82Q] Purkinje neurons is associated with normalization of BK channel membrane density and function
Fifteen-week-old ATXN1[82Q] Purkinje neurons have a restoration of repetitive spiking (Fig. 1K–O). This reflects a return of intrinsic spiking as repetitive firing was maintained in the presence of inhibitors of synaptic transmission (n = 10, data not shown). Because, calcium-activated potassium channels play a critical role in maintaining intrinsic spiking in Purkinje neurons (Raman and Bean, 1999; Cingolani et al., 2002; Womack and Khodakhah, 2003; Kasumu et al., 2012), we first determined whether repetitive spiking in atrophic ATXN1[82Q] Purkinje neurons relied on calcium-activated potassium channels. In cell attached, extracellular recordings in cerebellar slices, low doses of cadmium (20 μm) in the presence of synaptic inhibitors partially inhibited voltage-gated calcium channels, and in turn, calcium-activated potassium channels (Walter et al., 2006). Cadmium profoundly affected firing of ATXN1[82Q] Purkinje neurons, causing either an increase in firing frequency or membrane depolarization (Fig. 4A–C), suggesting that calcium-activated potassium channels continue to play a major role in spiking of atrophic ATXN1[82Q] Purkinje neurons. We next determined the role small-conductance calcium-activated potassium (SK) channels play in restoring spiking to ATXN1[82Q] Purkinje neurons. Apamin, a selective SK channel inhibitor, modestly increased firing irregularity without significantly increasing firing frequency (Figs. 4D–G), as has been shown in wild-type Purkinje neurons (Walter et al., 2006). IbTX induced burst firing in atrophic ATXN1[82Q] Purkinje neurons with an increase in predominant firing frequency (Figs. 4H–K). A combination of apamin and IbTX recapitulated the effect of cadmium on firing in ATXN1[82Q] Purkinje neurons, in either increasing the predominant firing frequency or inducing membrane depolarization (Fig. 4L–N). The effect of IbTX on firing of atrophic ATXN1[82Q] Purkinje neurons was unexpected given the even greater decline in BK channel expression by quantitative RT-PCR in ATXN1[82Q] cerebella at this age than at 5 weeks (Figs. 3J, 4O). Thus, despite the marked reduction in BK channel expression that accompanies neuronal atrophy, ATXN1[82Q] Purkinje neurons continue to rely on BK channels for intrinsic pacemaker firing.
Figure 4.

BK channels contribute to restoration of spiking in 15-week-old atrophic ATXN1[82Q] Purkinje neurons. A, Fifteen-week-old atrophic ATXN1[82Q] Purkinje neurons exhibit repetitive firing even in the presence of synaptic inhibitors (50 μm picrotoxin to block GABAA receptors in addition to 10 μm DNQX and 5 μm CPP to block glutamatergic transmission) B, Cadmium (20 μm), a blocker of voltage-gated calcium channels, and hence calcium-activated potassium channels, increases firing frequency leading to membrane depolarization (indicated by a * in C) in 15-week-old ATXN1[82Q] Purkinje neurons, summarized in C. Tonically firing 15-week-old ATXN1[82Q] Purkinje neurons (D) demonstrate no change in firing frequency following application of 100 nm apamin, a selective SK channel inhibitor (E), and summarized in F. Apamin increases the irregularity of spiking (G) in ATXN1[82Q] Purkinje neurons. H, Tonically firing 15-week-old ATXN1[82Q] Purkinje neurons demonstrate bursting and an increase in firing frequency following application of 200 nm iberiotoxin, a selective BK channel inhibitor (I), summarized in J. Iberiotoxin (K) markedly increases the irregularity of spiking. L, Tonically firing 15-week-old ATXN1[82Q] Purkinje neurons demonstrate bursting followed by membrane depolarization following application of a combination of apamin and iberiotoxin (M), summarized in N. The large effect of iberiotoxin on firing in 15-week-old ATXN1[82Q] Purkinje neurons (O) was despite an even greater reduction in BK channel expression in 15-week-old compared with 5-week-old ATXN1[82Q] cerebella as assessed by quantitative RT-PCR (n = 5–6 animals of each genotype. Data were analyzed by a one-way ANOVA with a Bonferroni post hoc test). Data are expressed as mean ± SEM.
We next determined whether the return of repetitive spiking in atrophic ATXN1[82Q] Purkinje neurons was associated with a restoration of the AHP amplitude. The AHP amplitude in 15-week-old ATXN1[82Q] Purkinje neurons proved to be similar to that of wild-type neurons (Fig. 5A,B), suggesting that the AHP is necessary for Purkinje neuron pacemaking. Fifteen-week-old wild-type Purkinje neurons continued to have a significant IbTX-sensitive AHP (Fig. 5D). Remarkably, at 15 weeks the amplitude of the IbTX-sensitive AHP was greater in ATXN1[82Q] Purkinje neurons than in wild-type neurons (Fig. 5C,D), suggesting that atrophic ATXN1[82Q] Purkinje neurons are even more dependent on BK channels for spiking than wild-type neurons. The molecular layer of 15 week ATXN1[82Q] cerebella showed significantly reduced calbindin staining (Figs. 5E,F), consistent with dendritic loss. BK channel staining in the molecular layer was similar to that in wild-type cerebella when normalized to account for dendritic loss (Fig. 5G–K). We next used RNA sequencing in ATXN1[82Q] cerebella to assess the expression of ion-channels potentially important for pacemaking. We measured the correlation between the expression of specific ion-channels and Purkinje neuron capacitance, a measure of cell atrophy, In ATXN1[82Q] mice, the change in expression of six of 110 ion-channel transcripts significantly correlated with the reduction in ATXN1[82Q] Purkinje neuron capacitance between 5 and 12 weeks (Fig. 5L; data not shown). Although the expression of other potassium channels was reduced in this dataset (Table 1), the BK channel was the only potassium channel transcript that significantly correlated with the reduction in ATXN1[82Q] Purkinje neuron capacitance. These data suggest that the longitudinal reduction in BK channel gene expression correlates with progressive Purkinje cell atrophy, and suggest that BK channels could play a significant role in Purkinje neuron remodeling in ATXN1[82Q] mice.
Figure 5.
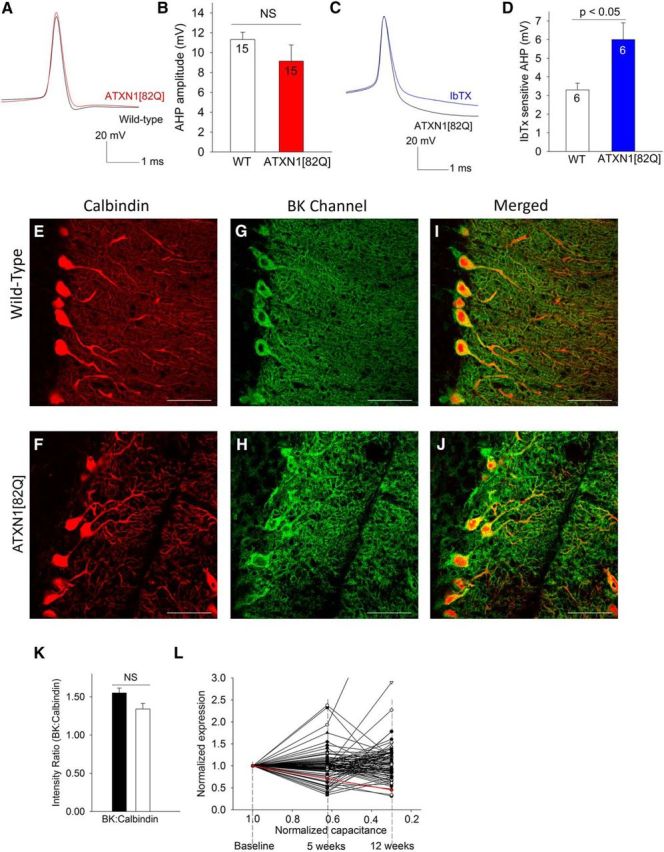
BK channel function is maintained in atrophic 15-week-old ATXN1[82Q] Purkinje neurons despite reduced BK channel gene expression. A, The amplitude of the AHP is similar in 15-week-old wild-type and ATXN1[82Q] Purkinje neurons, summarized in B. The iberiotoxin-sensitive AHP (C) is re-established in 15-week-old ATXN1[82Q] Purkinje neurons, and is now larger than that in wild-type neurons (D). E, Calbindin immunostaining of wild-type cerebella. F, Calbindin immunostaining of ATXN1[82Q] cerebella showing marked thinning of the molecular layer. G, Immunostaining for BK channels in wild-type cerebella shows prominent staining of Purkinje neurons. H, There is restoration of BK channel staining in 15-week-old ATXN1[82Q] Purkinje neurons. I, Merged images of calbindin and BK immunostaining in wild-type and ATXN1[82Q] (J) cerebella confirming that the BK channel staining corresponds to calbindin-positive Purkinje neurons. K, BK staining of ATXN1[82Q] Purkinje neurons is similar to wild-type when normalized for calbindin immunostaining (N = 3 animals of each genotype. Data were analyzed by a one-way ANOVA with a Bonferroni post hoc test). L, Correlation analysis of ion-channel transcripts as assessed by RNA sequencing of ATXN1[82Q] and wild-type cerebella at 5 and 12 weeks and cell size as assessed by membrane capacitance. For purposes of assessing the correlation, an arbitrary baseline was chosen, where normalized transcript expression and capacitance were assumed to be similar in wild-type and ATXN1[82Q] cerebella. The reduction in BK channel transcript, shown in red, is highly correlated with the reduction in cell size (R2 = 1, p < 0.001). Data are expressed as mean ± SEM.
In addition to BK channels, a reduction in subthreshold-activated potassium channels also contributes to adaptive ATXN1[82Q] Purkinje neuron remodeling
In addition to BK channels, the reduction in subthreshold-activated potassium channels is also associated with membrane depolarization in 5-week-old ATXN1[82Q] Purkinje neurons (Fig. 2P–R). We therefore determined whether abnormalities in these other subthreshold-activated potassium channels accompanies Purkinje neuron atrophy in ATXN1[82Q] mice. The barium-sensitive subthreshold-activated current in atrophic 15-week-old ATXN1[82Q] Purkinje neurons, when normalized for cell capacitance (a surrogate for cell size; Fig. 6A–C), was significantly larger than in wild-type neurons. To determine the functional significance of these changes in barium-sensitive current-amplitude, we determined the effect of a low dose of barium (50 μm) that relatively selectively inhibits Kir channels (Quayle et al., 1993; Franchini et al., 2004; Hibino et al., 2010; Sepúlveda et al., 2015) on Purkinje neuron firing. The firing frequency of 15-week-old wild-type Purkinje neurons increased only modestly following bath application of barium chloride (Fig. 6D–G). In contrast, barium had a profound effect on firing in 15-week-old ATXN1[82Q] Purkinje neurons, with an increased firing frequency, followed by burst firing and membrane depolarization (Fig. 6H–K). These results suggest that the Kir class of subthreshold-activated potassium channels play an important role in reestablishing repetitive spiking in atrophic ATXN1[82Q] Purkinje neurons and suggests that cell atrophy compensates for the reduction of not just BK channels but also the reduction in other subthreshold-activated potassium channels.
Figure 6.

Subthreshold-activated potassium channels contribute to restoration of spiking in ATXN1[82Q] Purkinje neurons. A, Fifteen-week-old wild-type Purkinje neurons exhibit a barium-sensitive current, obtained by subtraction, and normalization for cell capacitance (inset shows the voltage step protocol used to elicit currents). B, When normalized for cell size (capacitance), the current density of the barium-sensitive subthreshold-activated current in 15-week-old atrophic ATXN1[82Q] Purkinje neurons is significantly larger than in wild-type Purkinje neurons (summarized in C). Because the barium-sensitive current was similar in 5- and 15-week-old wild-type neurons, the data were pooled. Data were analyzed with a Student's t test. D, Fifteen-week-old wild-type Purkinje neurons exhibit repetitive spiking that is only modestly affected by 50 μm barium chloride (E), summarized in F. Barium does not change the regularity of spiking (G) of 15-week-old wild-type Purkinje neurons. H, Fifteen-week-old ATXN1[82Q] Purkinje neurons exhibit tonic repetitive spiking. I, Following application of 50 μm barium chloride, ATXN1[82Q] Purkinje neurons exhibit burst firing followed by membrane depolarization (J), summarized in K. Data are expressed as mean ± SEM.
Restoring BK channel expression in ATXN1[82Q] mice improves motor dysfunction and partially restores dendritic morphology
To determine the functional significance of reduced BK channel expression, we sought to restore expression of BK channels in Purkinje neurons of ATXN1[82Q] mice. MBr5, a brain-specific isoform of the BK channel (Butler et al., 1993) was cloned into a plasmid for packaging into AAV2/5, a delivery vector that effectively transduces Purkinje neurons (Keiser et al., 2014). Two weeks following viral delivery of BK channels to 3.5- to 4-week-old ATXN1[82Q] cerebella, we observed a robust increase in BK channel transcript in cerebella of injected mice (Fig. 7A). Injection of BK-AAV to one hemisphere of the cerebellum increased immunostaining for BK channels in the injected hemisphere (Fig. 7B,C). Two weeks following cerebellar delivery of AAV-BK, the majority of ATXN1[82Q] Purkinje neurons now exhibited tonic repetitive spiking with a firing frequency similar to wild-type neurons (Fig. 7D,E) accompanied by a normalization of AHP amplitude (Fig. 7F). This result supports the view that the early membrane depolarization in ATXN1[82Q] Purkinje neurons is caused in part by the reduction of BK channels. Although increasing BK channel expression in ATXN1[82Q] Purkinje neurons restored the AHP and spontaneous spiking, Purkinje neurons from BK-AAV-injected mice could not sustain trains of spikes in response to increasing depolarizing current injection (Fig. 7G–I), consistent with a role for other subthreshold-activated potassium channels in maintaining the membrane potential of ATXN1[82Q] Purkinje neurons. We next sought to determine to what extent, the reduction in BK channels contributes to motor dysfunction and dendritic remodeling in ATXN1[82Q] mice. BK-AAV-treated animals showed significant improvement in motor function 10 weeks following injection, approaching the performance of wild-type mice (Fig. 7J). There was also a modest improvement in the density of Purkinje neuron dendritic branches in BK-AAV-treated ATXN1[82Q] mice (Fig. 7K–M) but without any improvement in molecular layer thickness (Fig. 7N). These data suggest that the reduction of BK channel expression in Purkinje neurons contributes to motor dysfunction and contributes to maintaining Purkinje neuron dendritic morphology.
Figure 7.

Viral-mediated BK channel expression in ATXN1[82Q] cerebella improves motor dysfunction and partially rescues dendritic atrophy. A, Delivery of BK-AAV to 3.5–4 week ATXN1[82Q] cerebella normalizes BK channel transcripts 2 weeks following injection as assessed by quantitative RT-PCR and normalized to wild-type mice. B, Immunostaining for BK channels reveals increased BK channel staining in the left hemisphere injected with BK-AAV (arrows), summarized in C. In ATXN1[82Q] mice injected with BK-AAV (D) at 3.5–4 weeks, the majority of Purkinje neurons exhibit tonic repetitive spiking with a normal firing frequency 2 weeks following injection (N = 3 mice; E) The amplitude of the AHP in BK-AAV-injected ATXN1[82Q] mice (F) is similar to wild-type mice (data for uninjected ATXN1[82Q] and wild-type mice are reproduced from Figs. 1J and 2L). G, Wild-type Purkinje neurons exhibit trains of repetitive spiking in response to injected current form a negative holding potential. H, In ATXN1[82Q] mice injected with BK-AAV at 3.5–4 weeks, a Purkinje neuron undergoes depolarization block of repetitive spiking for the same amount of injected current as that shown in G, 2 weeks following BK-AAV injection (N = 3 mice), summarized in I. J, BK-AAV-injected mice have a significant improvement in motor performance, approaching the performance of wild-type mice, unlike the motor impairment seen in GFP-AAV-injected ATXN1[82Q] mice. Data were analyzed with a two-way repeated-measures ANOVA. K, BK-AAV-injected mice have a preservation of the density of calbindin staining intensity in the cerebellar molecular layer compared with GFP-AAV-injected mice (L), summarized in M. There was, however, no rescue of the reduction in molecular layer thickness in BK-AAV-injected mice (N). Data are expressed as mean ± SEM.
Activating potassium channels improves Purkinje neuron atrophy in ATXN1[82Q] mice
We sought to determine whether restoring the normal membrane potential by activating potassium channels could prevent the dendritic remodeling associated with reduced BK and subthreshold-activated potassium channel function. FFA has previously been reported to activate both BK and subthreshold-activated potassium channels (Ottolia and Toro, 1994; Takahira et al., 2005; X. Li et al., 2013). Application of FFA to depolarized Purkinje neurons in cerebellar slices from 5-week-old ATXN1[82Q] mice resulted in membrane potential repolarization in a dose-dependent manner even in the absence of TTX (Fig. 8A,B). FFA, however, was unable to restore spiking and attenuated repetitive spiking in ATXN1[82Q] Purkinje neurons (Fig. 8C). In association with repolarization of the membrane, FFA reduced the input resistance of the cell, suggesting that it activates a potassium conductance at a subthreshold voltage (Fig. 8D). Because it is not physically possible to clamp the membrane of 5-week-old ATXN1[82Q] Purkinje neurons at a more positive voltage, to determine the molecular identity of this potassium conductance, we exploited the reduced size of atrophic 15-week-old Purkinje neurons, where adequate voltage-clamp can be achieved. FFA activated a large subthreshold-activated potassium current in 15-week-old ATXN1[82Q] Purkinje neurons that was weakly inwardly rectifying (Fig. 8E,F). The FFA activated current was barium-sensitive, and barium inhibited the membrane repolarization mediated by FFA (data not shown). We next determined whether the FFA mediated reduction in intrinsic membrane excitability in ATXN1[82Q] Purkinje neurons could prevent dendritic atrophy. Oral and intraperitoneally administered FFA did not achieve significant brain concentrations (data not shown). Five-week old ATXN1[82Q] mice were therefore implanted for 6 weeks with an osmotic pump and cerebellar cannula to perfuse FFA or vehicle. An inert dye, fast green, was introduced into the osmotic pump to reveal the extent of cerebellar perfusion (Fig. 8G). Perfusion was very focal in the midline cerebellum, restricted to lobules III–V (Fig. 8H). Due to variable destruction of lobules IV and V (Fig. 8I) from pump placement, the effect of FFA on molecular layer thickness was assessed in the posterior aspect of lobule III. There was a significant preservation of molecular layer thickness in lobule III of FFA perfused animals compared with vehicle perfused ATXN1[82Q] mice (Fig. 8I–K). Higher-magnification images revealed a preservation of complexity of the dendritic arbor of ATXN1[82Q] Purkinje neurons in FFA perfused lobule III (Fig. 8L,M). Molecular layer thickness in lobule II, which was not perfused by the osmotic pump, was similar in FFA and vehicle-treated ATXN1[82Q] mice (Fig. 8I–K). These data suggest that activation of potassium currents and reducing Purkinje neuron membrane excitability in ATXN1[82Q] mice prevents neuronal atrophy.
Figure 8.

Flufenamic acid, an agent that activates potassium channels and restores the resting membrane potential of ATXN1[82Q] Purkinje neurons, prevents dendritic atrophy. A, Five-week-old ATXN1[82Q] Purkinje neurons have a depolarized membrane potential. A combination of TTX and 100 μm FFA hyperpolarizes the membrane. B, FFA hyperpolarizes the membrane, even in the absence of TTX in a dose-dependent manner (3–12 neurons at each concentration). C, FFA, even at the lowest dose of 25 μm prevented ATXN1[82Q] Purkinje neurons from maintaining trains of spikes in response to injected current. D, The current activated by 100 μm FFA in depolarized ATXN1[82Q] Purkinje neurons, obtained by subtraction, is consistent with a potassium current (inset shows the voltage step protocol used to elicit currents). E, FFA activates a weakly inwardly rectifying potassium current in 15-week-old ATXN1[82Q] Purkinje neurons (inset shows the voltage step protocol used to elicit currents), summarized in F. Perfusion of FFA or vehicle to the midline cerebellum (G) is focal and restricted to lobules III–VI (H). I, Perfusion of the cerebellum with vehicle does not rescue molecular layer thinning in the area perfused (lobule III, red asterisk) or in the area not perfused (lobule II, white asterisk). J, Perfusion of the cerebellum with FFA results in preservation of molecular layer thickness in lobule III which is perfused (red asterisk), but not lobule II (white asterisk), which is not perfused, summarized in K (N = 5 mice for each treatment; data were analyzed with a one-way ANOVA with a Bonferroni post hoc test). L, Higher-magnification images from vehicle and M, FFA perfused lobule III with preservation of the complexity of dendritic branching in the FFA perfused areas. Data are expressed as mean ± SEM.
Discussion
Our results suggest that expression of ATXN1[82Q] in Purkinje neurons causes an early membrane depolarization with relatively intact morphology, followed by Purkinje neuron atrophy and subsequent restoration of pacemaker firing. We show that the reduction in cell size that accompanies cell atrophy allows the cell to maintain the membrane density of potassium channels essential for maintaining normal membrane excitability, despite a reduction in the expression of these channels. Purkinje neuron atrophy could therefore be viewed as an active adaptive response to counteract the effect of a reduction in potassium channels essential for maintaining normal membrane potential and pacemaker spiking. Homeostatic compensation after ion-channel deletion has been predicted to occur through using differential ion channel expression rates (O'Leary et al., 2014). We show that physical remodeling of the cell is an additional mechanism for homeostatic compensation for altered ion channel expression. The restoration in spiking and improvement in Purkinje neuron physiology that accompanies atrophy would, however, appear to be inconsistent with the progressive motor dysfunction seen in this mouse model of SCA1 (Clark et al., 1997; Hourez et al., 2011). It is important, however, to note that at the onset of motor dysfunction, although the majority of Purkinje neurons have a depolarized membrane potential, up to 35% of Purkinje neurons have intact firing, along with a retained dendritic arbor. Others have noted significant redundancy in Purkinje neurons, with up to a 50% loss of neurons before detectable motor dysfunction (Elrick et al., 2010). At the time point corresponding to a relative restoration of Purkinje neuron intrinsic firing, there is marked dendritic atrophy. The spontaneous firing of Purkinje neurons is modulated by extensive excitatory and inhibitory dendritic synaptic input from the cerebral cortex, vestibular system, and periphery (Armstrong and Rawson, 1979; Häusser and Clark, 1997). The reduction in dendritic arbor in the ATXN1[82Q] mice would be expected to greatly reduce the number of synaptic contacts (Inoue et al., 2001; Barnes et al., 2011) and thereby compromise the ability of Purkinje neurons to modulate their intrinsic firing for motor control. It would be anticipated that early in the neurodegenerative process, sacrificing membrane surface area to maintain intrinsic excitability would be adaptive in maintaining normal motor function. Beyond a certain point, however, progressive dendrite loss would compromise the ability of Purkinje neurons to participate effectively in the cerebellar circuit, leading to progressive motor dysfunction.
In SCA1, as in many inherited ataxias, the respective disease genes are expressed widely in neuronal and non-neuronal tissues (Servadio et al., 1995), yet preferentially affect a relatively small subset of neurons in the brain (Dürr, 2010). Although a great deal about the cellular roles of these disease proteins has been identified, the basis for the preferential vulnerability of specific neuronal populations remains unclear. Intriguingly, many of the most affected neurons in SCA1 (Rüb et al., 2013) exhibit autonomous pacemaker firing, including Purkinje neurons, neurons of the deep cerebellar nuclei, substantia nigra neurons, vestibular neurons, and inferior olivary neurons (Aizenman and Linden, 1999; Raman and Bean, 1999; Wolfart et al., 2001; Nelson et al., 2003). Other affected neurons maintain high rates of firing, such as motor cranial nerve nuclei (Fuchs and Luschei, 1970). The predicted role of ATXN1 is in transcriptional regulation, and mutant ATXN1 must translocate to the nucleus to mediate its toxicity (Klement et al., 1998; Lin et al., 2000; Lam et al., 2006). In this study, we demonstrate that alterations in expression of ion-channel genes important for rapid spiking neurons are affected in SCA1. Although transcriptional changes caused by mutant ATXN1 would be expected to be widespread, it is likely that the vulnerability of cerebellar Purkinje neurons and possibly other pacemaker neurons reflects involvement of the proteins encoded by these genes, which are vital for the physiology of these neurons.
In degenerative nervous system disorders, cellular atrophy is often present before loss of neurons. In disease models, increased synaptically induced excitability is thought to contribute to neuronal remodeling. In this study, we demonstrate that alterations in intrinsic excitability are a cause for neuronal atrophy. Intrinsic firing of Purkinje neurons relies on a balance between depolarizing sodium currents and repolarizing potassium currents in the interspike interval (Raman and Bean, 1997, 1999). Calcium-activated potassium channels play a major role in preventing the spiking of Purkinje neurons from undergoing depolarization block (Raman and Bean, 1999; Womack and Khodakhah, 2003; Sausbier et al., 2004). In this study, we identified a reduction in BK-dependent AHP, which is responsible for membrane depolarization in ATXN1[82Q] Purkinje neurons similar to that seen in the BK channel knock-out mouse (Sausbier et al., 2004). We suggest that the increased intrinsic excitability from the reduction of BK channels alone, however, is insufficient in initiating Purkinje neuron remodeling. The disruption of membrane repolarization and repetitive spiking in ATXN1[82Q] Purkinje neurons is due to the loss of both BK channels and other subthreshold activated potassium channels. The role of potassium channels active at subthreshold voltages in Purkinje neurons remains poorly understood. Purkinje neurons express several potassium channels of the Kcnj (Kir or inwardly rectifying channels; Prüss et al., 2005; Aguado et al., 2008; Fernández-Alacid et al., 2009) and Kcnk (2-pore potassium channels) class (Kindler et al., 2000; Kanjhan et al., 2004). A tetraethylammonium insensitive, barium-sensitive background current has previously been reported in Purkinje neurons (Bushell et al., 2002), although the molecular identity of these channels remains unclear. If in human SCA1, as in ATXN1[82Q] Purkinje neurons, neuronal intrinsic excitability is increased secondary to a net loss of more than one potassium current important for membrane repolarization, it may be important to target more than one class of potassium channels for the treatment of disease.
Onset of symptoms in patients with SCA1 typically occurs in the fourth decade of life despite lifelong expression of the mutant ATXN1 gene. Before symptoms, individuals harboring the SCA1 mutation have been reported to show some cerebellar atrophy (Jacobi et al., 2012). In addition to redundancy in cerebellar neurons, it is intriguing to speculate whether the late onset of symptoms in SCA1 is secondary to adaptive changes in neurons that enable them to maintain function, despite progressive transcriptional dysfunction in genes vital for the physiology of these neurons. Compensatory mechanisms likely fail once transcriptional changes reach a critical level. These findings may have important therapeutic implications. Although alterations in neuronal firing properties and the dendritic atrophy accompanying neurodegeneration likely contribute independently to motor symptoms, altered physiology also contributes to dendritic atrophy. Thus, correction of aberrant physiology could have both symptomatic and neuroprotective benefits.
Footnotes
This work was supported by the NIH K08NS072158 (V.G.S.), R01NS085054 (V.G.S.) R37NS0022920 (H.T.O.), R01NS062051 (P.O.), and R01NS082351 (P.O.). We thank Drs Henry Paulson and William Dauer for helpful comments and suggestions, and Alexi Vasbinder and Allison Sylvia for technical support.
The authors declare no competing financial interests.
References
- Aguado C, Colón J, Ciruela F, Schlaudraff F, Cabañero MJ, Perry C, Watanabe M, Liss B, Wickman K, Luján R. Cell type-specific subunit composition of G-protein-gated potassium channels in the cerebellum. J Neurochem. 2008;105:497–511. doi: 10.1111/j.1471-4159.2007.05153.x. [DOI] [PubMed] [Google Scholar]
- Aizenman CD, Linden DJ. Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J Neurophysiol. 1999;82:1697–1709. doi: 10.1152/jn.1999.82.4.1697. [DOI] [PubMed] [Google Scholar]
- Armstrong DM, Rawson JA. Activity patterns of cerebellar cortical neurones and climbing fibre afferents in the awake cat. J Physiol. 1979;289:425–448. doi: 10.1113/jphysiol.1979.sp012745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkali A, Corta E, Berrueta LA, Gallo B, Vicente F. Study of the solid-phase extraction of diclofenac sodium, indomethacin and phenylbutazone for their analysis in human urine by liquid chromatography. J Chromatogr B Biomed Sci Appl. 1999;729:139–145. doi: 10.1016/S0378-4347(99)00143-7. [DOI] [PubMed] [Google Scholar]
- Barnes JA, Ebner BA, Duvick LA, Gao W, Chen G, Orr HT, Ebner TJ. Abnormalities in the climbing fiber-Purkinje cell circuitry contribute to neuronal dysfunction in ATXN1[82Q] mice. J Neurosci. 2011;31:12778–12789. doi: 10.1523/JNEUROSCI.2579-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burright EN, Clark HB, Servadio A, Matilla T, Feddersen RM, Yunis WS, Duvick LA, Zoghbi HY, Orr HT. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. 1995;82:937–948. doi: 10.1016/0092-8674(95)90273-2. [DOI] [PubMed] [Google Scholar]
- Bushell T, Clarke C, Mathie A, Robertson B. Pharmacological characterization of a non-inactivating outward current observed in mouse cerebellar Purkinje neurones. Br J Pharmacol. 2002;135:705–712. doi: 10.1038/sj.bjp.0704518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Tsunoda S, McCobb DP, Wei A, Salkoff L. mSlo, a complex mouse gene encoding “maxi” calcium-activated potassium channels. Science. 1993;261:221–224. doi: 10.1126/science.7687074. [DOI] [PubMed] [Google Scholar]
- Chen X, Kovalchuk Y, Adelsberger H, Henning HA, Sausbier M, Wietzorrek G, Ruth P, Yarom Y, Konnerth A. Disruption of the olivo-cerebellar circuit by Purkinje neuron-specific ablation of BK channels. Proc Natl Acad Sci U S A. 2010;107:12323–12328. doi: 10.1073/pnas.1001745107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Gymnopoulos M, Boccaccio A, Stocker M, Pedarzani P. Developmental regulation of small-conductance Ca2+-activated K+ channel expression and function in rat Purkinje neurons. J Neurosci. 2002;22:4456–4467. doi: 10.1523/JNEUROSCI.22-11-04456.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, Matilla A, Zoghbi HY, Orr HT. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. 1997;17:7385–7395. doi: 10.1523/JNEUROSCI.17-19-07385.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B. Molecular diversity of K+ channels. Ann NY Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- Duarri A, Jezierska J, Fokkens M, Meijer M, Schelhaas HJ, den Dunnen WF, van Dijk F, Verschuuren-Bemelmans C, Hageman G, van de Vlies P, Küsters B, van de Warrenburg BP, Kremer B, Wijmenga C, Sinke RJ, Swertz MA, Kampinga HH, Boddeke E, Verbeek DS. Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann Neurol. 2012;72:870–880. doi: 10.1002/ana.23700. [DOI] [PubMed] [Google Scholar]
- Dürr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885–894. doi: 10.1016/S1474-4422(10)70183-6. [DOI] [PubMed] [Google Scholar]
- Dürr A, Stevanin G, Cancel G, Duyckaerts C, Abbas N, Didierjean O, Chneiweiss H, Benomar A, Lyon-Caen O, Julien J, Serdaru M, Penet C, Agid Y, Brice A. Spinocerebellar ataxia 3 and Machado–Joseph disease: clinical, molecular, and neuropathological features. Ann Neurol. 1996;39:490–499. doi: 10.1002/ana.410390411. [DOI] [PubMed] [Google Scholar]
- Duvick L, Barnes J, Ebner B, Agrawal S, Andresen M, Lim J, Giesler GJ, Zoghbi HY, Orr HT. SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron. 2010;67:929–935. doi: 10.1016/j.neuron.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrick MJ, Pacheco CD, Yu T, Dadgar N, Shakkottai VG, Ware C, Paulson HL, Lieberman AP. Conditional Niemann–Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum Mol Genet. 2010;19:837–847. doi: 10.1093/hmg/ddp552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Alacid L, Aguado C, Ciruela F, Martín R, Colón J, Cabañero MJ, Gassmann M, Watanabe M, Shigemoto R, Wickman K, Bettler B, Sánchez-Prieto J, Luján R. Subcellular compartment-specific molecular diversity of pre- and post-synaptic GABA-activated GIRK channels in Purkinje cells. J Neurochem. 2009;110:1363–1376. doi: 10.1111/j.1471-4159.2009.06229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Genís D, Dávalos A, Bernadó L, Sant F, Serrano T. The Purkinje cell in olivopontocerebellar atrophy: a Golgi and immunocytochemical study. Neuropathol Appl Neurobiol. 1994;20:38–46. doi: 10.1111/j.1365-2990.1994.tb00955.x. [DOI] [PubMed] [Google Scholar]
- Franchini L, Levi G, Visentin S. Inwardly rectifying K+ channels influence Ca2+ entry due to nucleotide receptor activation in microglia. Cell Calcium. 2004;35:449–459. doi: 10.1016/j.ceca.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Fuchs AF, Luschei ES. Firing patterns of abducens neurons of alert monkeys in relationship to horizontal eye movement. J Neurophysiol. 1970;33:382–392. doi: 10.1152/jn.1970.33.3.382. [DOI] [PubMed] [Google Scholar]
- Häusser M, Clark BA. Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron. 1997;19:665–678. doi: 10.1016/S0896-6273(00)80379-7. [DOI] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010;90:291–366. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- Hourez R, Servais L, Orduz D, Gall D, Millard I, de Kerchove d'Exaerde A, Cheron G, Orr HT, Pandolfo M, Schiffmann SN. Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci. 2011;31:11795–11807. doi: 10.1523/JNEUROSCI.0905-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Lin X, Kohlmeier KA, Orr HT, Zoghbi HY, Ross WN. Calcium dynamics and electrophysiological properties of cerebellar Purkinje cells in SCA1 transgenic mice. J Neurophysiol. 2001;85:1750–1760. doi: 10.1152/jn.2001.85.4.1750. [DOI] [PubMed] [Google Scholar]
- Jacobi H, Hauser TK, Giunti P, Globas C, Bauer P, Schmitz-Hübsch T, Baliko L, Filla A, Mariotti C, Rakowicz M, Charles P, Ribai P, Szymanski S, Infante J, van de Warrenburg BP, Dürr A, Timmann D, Boesch S, Fancellu R, Rola R, et al. Spinocerebellar ataxia types 1, 2, 3 and 6: the clinical spectrum of ataxia and morphometric brainstem and cerebellar findings. Cerebellum. 2012;11:155–166. doi: 10.1007/s12311-011-0292-z. [DOI] [PubMed] [Google Scholar]
- Kanamori T, Kanai MI, Dairyo Y, Yasunaga K, Morikawa RK, Emoto K. Compartmentalized calcium transients trigger dendrite pruning in Drosophila sensory neurons. Science. 2013;340:1475–1478. doi: 10.1126/science.1234879. [DOI] [PubMed] [Google Scholar]
- Kanjhan R, Anselme AM, Noakes PG, Bellingham MC. Postnatal changes in TASK-1 and TREK-1 expression in rat brain stem and cerebellum. Neuroreport. 2004;15:1321–1324. doi: 10.1097/01.wnr.0000127462.15985.dc. [DOI] [PubMed] [Google Scholar]
- Kasumu AW, Hougaard C, Rode F, Jacobsen TA, Sabatier JM, Eriksen BL, Strøbæk D, Liang X, Egorova P, Vorontsova D, Christophersen P, Rønn LC, Bezprozvanny I. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem Biol. 2012;19:1340–1353. doi: 10.1016/j.chembiol.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser MS, Boudreau RL, Davidson BL. Broad therapeutic benefit after RNAi expression vector delivery to deep cerebellar nuclei: implications for spinocerebellar ataxia type 1 therapy. Mol Ther. 2014;22:588–595. doi: 10.1038/mt.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindler CH, Pietruck C, Yost CS, Sampson ER, Gray AT. Localization of the tandem pore domain K+ channel TASK-1 in the rat central nervous system. Brain Res Mol Brain Res. 2000;80:99–108. doi: 10.1016/S0169-328X(00)00136-4. [DOI] [PubMed] [Google Scholar]
- Kirov SA, Harris KM. Dendrites are more spiny on mature hippocampal neurons when synapses are inactivated. Nat Neurosci. 1999;2:878–883. doi: 10.1038/13178. [DOI] [PubMed] [Google Scholar]
- Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/S0092-8674(00)81781-X. [DOI] [PubMed] [Google Scholar]
- Lam YC, Bowman AB, Jafar-Nejad P, Lim J, Richman R, Fryer JD, Hyun ED, Duvick LA, Orr HT, Botas J, Zoghbi HY. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell. 2006;127:1335–1347. doi: 10.1016/j.cell.2006.11.038. [DOI] [PubMed] [Google Scholar]
- Lee YC, Durr A, Majczenko K, Huang YH, Liu YC, Lien CC, Tsai PC, Ichikawa Y, Goto J, Monin ML, Li JZ, Chung MY, Mundwiller E, Shakkottai V, Liu TT, Tesson C, Lu YC, Brice A, Tsuji S, Burmeister M, et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann Neurol. 2012;72:859–869. doi: 10.1002/ana.23701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Blankenship ML, Baccei ML. Inward-rectifying potassium (Kir) channels regulate pacemaker activity in spinal nociceptive circuits during early life. J Neurosci. 2013;33:3352–3362. doi: 10.1523/JNEUROSCI.4365-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XZ, Ma KT, Guan BC, Li L, Zhao L, Zhang ZS, Si JQ, Jiang ZG. Fenamates block gap junction coupling and potentiate BKCa channels in guinea pig arteriolar cells. Eur J Pharmacol. 2013;703:74–82. doi: 10.1016/j.ejphar.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Antalffy B, Kang D, Orr HT, Zoghbi HY. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci. 2000;3:157–163. doi: 10.1038/72101. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−δ δ CT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Magistretti J, Mantegazza M, Guatteo E, Wanke E. Action potentials recorded with patch-clamp amplifiers: are they genuine? Trends Neurosci. 1996;19:530–534. doi: 10.1016/S0166-2236(96)40004-2. [DOI] [PubMed] [Google Scholar]
- Mavroudis IA, Fotiou DF, Manani MG, Njaou SN, Frangou D, Costa VG, Baloyannis SJ. Dendritic pathology and spinal loss in the visual cortex in Alzheimer's disease: a Golgi study in pathology. Int J Neurosci. 2011;121:347–354. doi: 10.3109/00207454.2011.553753. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Kubo Y. Localization and developmental changes of the expression of two inward rectifying K(+)-channel proteins in the rat brain. Brain Res. 1997;750:251–263. doi: 10.1016/S0006-8993(96)01365-0. [DOI] [PubMed] [Google Scholar]
- Morimoto T. Role of electrical activity of neurons for neuroprotection. Int Rev Neurobiol. 2012;105:19–38. doi: 10.1016/B978-0-12-398309-1.00003-2. [DOI] [PubMed] [Google Scholar]
- Nelson AB, Krispel CM, Sekirnjak C, du Lac S. Long-lasting increases in intrinsic excitability triggered by inhibition. Neuron. 2003;40:609–620. doi: 10.1016/S0896-6273(03)00641-X. [DOI] [PubMed] [Google Scholar]
- O'Leary T, Williams AH, Franci A, Marder E. Cell types, network homeostasis, and pathological compensation from a biologically plausible ion channel expression model. Neuron. 2014;82:809–821. doi: 10.1016/j.neuron.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/S0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- Orr HT, Zoghbi HY. SCA1 molecular genetics: a history of a 13 year collaboration against glutamines. Hum Mol Genet. 2001;10:2307–2311. doi: 10.1093/hmg/10.20.2307. [DOI] [PubMed] [Google Scholar]
- Ottolia M, Toro L. Potentiation of large conductance KCa channels by niflumic, flufenamic, and mefenamic acids. Biophys J. 1994;67:2272–2279. doi: 10.1016/S0006-3495(94)80712-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz G, Nelson CD, Koski DM, Henry PG, Marjanska M, Deelchand DK, Shanley R, Eberly LE, Orr HT, Clark HB. Noninvasive detection of presymptomatic and progressive neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci. 2010;30:3831–3838. doi: 10.1523/JNEUROSCI.5612-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patt S, Gertz HJ, Gerhard L, Cervós-Navarro J. Pathological changes in dendrites of substantia nigra neurons in Parkinson's disease: a Golgi study. Histol Histopathol. 1991;6:373–380. [PubMed] [Google Scholar]
- Prüss H, Derst C, Lommel R, Veh RW. Differential distribution of individual subunits of strongly inwardly rectifying potassium channels (Kir2 family) in rat brain. Brain Res Mol Brain Res. 2005;139:63–79. doi: 10.1016/j.molbrainres.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Quayle JM, McCarron JG, Brayden JE, Nelson MT. Inward rectifier K+ currents in smooth muscle cells from rat resistance-sized cerebral arteries. Am J Physiol. 1993;265:C1363–1370. doi: 10.1152/ajpcell.1993.265.5.C1363. [DOI] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19:1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüb U, Schöls L, Paulson H, Auburger G, Kermer P, Jen JC, Seidel K, Korf HW, Deller T. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog Neurobiol. 2013;104:38–66. doi: 10.1016/j.pneurobio.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H, Sausbier U, Sailer CA, Feil R, Hofmann F, Korth M, Shipston MJ, Knaus HG, Wolfer DP, Pedroarena CM, Storm JF, Ruth P. Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci U S A. 2004;101:9474–9478. doi: 10.1073/pnas.0401702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott SA. Dendritic atrophy and remodeling of amygdaloid neurons in Alzheimer's disease. Dementia. 1993;4:264–272. doi: 10.1159/000107332. [DOI] [PubMed] [Google Scholar]
- Sepúlveda FV, Pablo Cid L, Teulon J, Niemeyer MI. Molecular aspects of structure, gating, and physiology of pH-sensitive background K2P and Kir K+-transport channels. Physiol Rev. 2015;95:179–217. doi: 10.1152/physrev.00016.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servadio A, Koshy B, Armstrong D, Antalffy B, Orr HT, Zoghbi HY. Expression analysis of the ataxin-1 protein in tissues from normal and spinocerebellar ataxia type 1 individuals. Nat Genetics. 1995;10:94–98. doi: 10.1038/ng0595-94. [DOI] [PubMed] [Google Scholar]
- Shakkottai VG, do Carmo Costa M, Dell'Orco JM, Sankaranarayanan A, Wulff H, Paulson HL. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci. 2011;31:13002–13014. doi: 10.1523/JNEUROSCI.2789-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen AM, Bean BP. Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci. 2003;23:9650–9663. doi: 10.1523/JNEUROSCI.23-29-09650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahira M, Sakurai M, Sakurada N, Sugiyama K. Fenamates and diltiazem modulate lipid-sensitive mechano-gated 2P domain K(+) channels. Pflugers Arch. 2005;451:474–478. doi: 10.1007/s00424-005-1492-5. [DOI] [PubMed] [Google Scholar]
- Urabe M, Ding C, Kotin RM. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther. 2002;13:1935–1943. doi: 10.1089/10430340260355347. [DOI] [PubMed] [Google Scholar]
- Walter JT, Alviña K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- Waters MF, Minassian NA, Stevanin G, Figueroa KP, Bannister JP, Nolte D, Mock AF, Evidente VG, Fee DB, Müller U, Dürr A, Brice A, Papazian DM, Pulst SM. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat Genetics. 2006;38:447–451. doi: 10.1038/ng1758. [DOI] [PubMed] [Google Scholar]
- Wolfart J, Neuhoff H, Franz O, Roeper J. Differential expression of the small-conductance, calcium-activated potassium channel SK3 is critical for pacemaker control in dopaminergic midbrain neurons. J Neurosci. 2001;21:3443–3456. doi: 10.1523/JNEUROSCI.21-10-03443.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K. Somatic and dendritic small-conductance calcium-activated potassium channels regulate the output of cerebellar purkinje neurons. J Neurosci. 2003;23:2600–2607. doi: 10.1523/JNEUROSCI.23-07-02600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT. Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1. J Biol Chem. 2009;284:7425–7429. doi: 10.1074/jbc.R800041200. [DOI] [PMC free article] [PubMed] [Google Scholar]