

Abstract
Myelin pallor in HIV+ individuals can occur very early during the disease process. While myelin damage might partly originate from HIV-induced vascular changes, the timing suggests that myelin and/or oligodendrocytes (OLs) may be directly affected. Histological (Golgi–Kopsch, electron microscopy) and biochemical studies have revealed an increased occurrence of abnormal OL/myelin morphology and dysregulated myelin protein expression in transgenic mice expressing the HIV-1 transactivator of transcription (Tat) protein. This suggests that viral proteins by themselves might cause OL injury. Since Tat interacts with NMDARs, we hypothesized that activation of NMDARs and subsequent disruption of cytoplasmic Ca2+ ([Ca2+]i) homeostasis might be one cause of white matter injury after HIV infection. In culture, HIV-1 Tat caused concentration-dependent death of immature OLs, while more mature OLs remained alive but had reduced myelin-like membranes. Tat also induced [Ca2+]i increases and Thr-287 autophosphorylation of Ca2+/calmodulin-dependent protein kinase II β (CaMKIIβ) in OLs. Tat-induced [Ca2+]i was attenuated by the NMDAR antagonist MK801, and also by the AMPA/kainate receptor antagonist CNQX. Importantly, both MK801 and CNQX blocked Tat-induced death of immature OLs, but only MK801 reversed Tat effects on myelin-like membranes. These results suggest that OLs can be direct targets of HIV proteins released from infected cells. Although viability and membrane production are both affected by glutamatergic receptor-mediated Ca2+ influx, and possibly the ensuing CaMKIIβ activation, the roles of AMPARs and NMDARs appear to be different and dependent on the stage of OL differentiation.
SIGNIFICANCE STATEMENT Over 33 million individuals are currently infected by HIV. Among these individuals, ∼60% develop HIV-associated neurocognitive disorders. Myelin damage and white matter injury have been frequently reported in HIV patients but not extensively studied. Clinical studies using combined antiretroviral therapy (cART) together with adjunctive “anti-inflammatory” drugs show no improvement over cART alone, suggesting existence of injury mechanisms in addition to inflammation. In our studies, oligodendrocytes exhibited rapid increases in intracellular Ca2+ level upon HIV-1 transactivator of transcription (Tat) exposure. Thus, immature and mature oligodendrocytes can be direct targets of Tat. Since ionotropic glutamate receptor antagonists can partially or fully reverse the detrimental effects of Tat, glutamate receptors could be a potential therapeutic target for white matter damage in HIV patients.
Keywords: calcium, HIV, neuroAIDS, NMDA, oligodendrocyte, Tat
Introduction
Although combined antiretroviral therapy (cART) dramatically decreases mortality and neurological deficits, 60% of the HIV-infected population still experience HIV-associated neurocognitive disorders (HAND; Williams et al., 2009; Power et al., 2012). Despite viral suppression, infected brain cells, including microglia and astrocytes, continuously express and secrete viral proteins. HIV-1 transactivator of transcription (Tat) is a highly conserved HIV protein that can be detected in blood (Xiao et al., 2000), extracellular matrix (Urbinati et al., 2005a), and CSF (Wang et al., 2014) in HIV+ individuals, even with cART (Johnson et al., 2013). In addition to its essential role in viral replication and efficient viral gene transcription, Tat has also been shown to be a toxic factor in the CNS that leads to glial inflammatory responses (Nath et al., 2000; El-Hage et al., 2006, 2008a) and neuronal cell death (Zou et al., 2011; Rumbaugh et al., 2013).
Oligodendrocytes (OLs) are myelinating CNS cells. OL dysfunction can lead to abnormal myelination, impaired neuronal signaling, and axon degeneration, resulting in serious neurological deficits. OLs do not express the CD4 receptor, which is the main cellular receptor for HIV entry (Sattentau et al., 1986). However, OLs from postmortem HIV-infected tissues show upregulated expression of p53 and BAX (Jayadev et al., 2007), indicating activation of death pathways. Early studies using Tat transgenic mice showed that OLs exhibit caspase-3 activation and decreased process length when Tat expression was induced for 7 d (Hauser et al., 2009), suggesting that Tat may affect OL morphology and function in vivo.
OL lineage cells express functional ionotropic glutamate receptors (iGluRs), including AMPARs and NMDARs, both in vivo and in vitro (Káradóttir et al., 2005; Salter and Fern, 2005; Micu et al., 2006; Alix and Fern, 2009). The expression of these receptors on OLs is highly heterogeneous. Individual cells express different levels of iGluRs, and the distribution, subunit components, and downstream signaling pathways of different iGluRs may vary (Káradóttir et al., 2005; Matute et al., 2006; Micu et al., 2006). Adding further complexity, iGluR expression in OLs is also developmentally regulated. AMPARs are expressed at all developmental stages, while NMDARs show later expression (Salter and Fern, 2005). In addition, the majority of NMDARs expressed on OLs are clustered on processes and the myelin sheath while AMPARs are evenly distributed on the cell body (Micu et al., 2006). Activation of iGluRs results in immature OL death (Deng et al., 2003; Follett et al., 2004) and myelin disruption (Micu et al., 2006). Importantly, since OL NMDARs are less susceptible to Mg2+ blockade (Káradóttir et al., 2005), and OL AMPARs lack the Ca2+-impermeable GluR2 subunit (Hollmann and Heinemann, 1994; Matute et al., 2002), the level of extracellular glutamate necessary to injure OLs may be much lower than required for excitotoxic neuronal injury.
Using the Golgi–Kopsch procedure, we observed an increased occurrence of OLs with aberrant morphology in the corpus callosum and anterior commissure of transgenic mice expressing HIV-1 Tat. Electron microscopy (EM) also demonstrated disrupted myelin structure in the caudate–putamen. In Western blots of tissues from the same mice, myelin protein expression was abnormal. Since Tat-induced activation of iGluRs and resulting Ca2+ influx cause neuronal injury (King et al., 2006; Kim et al., 2008), we hypothesized a similar mechanism for OLs. Increased cytoplasmic Ca2+ ([Ca2+]i) and Ca2+/calmodulin-dependent protein kinase II β (CaMKIIβ) activation occurred in immature and mature OLs with Tat treatment; both were attenuated by NMDAR and AMPA/kainate receptor (AMPA/KAR) antagonists. Additionally, Tat caused immature OL death and reduced myelin-like membrane production by mature OLs, both in a dose-dependent manner. Both effects were blocked by MK801, while CNQX only blocked immature OL death. Thus, iGluRs are potential mediators of HIV-induced white matter (WM) damage through CaMKIIβ activation. Since these experiments were performed using a highly purified OL culture model, the effects likely reflect direct actions of Tat, rather than a response to Tat-induced inflammation.
Materials and Methods
All experiments involving animals were performed in compliance with procedures reviewed and approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee.
Tat transgenic mice and in vivo Tat expression.
Experiments used a transgenic mouse engineered to inducibly express HIV-1IIIB Tat1–86. Tat expression is controlled by a tet responsive element, whose activation is controlled by a reverse tetracycline transactivator (rtTA) transgene driven by a human glial fibrillary acidic protein (GFAP) promoter (Hauser et al., 2009; Hahn et al., 2012). Within the CNS, Tat expression is induced in astroglia by administration of doxycycline (DOX). HIV transgenic (Tat+) and Tat− control littermates were fed DOX-containing chow (6 g/kg; Harlan Laboratories) ad libitum to induce expression of Tat. Both acute and chronic time periods were chosen for Tat induction to visualize early changes in OL processes (Golgi impregnation, 7 d) that might precede later myelin changes (EM, 12 weeks) in Tat+ mice. The Tat− mice express rtTA but not Tat and are a control for possible effects of the foreign transcription factor. The expression of the rtTA transgene in Tat− mice and rtTA and Tat transgenes in Tat+ mice was confirmed by genotyping. While our studies were not designed to determine whether defects were specific to particular regions, we observed OL and myelin injury in three different CNS regions: caudate–putamen (observed by EM), corpus callosum (observed by Golgi impregnation), and anterior commissure (observed by Golgi impregnation).
Oligodendroglial cultures.
Primary cultures were prepared from mice (CD-1, Charles River Laboratory) of both sexes at postnatal day 0–1 as previously published (Zou et al., 2011). In brief, whole brains were dissected and minced before being incubated (37°C, 5% CO2) with 2.5 mg/ml trypsin (Sigma-Aldrich) and 0.015 mg/ml DNase (Sigma-Aldrich) in DMEM (Life Technologies) for 30 min. Tissue was triturated and resuspended in DMEM supplemented with 10% fetal bovine serum (Thermo Scientific Hyclone), 6 g/L glucose (Sigma-Aldrich), sodium bicarbonate, and penicillin/streptomycin (Life Technologies), filtered through nylon mesh with pore sizes 100 and 40 μm, and then plated in T25 flasks (Corning) precoated with poly-l-lysine (1 mg/ml; Sigma-Aldrich) at a density of 2 brains/flask.
At day 8, primary glial cultures were put on an orbital shaker (100 rpm, 20 min) to dislodge microglia loosely attached to the culture surface. Medium was replaced and the flasks were hit sharply against the table 5–10 times to release as many O2A/glial progenitor cells as possible. The suspension was panned on a plastic, nontissue culture dish for 2 h to remove adherent astroglia and microglia. Progenitors were plated in serum-free medium supplied with 10 ng/ml CNTF (Peprotech), 5 μg/ml N-acetyl-L-cysteine (Sigma-Aldrich), and 15 nm triiodothyronine (Sigma-Aldrich) on culture surfaces coated with poly-l-lysine as described above. Medium was changed every 3 d.
Viral protein and receptor antagonist treatments.
OL cultures were treated with HIV-1 Tat1–86 (clade B, ImmunoDX) at 1, 10, or 100 nm ± MK801 (20 μm; Tocris Bioscience) and CNQX (20 μm; Tocris Bioscience), at day 2 or 7 after plating the enriched cultures. In selected studies, heat-inactivated Tat1–86 was used as a control. If the cells were treated concurrently with Tat and MK801 or CNQX, the receptor antagonists were added 30 min before Tat.
The concentrations of Tat used in our experiments (1–100 nm) are a reasonable approximation of titers in HIV-infected brains. The serum Tat concentration in HIV patients is between 2 and 40 ng/ml (Xiao et al., 2000). Tat has a high affinity for cell-surface proteins (e.g., heparin sulfate proteoglycan; Liu et al., 2000) and extracellular cellular matrix components (e.g., glycosaminoglycans; Xiao et al., 2000). Portions of cells adjacent to productively infected cells may be subjected to higher levels of Tat within the restricted confine of the extracellular space. Further, the potency of recombinant Tat, as used in our experiments, has been reported to be much less than that of secreted Tat (Li et al., 2008).
MK801 and CNQX concentrations were chosen to maximally block the NMDAR and AMPA//KAR, respectively, as previously reported (Salter and Fern, 2005). Nimodipine (10 μm), dantrolene (10 μm), dl-threo-β-benzyloxyaspartate (dl-TBOA; 100 μm), RS102895 (10 μm), SB328437 (10 μm), and the arginyl-glycyl-aspartyl-serine (RGDS) peptide (10 μm; all from Tocris Bioscience) were used when assessing effects of different inhibitors on Tat-induced [Ca2+]i change in OLs and were added 20 min before Tat treatment and Ca2+ imaging.
Time-lapse analysis of individual OLs.
Enriched immature OLs were plated on a 12-well plate at a density of 1.5 × 104 cells/well. After Tat ± MK801/CNQX treatment, the plate was transferred to a Heating Insert M12 (PeCon) and put on the scanning stage of a Zeiss Axio Observer Z1 microscope (Carl Zeiss Microscopy) equipped with an environmental incubator (PeCon) that keeps a constant temperature (37°C) and CO2 level (5%) during the experimental period. Both the heating insert and the environmental incubator were prewarmed to 37°C ≥1 h before the start of the experiment. For each treatment, >25 OLs were systematically yet arbitrarily chosen, and repeatedly imaged every hour using an automated stage controlled by the Zeiss Axiovision 4.8 software (Carl Zeiss). Viability of cells was confirmed at the end of each experiment using calcein-acetoxy methylester (calcein-AM; Life Technologies) staining. Time of cell death was determined by carefully assessing all preselected OLs in hourly digital images, using rigorous morphological criteria, including but not restricted to (1) loss of phase brightness, (2) degenerating processes, and (3) involution or fragmentation of cell soma. Data were analyzed at 4 h intervals and presented as mean percentage of survival ± SEM from ≥4 individual experiments (n ≥ 4; ≥25 cells analyzed per treatment, per N). Repeated-measures ANOVA and post hoc Bonferroni's test were used to compare whether differences between treatments were significant (p ≤ 0.05) or not significant (p > 0.05).
Live–dead assay.
Cell viability after Tat treatment was confirmed using the LIVE/DEAD Viability/Cytotoxicity Kit (Life Technologies). To make the working solution, 20 μl of 2 mm ethidium homodimer-1 (EthD-1) stock solution and 5 μl of 4 mm calcein-AM stock solution were added to 10 ml of sterile, tissue culture-grade Dulbecco's modified PBS (D-PBS) sequentially and vortexed for 20 s. Five hundred microliters of working solution were added to each well of a sterile 24-well plate. OLs were plated on 12-mm-diameter German glass coverslips (Chemglass Life Sciences) and treated with vehicle or 100 nm Tat for 24, 48, and 72 h. After treatment, coverslips were quickly rinsed in D-PBS once and transferred to individual wells of a 24-well plate to incubate in working solution (30 min, room temperature). Coverslips were then carefully inverted onto a clean Fisherbrand Superfrost Plus Gold microscope slide (Fisher Scientific) with 10 μl of fresh D-PBS and sealed with clear fingernail polish. Fluorescent (excitation/emission wavelengths: ∼495/∼515 nm for calcein-AM; ∼528/∼617 nm for EthD-1) and phase images were taken using a Zeiss Axio Observer Z1 microscope with Zeiss Axiovision 4.8 software (Carl Zeiss). Live or dead cells were defined by green (calcein-AM) or red (EthD-1) fluorescence, respectively. For each n (individual experimental group), 20 images were systematically, yet arbitrarily taken and analyzed, and the relative ratio of live/dead cells was quantified by numbers of green/red fluorescent pixels using ImageJ software (National Institutes of Health).
Intracellular calcium assessment.
After 2 or 7 d culture, OLs plated on 35 mm glass-bottom culture dishes (MatTek) were loaded with 2.5 μm fura-2-AM (Life Technologies) for 30 min at 37°C. Cells were washed twice and incubated in DMEM for another 30 min to ensure de-esterification of the AM group before transfer to the environmental incubator of the Zeiss Axio Observer microscope. Cytosolic Ca2+ concentration was assessed using the physiology module of the AxioVision software. Fura-2-AM was excited at 340 and 380 nm, and emissions collected at 510–540 nm. Data were collected every second for the first 90 s, then every 5 s for the next minute, then every 30 s for the rest of the 20 min experimental period. The F340/F380 ratio was converted to calcium concentration via a standard curve generated using a calcium calibration buffer kit (Life Technologies). Data were presented as mean [Ca2+]i ± SEM from ≥4 individual experiments (>25 cells analyzed per n).
CM-DiI live staining.
Cells from day 8 primary cultures were plated on 35 mm glass-bottom culture dishes (1.5 × 104 cells/dish) and incubated for 6 d in growth medium as described above. Two micrograms per milliliter of CM-DiI (Life Technologies) was added to the medium, and cells were incubated for 5 min before being transferred to 4°C for another 15 min. Cells were washed with prewarmed culture medium and placed back in the incubator. Treatments were applied the next day, before placement in the stage incubator. Individual OLs were selected as described in the repeated-measure paradigm. Fluorescent (excitation, 560/40 nm; emission, 630/75 nm) and phase images were taken at 0, 24, and 48 h after treatment. The membrane area of individual cells, defined as the total red fluorescent area surrounding the cell body minus the area of the cell body (Dennis et al., 2008), was quantified using IPLab imaging software (BD Biosciences).
Immunostaining.
Cells cultured on glass coverslips were fixed with 4% paraformaldehyde and permeabilized with Triton X-100. Neurons, astroglia, microglia, and OLs, were identified with antibodies specific to microtubule-associated protein 2 (1:1000; Abcam), GFAP (1:1000; Millipore), ionized calcium-binding adaptor molecule 1 (1:500; Wako Chemicals), and myelin basic protein (MBP, 1:1000; Abcam), and the O4 monoclonal antibody [grown in our laboratory from hybridoma cells (Knapp and Hauser, 1996), 1:20], respectively. Antibodies specific to αv or β3 subunits (1:1000; Abcam) were used to identify integrins on OLs. Corresponding secondary antibodies were conjugated to Alexa 488 and 594 (1:2000; Life Technologies). Cell nuclei were visualized with Hoechst 33342 dye (1:2000; Life Technologies); coverslips were mounted with Prolong Gold Antifade reagent (Life Technologies). Images were acquired using a Zeiss LSM 700 laser scanning confocal microscope and processed using the maximum projection module of Zen 2010 software (Carl Zeiss) to show cells in their entirety.
Western blotting.
Day 2 or 7 OLs were treated with vehicle or Tat for 30 min. Proteins from treated cells were extracted using RIPA buffer (Sigma-Aldrich) with protease and phosphatase inhibitors (Thermo Scientific). Striatal tissue from Tat transgenic mice was homogenized with three strokes (15 s/stroke) in a 2 ml ceramic bead tube (MO BIO Laboratories) using a Precellys 24 Homogenizer (Bertin Technologies) before protein extraction using RIPA buffer with protease and phosphatase inhibitors. The concentration of extracted proteins was assessed using the BCA assay (Thermo Scientific) before lysates were mixed at 1:1 ratio with 2× Laemmli buffer (Bio-Rad). Equal amounts of total protein (5–10 μg) from each sample were loaded on Criterion 4–20% SDS-polyacrylamide gels (Bio-Rad) and electrophoretically separated under constant voltage (130 V) for 1.5 h. Proteins were then transferred onto polyvinylidene difluoride membranes (Bio-Rad) with constant current (0.6 A, 2 h, 4°C). After transfer, membranes were blocked in 0.1% casein solution (Bio-Rad; 1 h, room temperature) before probing with primary antibodies for MBP (1:5000; Abcam), myelin associated glycoprotein (MAG, 1:3000; Abcam), phospho-CaMKII (1:1000; Abcam), CaMKIIβ (1:1000; Life Technology), and glyceraldehyde 3-phosphate dehydrogenase (1:3000; Abcam) at 4°C overnight. Membranes were incubated with appropriate IRDye secondary antibodies (1:3000; Li-COR) for 1 h at room temperature, and imaged using an Odyssey Imager (Li-COR). Protein bands were quantified using Li-COR image studio software. The phospho-CaMKII antibody binds both phospho-CaMKIIα and phospho-CaMKIIβ, but not phospho-CaMKIIγ or phospho-CaMKIIδ. Phosphor-CaMKIIα (∼50 kDa) and phosphor-CaMKIIβ (∼60 kDa) can be easily identified on the blot by their different sizes.
EM.
Tissue processing and EM were performed as previously published (Hahn et al., 2012). In brief, Tat+ mice fed with DOX-containing chow for 3 months were perfused with phosphate buffer, pH 7.4 (Sigma-Aldrich), containing 2% paraformaldehyde and 2% glutaraldehyde before fixation/staining in 1% osmium tetroxide for 1 h. Fixed tissues were dehydrated through graded ethanols and infiltrated overnight in Embed 812 (Electron Microscopy Sciences), then polymerized at 60°C for 2 d. Tissue blocks were sectioned at 600–700 Å thickness on a Leica EM UC6i ultramicrotome (Leica Microsystems), collected onto formvar-coated grids, and stained with uranyl acetate (5%) and Reynolds's lead citrate (Reynolds, 1963). EM images were taken on a JEOL JEM-1230 Transmission Electron Microscope (JEOL USA) at 10,000–200,000× magnification using a Gatan Ultrascan 4000 digital camera with DigitalMicrograph software (Gatan).
Golgi–Kopsch impregnation.
Twelve-week-old Tat transgenic mice received normal (Tat−) or DOX-containing chow (Tat− and Tat+) for 7 d. A modified Golgi–Kopsch procedure as previously published (Hauser et al., 1989) was used to randomly impregnate neurons and glia. In brief, mice were anesthetized by isoflurane inhalation and euthanatized by intracardiac perfusion with 2% potassium dichromate and 5% glutaraldehyde. Whole forebrains were taken out and immersed in perfusion solution for 5 d before being rinsed and replaced in aqueous 0.75% silver nitrate solution for another 5 d. After that, tissues were infiltrated with graded sucrose solutions containing 0.75% silver nitrate for 24 h and frozen on dry ice before cutting into 120-μm-thick serial coronal sections and thaw-mounting onto Superfrost-Plus slides (Fisher). Fluid to tissue volume ratios were ≥50:1, and nonmetallic devices were used throughout. Images of impregnated OLs were taken randomly from corpus callosum and anterior commissure. Impregnated OLs were identified using strict morphological criteria including small (∼10 μm diameter) cell body, myelin-forming processes aligned parallel to WM tracts, and appropriate localization. For each experimental condition (Tat−, Tat− + DOX, and Tat+ + DOX), four mice were used (N = 4) and ≥40 cells were examined per mouse.
Results
Oligodendrocyte morphology in Tat transgenic mice
Fibrous astrocytes and OLs, the major cell types with cell bodies in the corpus callosum and anterior commissure, can be easily distinguished from each other in Golgi–Kopsch-impregnated sections by their distinct morphologies. Significantly more OLs with abnormal morphology, including stunted or swollen processes, large (several micrometers in diameter), club-like cytoplasmic endings, and aberrant cell bodies were noted in Tat+ mice when compared with their Tat− siblings with/without DOX (Fig. 1).
Figure 1.

Tat induces aberrant OL morphology in vivo. Golgi–Kopsch impregnation was used to visualize the structure of OLs in Tat− mice receiving normal chow, and in Tat− and Tat+ mice fed DOX-containing chow for 7 d. A–H, Sample bright-field images are labeled with the genotype and treatment. A–E, The majority of OLs from corpus callosum and anterior commissure of Tat− mice fed normal or DOX-containing chow show normal morphology. These OLs have small (∼10 μm diameter) cell bodies with smooth edges and relatively thick and continuous processes extending from their cell body to the WM tracts. F–H, Although most OLs in DOX-induced Tat+ mice appear normal, a significantly higher percentage had an aberrant phenotype. These OLs exhibited abnormal varicosities along or at the end of their processes (arrowheads), thin and disrupted processes, and irregular cell bodies (arrows). I, Quantification of OLs with abnormal morphology in corpus callosum and anterior commissure of Tat− and Tat+ mice. In both regions, Tat induction resulted in significantly more OLs with pathological phenotype (*p < 0.05; 1-way ANOVA followed by post hoc Bonferroni's test; n = 4 individual experiments; ≥40 cells were counted for each n; Scale bar, 10 μm for all images).
Ultrastructure of Tat+ mice
We next asked whether Tat induction by DOX caused any injury at the subcellular level in the transgenic mice. EM revealed that many OLs in Tat+ mice had a seemingly normal morphology and subcellular structure after 3 month DOX induction (Fig. 2A), yet a subpopulation showed very clear deficits. Enlarged periaxonal collars with dark cytoplasm were relatively common, potentially indicating accumulation of periaxonal fluid due to a deficit in exuding ion/fluid in OLs (Byun and Delpire, 2007; Fig. 2B,C,E). Myelin splitting was also common and associated with both normal-appearing axons (Fig. 2C,D,M–P) and with axons exhibiting increased axoplasmic density (Fig. 2E). Abnormal membranous structures and membrane inclusions with apparent periodicity were often observed (Fig. 2D-F,H), indicating potential deficits in OL membrane turnover. Hypomyelinated axons (Fig. 2D,N) and myelin membranes without an associated axon (Fig. 2J,L) were also noted.
Figure 2.

Ultrastructural examination of caudate–putamen in Tat+ mice. A, Even after 3 months of continuous Tat expression, most OLs show a normal phenotype and subcellular structure (G, Golgi apparatus; RER, rough endoplasmic reticulum). B–P, Despite this observation, multiple abnormal structures were commonly observed and documented in a subset of Tat-exposed OLs. These include (1) enlarged periaxonal collars containing dark cytoplasm (B, C, E, black arrows), possibly due to accumulation of periaxonal fluid; (2) myelin splitting, suggesting a potential deficit of myelin compaction, found both with normal-appearing axons (M, O, P; C, D, N, arrowheads) and with axons exhibiting abnormally dense cytoplasm (E, arrowhead); (3) abnormal membranous structures (D, E, white arrows) and myelin-like membrane inclusions with obvious periodicity (F, H), indicating potential deficits in OL membrane turnover; (4) hypomyelination, as indicated by axons of similar calibers in the same region invested with myelin sheaths of different thickness (N, compare axon groups a–c vs a*, b*, c*, respectively; also, in D, note the axon marked with #, which is unmyelinated, while neighboring axons with similar diameters have normal compact myelin); and (5) axons with abnormally dense cytoplasm (E, Ax) and myelin membrane in the absence of axon (J, L).
Tat expression leads to downregulated myelin proteins in vivo
A deficiency in MAG, which mediates axon–glial contact, has been reported to cause enlarged periaxonal collars (Trapp et al., 1984; Montag et al., 1994); decreased levels of MBP are often found in transgenic mice with myelin defects (Popko et al., 1987; Imgrund et al., 2009). We thus examined the expression level of MAG and MBP in Tat transgenic mice. Western blots of lysates from striatum showed that Tat+ mice exhibit significantly decreased levels of MAG and MBP when compared with their Tat− littermates after 3 months of DOX induction (Fig. 3).
Figure 3.

In vivo Tat expression leads to decreased MBP and MAG. Western blot analysis of tissue lysate from transgenic mice after 3 month Tat induction showed decreased levels of both MBP and MAG. Protein expression was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH; n = 4; values are mean ± SEM; *p < 0.05, t test).
Characterization of OL cultures
We used dissociated murine OL cultures to study whether the abnormalities of OLs and myelin in vivo were a direct effect of Tat or an indirect effect of Tat-induced inflammation. The percentage of cells expressing specific neural and glial cell markers was examined in cultures at 2–10 d after enrichment. Identification of OLs was made by immunostaining with the O4 antibody, which primarily detects the sulfated galactolipid sulfatide. In the CNS, sulfatide is expressed only by OLs; it is synthesized by immature, proliferating OLs and its expression is maintained in mature cells. As shown in Table 1, <5% of cells are positive for cell markers other than O4 in day 2 and 5 OL cultures. This ratio increases to 7–10% at day 7 and 10. Hence, even after 10 d culture, ≥90% of cells in our culture are OLs. The majority of cells that are not O4+ are GFAP+ astrocytes (Table 1).
Table 1.
Percentage of cells expressing neuron/glial-specific markers at various times in vitro
| O4+ | GFAP+ | MAP2+ | Iba+ | |
|---|---|---|---|---|
| Day 2 (n = 4, 372 cells) | 98.76 ± 0.73% | 1.00 ± 0.58% | 0% | 0.24 ± 0.24% |
| Day 5 (n = 4, 413 cells) | 96.35 ± 0.49% | 3.15 ± 0.62% | 0% | 0.50 ± 0.29% |
| Day 7 (n = 4, 396 cells) | 93.18 ± 0.15% | 5.35 ± 0.61% | 0.25 ± 0.25% | 1.22 ± 0.46% |
| Day 10 (n = 4, 383 cells) | 90.60 ± 0.39% | 7.31 ± 0.37% | 0% | 2.10 ± 0.44% |
n, Number of individual cultures; Iba, ionized calcium-binding adaptor molecule 1.
Dose-dependent effect of Tat on viability of immature, but not mature, OLs in vitro
Day 2 and 7 OL cultures were treated with Tat or vehicle, and imaged hourly for 72 h. At day 2, almost all OLs showed an immature phenotype, with multiple fine processes extending from relatively condensed, ovoid cell bodies. In addition, there were no myelin-like membranes extended from processes (Fig. 4A,B). Over time, vehicle-treated cells showed signs of differentiation. The cell body became much larger with increased cytoplasmic area (Fig. 4A). The maturing OLs developed multiple, thick, primary processes from which emanated an extensive network of thinner processes with large, myelin-like membrane expansions that also were O4+. Compared with controls, the viability of immature OLs was significantly decreased in a dose-dependent manner after treatment with HIV-1 Tat (Fig. 4B,D–G). In addition, OLs surviving after 72 h lacked the extensive network of branching processes and myelin-like membrane extensions that are typical for healthy OLs at this age (Fig. 4A). A live/dead assay confirmed that OLs treated with Tat for 24–72 h exhibited significantly more EthD-1 staining and less calcein-AM staining than OLs in vehicle-treated groups (Fig. 4F,G). On the other hand, Tat treatment had no effect on the viability of mature OLs (day 7; Fig. 4C,D).
Figure 4.
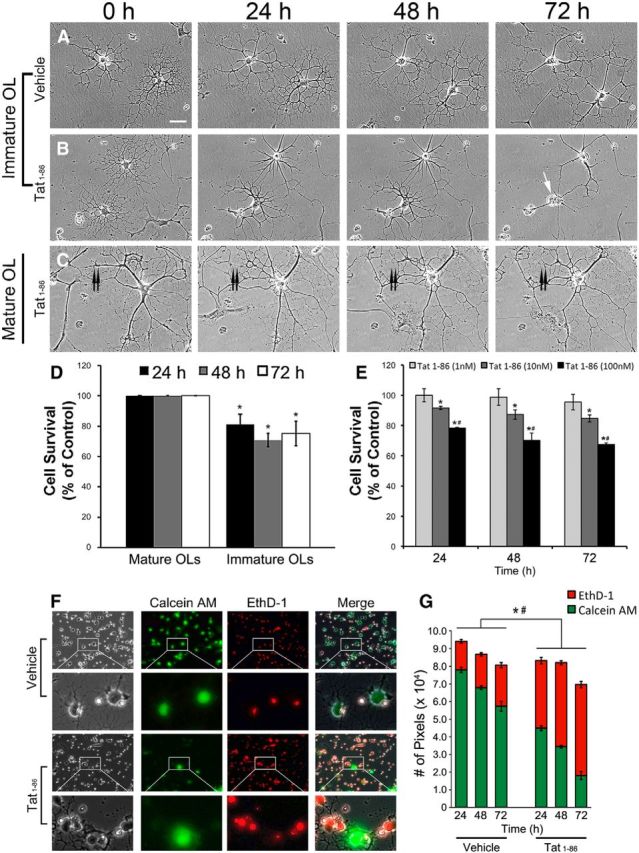
Survival of Tat-treated OLs. A–C, Sample images of OL cultures treated with vehicle or Tat (100 nm) after 0, 24, 48, and 72 h. A, Immature OLs treated with vehicle grow normally and differentiate over the 72 h period. At the end of 72 h, cells have complex process networks with myelin-like membranes (Scale bar, 100 μm). B, Immature OLs treated with 100 nm Tat. Both OLs in the field were healthy at 0 h. One OL was dead after 72 h (white arrow). Although the other OL survived, its process network was greatly reduced. An astrocyte at the lower right corner migrated out of the field by 24 h. C, A mature OL treated with 100 nm Tat appeared healthy and still maintained a complex process network after 72 h, even though it underwent extensive process reorganization. The double arrow marks a major process that was retracted. D, OL vulnerability to Tat was dependent on differentiation. Almost all mature OLs (>95%) survived for the 72 h experimental period, whether they were treated with vehicle or 100 nm Tat. In contrast, survival of immature OLs was significantly reduced by 100 nm Tat, even at 24 h. (*p < 0.05 vs immature OL controls; 1-way ANOVA followed by post hoc Bonferroni's test; n = 4 individual experiments; ≥25 cells were counted for each n). E, Concentration-dependent effect of Tat on immature OL survival. Immature OLs at day 2 were treated with 1, 10, and 100 nm Tat for 24, 48, and 72 h. Tat (1 nM) shows no obvious effect on OL survival, while 10 nm Tat reduces survival by 10–15% when compared with controls at 24, 48, and 72 h. The cytotoxic effect of 100 nm Tat is even more significant (22–35% reduction in survival; *p < 0.05 vs control; #p < 0.05 vs 10 nm Tat group; 2-way ANOVA followed by post hoc Bonferroni's test; n = 4 individual experiments; >40 cells were counted for each n). F, G, The live–dead assay shows that 100 nm Tat significantly increased immature OL death at 24, 48, and 72 h. F, Corresponding phase and fluorescent images of calcein-AM-labeled (live cells, green) and EthD-1-labeled (dead cells, red) day 2 immature OLs treated with vehicle or 100 nm Tat for 72 h. The majority of cells in the cultures appeared to shift from green to red with Tat treatment. G, At all time points, 100 nm Tat significantly increased the proportion of red pixels and reduced numbers of green pixels, indicating less OL survival with Tat and validating the visual observation in F (*p < 0.05 number of red pixels; Tat vs control of same time point; #p < 0.05, number of green pixels; Tat vs control of same time point; 1-way ANOVA followed by post hoc Bonferroni's test; n = 4 individual experiments; 20 randomly selected images were analyzed for each n).
Myelin-like membranes are reduced by Tat exposure
Since Tat did not reduce the viability of mature OLs, we examined whether it might affect their ability to form myelin-like membranes. Differentiating OLs in vitro produce characteristic membranes that are thought to be analogous to myelin membranes in the brain, and which contain all major lipids and proteins found in myelin within the CNS (Dubois-Dalcq et al., 1986; Knapp et al., 1987; Waggener et al., 2013). OL cultures at day 7 were labeled with CM-DiI before being treated with vehicle or Tat. Randomly selected cells with the morphology of maturing OLs were followed using a computer-controlled stage for 48 h; phase contrast and fluorescent images were taken at 0, 24, and 48 h. Calcein-AM was added to the cultures at the end of experiment to verify cell viability (Fig. 5A). Membrane area per OL, as indicated by CM-DiI labeling, was calculated using IPLab software as previously published (Dennis et al., 2008). In vehicle-treated cultures, individual OLs showed a ∼30% increase of membrane area at 24 h, suggesting new membrane production along with cell growth. This increased membrane size was maintained at 48 h (Fig. 5B). Tat (1 nM) did not have a significant effect on OL membrane area. However, adding 10 or 100 nm Tat to the medium significantly decreased OL membrane area at 24 h (∼75% of 0 h), and this decreased membrane size was maintained at 48 h (Fig. 5B).
Figure 5.

Tat reduces myelin-like membranes of mature OLs. A, Sample images showing changes in mature OL membrane area over 48 h. Cells were labeled with 2 μg/ml CM-DiI 1 d before Tat treatment. Phase contrast and fluorescent images of the same cell were taken at 0, 24, and 48 h. Phase and fluorescent images were merged to better visualize the myelin-like membrane structures. At the end of the experimental period, cells were labeled with calcein-AM to verify viability (green fluorescence). The yellow arrowhead indicates an area with an obvious reduction in the area of the cellular processes (loss of red fluorescence) over time. B, Quantification of the change in membrane area in vehicle and Tat-treated OLs. At both 24 and 48 h, vehicle-treated OLs showed ∼25% growth of membrane area. Tat (1 nM) did not have any significant effect on the membrane area when compared with controls. However, 10 or 100 nm Tat-treated OLs at 24 and 48 h exhibited ∼20% reductions in membrane area compared with 0 h (*p < 0.05, 2-way ANOVA followed by post hoc Bonferroni's test; n = 4 individual experiments; ≥25 cells were counted for each n).
Tat induces Ca2+ influx and CaMKIIβ activation in both mature and immature OLs in vitro
Since Tat has been reported to destabilize Ca2+ signaling in neurons (Kim et al., 2008; Perry et al., 2010), astroglia (El-Hage et al., 2005), and microglia (Sheng et al., 2000; Lokensgard et al., 2001), we hypothesized that the above-observed effects of Tat on OLs were mediated by changes in OL [Ca2+]i. To test this hypothesis, we first investigated whether Tat induces OL [Ca2+]i alterations. Figure 6 shows that Tat caused an increase in [Ca2+]i in both day 2 and 7 OLs in a dose-dependent manner. The Tat-induced Ca2+ influx seems to start from distant regions of processes upon Tat addition to the medium (Fig. 6B,D). When challenged with 100 nm Tat, [Ca2+]i levels rose to ∼150 nm in day 2 OLs and ∼230 nm in day 7 OLs, and these increased [Ca2+]i levels were maintained during a 20 min experimental period (Fig. 6B,D–F). The Tat-induced [Ca2+]i increase was dose dependent; with a lower Tat (1 nm) exposure, [Ca2+]i returned to control level after 5 min (Fig. 6E,F, cyan line). Furthermore, the Tat-induced [Ca2+]i increase was completely abolished in Ca2+-free medium (Fig. 6E,F, blue line) and greatly attenuated when Tat was heat-inactivated (Fig. 6E,F, black line).
Figure 6.
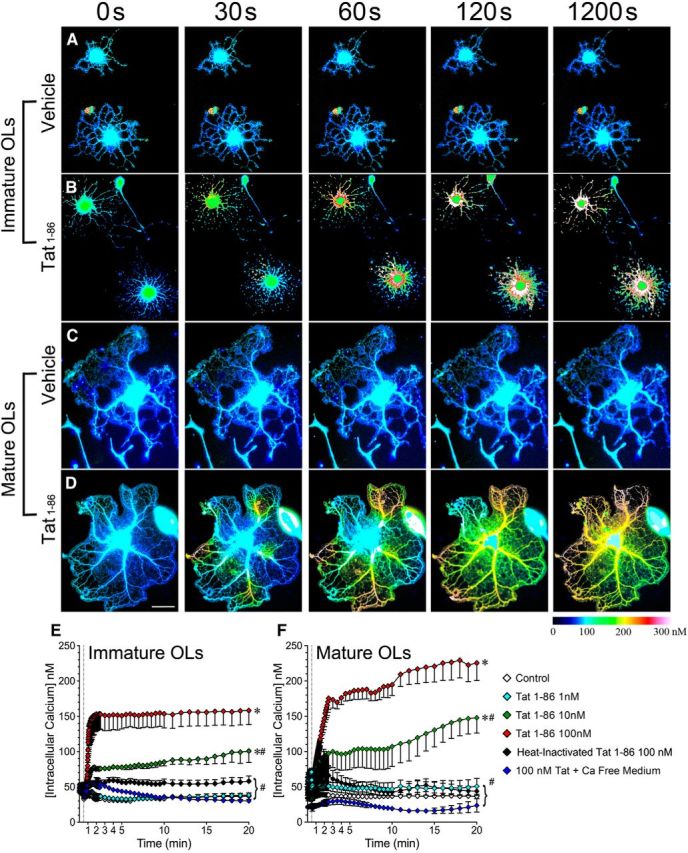
Tat induces Ca2+ influx in mature and immature OLs. A–D, Sample images showing changes of intracellular Ca2+ concentration over a 20 min period in immature (A, B) and mature (C, D) OLs treated with vehicle (A, C) or 100 nm Tat (B, D). [Ca2+]i was assessed by fura-2 AM 380/340 fluorescence ratio and displayed as pseudocolored images generated by Zeiss AxioVision 4.8 software. E, F, Tat induces dose-dependent [Ca2+]i increases in both immature (E) and mature (F) OLs. This effect is completely abolished in Ca2+-free medium, and greatly attenuated by heat inactivation of Tat [vertical dashed line indicates the time point (30 s) when treatment was added *p < 0.05 vs control; #p < 0.05, vs 100 nm Tat; 2-way ANOVA followed by post hoc Bonferroni's test; n = 4 individual experiments; ≥7 cells were counted for each n. Scale bar, 50 μm].
CaMKIIβ is a molecule central to OL intracellular Ca2+ signaling pathways (Derkach et al., 1999; Bayer and Schulman, 2001; Hudmon and Schulman, 2002) and has been reported to be important in maturation and myelination (Waggener et al., 2013). We investigated whether CaMKIIβ was activated as a consequence of the Tat-induced [Ca2+]i increase. Both immature and mature OLs exposed to 100 nm Tat for 30 min showed significantly increased levels of CaMKIIβ Thr-287 autophosphorylation, suggesting increased CaMKIIβ activation (Fig. 7). In either immature or mature OLs, the expression level of total CaMKIIβ was not affected by Tat treatment (data not shown).
Figure 7.

Tat induces CaMKIIβ Thr-287 phosphorylation in mature and immature OLs. Western blot analysis showed that Tat increased the level of CaMKIIβ Thr-287 phosphorylation in both immature and mature OLs by 30 min (n = 6; values are mean ± SEM; *p < 0.05, t test).
Blocking iGluRs attenuates the Tat-induced [Ca2+]i increases
Interactions between Tat and iGluRs lead to iGluR activation and Ca2+ influx, which have been reported extensively in neurons (Haughey et al., 2001; Kim et al., 2008). We therefore examined whether the Tat-induced Ca2+ influx in OLs is also mediated by iGluRs. Since iGluR expression and subunit composition is developmentally regulated and highly heterogeneous (Gallo and Ghiani, 2000; Itoh et al., 2002; Cavaliere et al., 2012) in OLs, we first verified the expression of functional iGluRs on our cultured OLs. In immature, day 2 OLs, NMDA at both 50 and 300 μm induced a minimum Ca2+ response (Fig. 8A, cyan and blue lines), while either 50 or 300 μm AMPA induced a significant Ca2+ influx lasting >20 min (Fig. 8A, yellow and brown lines). These results are consistent with the fact that the majority of the iGluRs expressed on immature OL somas are AMPA/KARs (Gallo and Ghiani, 2000; Itoh et al., 2002). In mature OLs, 50 μm NMDA invoked a small Ca2+ response (∼60 nm) that was significantly above control levels (∼40 nm; Fig. 6B, cyan line vs white line). A higher NMDA exposure (300 μm) elicited a sharp increase of [Ca2+]i to a level similar to that induced by Tat (Fig. 8B, blue line, vs Fig. 6F, red line). This high [Ca2+]i level came down to ∼60 nm within 10 min, but remained significantly higher than in vehicle-treated OLs. The AMPA responses of mature OLs were somewhat different. AMPA (50 μM) caused a moderate [Ca2+]i increase in mature OLs that recovered within 10 min (Fig. 8B, yellow line). AMPA (300 μM) induced an initial [Ca2+]i increase to ∼110 nm, which then rose consistently over the 20 min experimental period (Fig. 8B, brown line).
Figure 8.
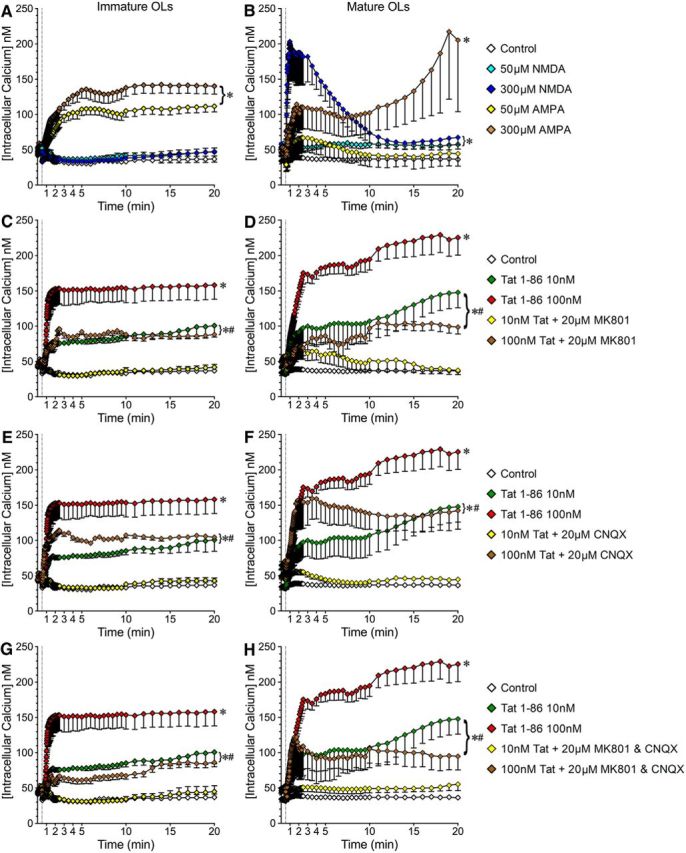
Tat-induced [Ca2+]i increases in mature and immature OLs are partially reversed by iGluR antagonists. A, B, Expression of functional NMDARs and AMPARs on OLs. A, In immature OLs, AMPA (at both 50 and 300 μm) causes [Ca2+]i increases while NMDA responses are minimal. B, In mature OLs, NMDA leads to a dose-dependent [Ca2+]i increase. AMPA elicits a [Ca2+]i increase only at the higher 300 μm concentration, and variability between cells is quite high. C–H, iGluR antagonists attenuate Tat-induced [Ca2+]i increases in OLs. C, E, G, In immature OLs, MK801 and/or CNQX blocks the [Ca2+]i increase induced by 10 nm Tat but only partially inhibits the [Ca2+]i increase elicited by 100 nm Tat. D, F, H, In mature OLs, the 10 nm Tat-induced [Ca2+]i increase is similarly abolished by MK801 and/or CNQX. In contrast, the [Ca2+]i increase induced by 100 nm Tat is only partially blocked by MK801 and/or CNQX (*p < 0.05 vs control; #p < 0.05 vs 100 nm Tat; 2-way ANOVA followed by post hoc Bonferroni's test; n = 4 individual experiments; ≥7 cells were counted per treatment for each n).
We next examined whether blocking iGluRs affected Tat-induced [Ca2+]i changes in OLs. In immature OLs, the [Ca2+]i increase induced by 10 nm Tat was blocked by both MK801 and CNQX (Fig. 8C,E, yellow line), but [Ca2+]i increases induced by 100 nm Tat were only reduced to ∼100 nm (Fig. 8C,E, brown line). MK801 and CNQX additively inhibited the effect of 100 nm Tat, but could not block the [Ca2+]i increase (Fig. 8G, brown line). The response in mature OLs was different. With 10 nm Tat, the initial [Ca2+]i level was significantly attenuated by MK801 or CNQX, and the [Ca2+]i level gradually returned to control over 5 min (Fig. 8D,F, yellow line). When MK801 and CNQX were added together, the effect of 10 nm Tat was blocked (Fig. 8H, yellow line). In contrast, only MK801 attenuated the initial [Ca2+]i rise caused by 100 nm Tat, which was reduced to ∼90 nm and maintained at this level over the 20 min experimental period. With CNQX, [Ca2+]i levels were reduced only at later time points (Fig. 8D,F, brown line). MK801 and CNQX together had no additive effect on 100 nm Tat-induced [Ca2+]i change in mature OLs (Fig. 8H, brown line). These results indicated that Tat-induced [Ca2+]i changes in OLs were mediated by both NMDAR and AMPA/KAR activation. Further, since iGluR antagonists (20 μm) did not block [Ca2+]i changes induced by the higher Tat concentration (100 nm), Tat may activate iGluRs in a way that cannot be completely inhibited by MK801/CNQX, or Tat may interact with other molecules on the surface of OLs that mediate [Ca2+]i change.
Blocking alternative, non-iGluR targets has no effect on Tat-induced increases in [Ca2+]i
Since Tat-induced [Ca2+]i increases were not blocked by inhibiting iGluRs, we tested a series of alternative Ca2+ modulators. The Tat-induced OL [Ca2+]i increase was abolished in Ca2+-free medium (Fig. 6E,F). To test whether the initial Ca2+ influx triggers Ca2+-induced Ca2+ release from internal stores, immature/mature OLs were treated with 10 μm dantrolene for 20 min before addition of Tat. This had no effect on Tat-induced [Ca2+]i change in OLs. OLs express abundant αvβ3 integrin (Fig. 9B), which has been reported to interact with Tat (Urbinati et al., 2005b, 2012). However, blocking integrin with the RGDS inhibitor peptide had no effect on Tat-induced [Ca2+]i increases in immature or mature OLs (Fig. 9A). Similarly, the L-type voltage-gated calcium channel blocker nimodipine (10 μm), CCR2 receptor inhibitor RS102895 (10 μm), CCR3 receptor inhibitor SB328437 (10 μm), and sodium-dependent glutamate transporter inhibitor dl-TBOA (100 μm) all had no effect on Tat-induced [Ca2+]i changes in either immature or mature OLs (Fig. 9A).
Figure 9.
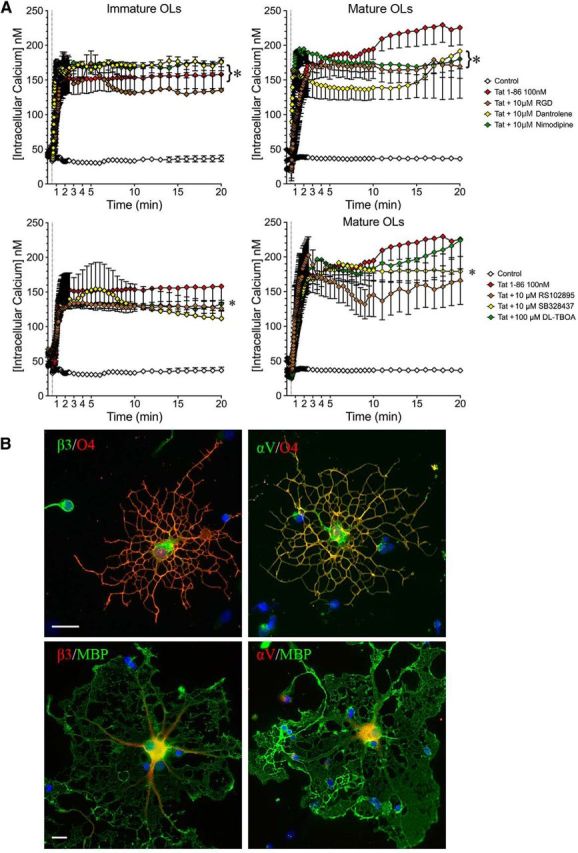
Blocking alternative, non-iGluR targets has no effect on Tat-induced increases in [Ca2+]i. A, Immature or mature OLs were exposed to nimodipine (L-type voltage-gated calcium channel inhibitor, 10 μm), dantrolene (ryanodine receptor inhibitor, 10 μm), dl-TBOA (sodium-dependent glutamate transporter inhibitor, 100 μm), RS102895 (CCR2 inhibitor, 10 μm), SB328437 (CCR3 inhibitor, 10 μm), or the RGDS peptide (integrin inhibitor, 10 μm) for 20 min before Tat treatment. None of these inhibitors affected Tat-induced OL [Ca2+]i increases (*p < 0.05 vs control; 2-way ANOVA followed by post hoc Bonferroni's test; n = 3 individual experiments; ≥7 cells were counted for each n). B, Expression of αV and β3 integrin on O4+ and MBP+ OLs (scale bar, 50 μm).
CNQX and MK801 effects on OL survival and membrane production
Since Tat-induced [Ca2+]i changes were attenuated by MK801 and/or CNQX, we examined whether blocking iGluRs would also reverse Tat effects on immature OL survival. Both CNQX and MK801 (20 μm) reversed the effect of Tat on OL viability at 24, 48, and 72 h. CNQX by itself had no effect on OL viability, while cells treated with MK801 alone had a significantly higher survival rate than controls (Fig. 10A). We next tested whether blocking iGluRs could also reverse the effect of Tat on the area of myelin-like membranes produced by mature OLs. Neither CNQX nor MK801 by themselves had any effect on the area of myelin-like membranes produced by mature OLs at either 24 or 48 h. Exposure to 100 nm Tat significantly decreased the size of the membrane area by >20% after 24 h. This effect was reversed by MK801, but not by CNQX. Results at 48 h were similar (Fig. 10B).
Figure 10.

Effects of iGluR antagonists MK801 and CNQX on Tat-induced functional effects. A, Immature OLs treated with 100 nm Tat exhibit a significantly reduced survival rate at all time points, compared with control. Either 20 μm CNQX or 20 μm MK801 completely reversed the Tat-induced cell death at all time points. CNQX (20 μM) by itself did not change the viability of immature OLs. On the other hand, at both 48 and 72 h, 20 μm MK801 significantly increased the viability of OLs (*p < 0.05, less survival vs control; #p < 0.05, greater survival vs control; 1-way ANOVA followed by post hoc Bonferroni's test; n = 4 individual experiments; ≥25 cells were counted for each n). B, At both 24 and 48 h, mature OLs treated with 100 nm Tat exhibit a significant, ∼20% reduction in total membrane area when compared with 0 h. In contrast, the membrane area of vehicle-treated OLs increased significantly by ∼25% at both time points. MK801 (20 μM), but not CNQX (20 μM), reversed the reduction in membrane area caused by Tat. Neither MK801 nor CNQX by itself had any effect on OL membrane production (*p < 0.05 vs 0 h; 1-way ANOVA followed by post hoc Bonferroni's test; n = 4 individual experiments; ≥25 cells were counted for each n).
Discussion
Our findings establish that HIV-1 Tat, through activation of iGluRs, increases [Ca2+]i and CaMKIIβ activation in OLs. In vivo, transgenic mice with inducible Tat expression had higher numbers of OLs with an abnormal phenotype in Golgi-impregnated cells, disturbances in myelin ultrastructure, and decreased myelin protein (MBP and MAG) expression. In highly purified murine OL cultures, the outcome of Tat exposure varied with the stage of differentiation. At Tat levels that reduced immature OL survival, mature OLs remained viable despite persistent elevations in [Ca2+]i, but had reduced myelin-like membrane areas.
Effects of Tat in vivo
Although cART suppresses CSF HIV RNA levels to below clinical detection limits (Staprans et al., 1999; Yilmaz et al., 2006), HAND still occurs in ∼60% of patients, suggesting that CNS injury can occur without viral replication. Tat may be an important mediator of HAND, as it can be produced in the absence of viral replication, and has been shown to be at high titers in the CSF of cART-controlled patients (Johnson et al., 2013). Multiple cellular abnormalities have been reported in Tat transgenic mice, including reactive astrogliosis and microgliosis (Bruce-Keller et al., 2008), elevated caspase-3 activation in OLs (Hauser et al., 2009), and synaptodendritic injury (Kim et al., 2003; Fitting et al., 2014), which coincides with deficits in behavior, including learning and memory (Carey et al., 2012; Fitting et al., 2013), and cognitive/motor impairments (Hahn et al., 2015). Most studies suggest Tat-induced inflammation as the predominant cause of CNS injury. However, controlled trials of cART with adjunctive “anti-inflammatory” agents do not show greater efficacy against HAND than cART alone (Tan and McArthur, 2012), suggesting neurotoxic effectors in addition to inflammation. Our in vivo studies showed that Tat caused myelin damage and OL injury, and we showed direct Tat toxicity to OLs using a highly purified culture system (>90% OLs) and Tat concentrations that reasonably approximate titers in HIV-infected brains (see Materials and Methods).
Tat-induced [Ca2+]i increases
Tat disrupts Ca2+ homeostasis in astroglia (El-Hage et al., 2005, 2008b), microglia (Sheng et al., 2000; Lokensgard et al., 2001), macrophages (Contreras et al., 2005), and neurons (Haughey and Mattson, 2002; Zhu et al., 2009). We found that Tat induced a concentration-dependent [Ca2+]i increase in both immature and mature OLs. The primary source of Ca2+ was extracellular, since the [Ca2+]i increases were abolished in Ca2+-free medium. Transient [Ca2+]i increases occurred at 1 nm Tat with more sustained [Ca2+]i elevations at 10–100 nm Tat. Higher Tat concentrations (≥10 nm) may elicit an initial [Ca2+]i increase that depolarizes the membrane, leading to secondary Ca2+ influx via voltage-gated calcium channels. Such channels are expressed by OLs and activated by cation influx via glycine receptors and Na+-dependent transporters (Kirischuk et al., 1995; Belachew et al., 2000). Alternatively, the [Ca2+]i increase induced by 1 nm Tat may be below the threshold required to activate Ca2+-induced Ca2+ release from internal Ca2+ stores. Neither the L-type voltage-gated calcium channel blocker nimodipine nor the ryanodine receptor inhibitor dantrolene affected Tat-induced [Ca2+]i increases (Fig. 9A), indicating involvement of alternative molecular targets (see below). Notably, 1 nm Tat does not affect immature OL viability or mature OL membrane area. These results strongly suggest the existence of a [Ca2+]i threshold above which downstream signaling molecules involved in functional effects, such as CaMKIIβ, will be activated.
iGluRs expression on OLs
We found that stage of development was a significant determinant in the OL response to Tat, possibly due to developmentally regulated expression of iGluRs on OLs (Deng et al., 2003). In OL cultures, AMPA elicited a dose-dependent [Ca2+]i increase in immature OLs, while even high NMDA concentrations elevated [Ca2+]i minimally (Fig. 6A). Mature OLs responded to both AMPA and NMDA with prominent [Ca2+]i increases. These results are consistent with earlier studies showing that NMDA-evoked currents were detected in precursor, immature, and mature OLs isolated from murine cerebellum and corpus callosum (Káradóttir et al., 2005; Salter and Fern, 2005). A more recent study by Alix and Fern (2009) also demonstrated the colocalization of NMDAR NR1 subunit and CNPase+ oligodendroglial processes in postnatal day 10 rat optic nerve. At odds with these reports, De Biase et al. (2010) assessed gene expression in acutely purified/sorted CNS cells (Cahoy et al., 2008) and concluded that levels of NMDARs and AMPARs are high in NG2+ OL progenitors, but decline markedly with maturation. Different receptor levels might reflect the effect of isolation procedures, although this has not been explored.
NMDARs on myelinating OLs in vivo were found clustered in processes and on myelin (Salter and Fern, 2005; Micu et al., 2006). Consistent with this localization, the [Ca2+]i increases mediated by Tat–NMDAR interactions began at distal processes and traveled proximally (Fig. 6). De Biase et al. (2010) released caged glutamate by UV photolysis in a 100 μm circle centered on the cell body, and only detected slight NMDA currents. Glutamate released in this way may be sequestered by neighboring cells before stimulating NMDARs on more distal OL processes. It is noteworthy that Ca2+ responses to AMPA in immature OLs are more homogeneous than in mature OLs, suggesting a higher variability of AMPAR expression or a greater diversity in the composition of AMPAR subunits with maturation, as seen in vivo (Matute et al., 2002; Williams et al., 2009).
iGluRs mediate Tat-induced [Ca2+]i increases
It is not surprising that Tat interacts with NMDARs on OLs, since Tat–NMDAR interactions occur in neurons (King et al., 2006; Kim et al., 2008; Capone et al., 2013; Rumbaugh et al., 2013;). However, since NMDA minimally affected [Ca2+]i in immature OLs, it was intriguing that Tat-induced [Ca2+]i increases were attenuated by MK801. Tat and NMDA appear to interact differently with NMDARs, since Tat–NMDAR interactions cause persistent, rather than transient, [Ca2+]i increases (Fig. 8; Fitting et al., 2014). CNQX also attenuated the effects of Tat on [Ca2+]i, which was unexpected since direct Tat–AMPA/KAR interactions have never been reported. In neurons, activation of cytoplasmic CaMKII recruits AMPARs from intracellular vesicles to cytoplasmic membranes, and increases the likelihood of channel opening by phosphorylating Ser-831 of AMPAR GluA1 subunits (Milstein and Nicoll, 2008; Derkach, 2011; Kristensen et al., 2011). We found that exposure to Tat leads to phosphorylation and activation of CaMKIIβ in OLs (Fig. 7). Since their expression of the major cytoplasmic Ca2+ binding protein calbindin is extremely low compared with other CNS cells (Baimbridge et al., 1992; Zhang et al., 2014), we propose that Tat–NMDAR interactions lead to an initial Ca2+ influx that activates CaMKIIβ, which then recruits and phosphorylates AMPARs and raises [Ca2+]i levels. Combined MK801 and CNQX had no additive effect, implicating additional, non-iGluR cellular components in Ca2+ responses to Tat. Blocking experiments preclude a role for integrins, chemokine receptors CCR3 and CCR2, sodium-dependent glutamate transporters, and ryanodine receptors in Tat-induced [Ca2+]i increases (Fig. 9). Similar to neurons (Fitting et al., 2014), Tat-activated NMDARs might cause Na+ influx, reversing Na+/Ca2+ exchange and destabilize mitochondrial inner membrane potential.
Effects of Tat on OL viability and function
Ca2+ influx via AMPA/KARs can activate various death pathways in OLs (Matute, 2011). AMPAR activation leads to caspase-8 recruitment and Bid truncation, while KAR activation has been linked to caspase-3 and caspase-9 activation (Sanchez-Gomez et al., 2003; Matute et al., 2006). We found that CNQX completely reversed Tat-induced death of immature OLs, implicating activation of AMPA/KAR-mediated death pathways. Surprisingly, MK801 had a similar effect, suggesting a requirement for both NMDARs and AMPA/KARs. This further implicates NMDAR-mediated AMPA/KAR activation. Interestingly, MK801 increased baseline survival of immature OLs, suggesting that some glutamate is released from OLs injured during the culture process.
Mature OLs remained viable when exposed to Tat, even when [Ca2+]i was elevated to a level that resulted in the death of immature OLs, consistent with a previous finding that Ca2+-dependent excitotoxicity decreases with OL maturation (McDonald et al., 1998; Kavanaugh et al., 2000; Itoh et al., 2002; Deng et al., 2003). Rumbaugh et al. (2013) have proposed that the peptide Tat31–61 directly binds the NR1 subunit resulting in NMDAR activation. In vivo, NMDARs are mainly clustered on cytoplasmic processes, including the myelin sheath, and their activation causes process degeneration and myelin injury (Micu et al., 2006). Consistent with this subcellular distribution of NMDARs, our experiments showed that Tat-induced [Ca2+]i increases in mature OLs originate at the distal processes and/or the peripheral edge of the myelin-like membranes (Fig. 6D). Importantly, the resultant reduction in myelin-like membranes can be reversed by MK801, but not CNQX (Fig. 10), suggesting the membrane retraction/OL injury is mediated by a common mechanism involving NMDAR activation and Ca2+ influx. It is possible that Ca2+ influx due to Tat–NMDAR interactions will generate a highly localized increase in [Ca2+]i within distal processes/myelin membranes that leads to local CaMKIIβ activation (Fig. 7). Waggener et al. (2013) showed that CaMKIIβ regulates OL maturation and CNS myelination by stabilizing the actin cytoskeleton. Our findings suggest that CaMKIIβ activation by Tat may also underlie some myelin damage with HIV infection.
Myelin damage and WM injury in HIV patients have been attributed to blood–brain barrier damage and to secondary effects mediated by inflammatory responses. Given the enriched nature of OL cultures in our studies, and their rapid increases in [Ca2+]i upon Tat exposure, we propose that immature and mature OLs can be direct targets of Tat. Since iGluR antagonists can partially or fully reverse the detrimental effects of Tat, glutamate receptors could be a potential therapeutic target for WM damage in HIV patients.
Footnotes
This work was supported by the National Institutes of Health [Grants F31 NS084838 (S.Z.), R21 NS069216 (P.E.K.), and K02 DA027374 (K.F.H.)]. Some fluorescent microscopy was performed at the Virginia Commonwealth University Department Anatomy and Neurobiology Microscopy Facility, supported in part with funding from National Institutes of Health–National Institute of Neurological Disorders and Stroke Center Core Grant 5P30NS047463.
The authors declare no competing financial interests.
References
- Alix JJ, Fern R. Glutamate receptor-mediated ischemic injury of premyelinated central axons. Ann Neurol. 2009;66:682–693. doi: 10.1002/ana.21767. [DOI] [PubMed] [Google Scholar]
- Baimbridge KG, Celio MR, Rogers JH. Calcium-binding proteins in the nervous system. Trends Neurosci. 1992;15:303–308. doi: 10.1016/0166-2236(92)90081-I. [DOI] [PubMed] [Google Scholar]
- Bayer KU, Schulman H. Regulation of signal transduction by protein targeting: the case for CaMKII. Biochem Biophys Res Commun. 2001;289:917–923. doi: 10.1006/bbrc.2001.6063. [DOI] [PubMed] [Google Scholar]
- Belachew S, Malgrange B, Rigo JM, Rogister B, Leprince P, Hans G, Nguyen L, Moonen G. Glycine triggers an intracellular calcium influx in oligodendrocyte progenitor cells which is mediated by the activation of both the ionotropic glycine receptor and Na+-dependent transporters. Eur J Neurosci. 2000;12:1924–1930. doi: 10.1046/j.1460-9568.2000.00085.x. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Turchan-Cholewo J, Smart EJ, Geurin T, Chauhan A, Reid R, Xu R, Nath A, Knapp PE, Hauser KF. Morphine causes rapid increases in glial activation and neuronal injury in the striatum of inducible HIV-1 Tat transgenic mice. Glia. 2008;56:1414–1427. doi: 10.1002/glia.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun N, Delpire E. Axonal and periaxonal swelling precede peripheral neurodegeneration in KCC3 knockout mice. Neurobiol Dis. 2007;28:39–51. doi: 10.1016/j.nbd.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capone C, Cervelli M, Angelucci E, Colasanti M, Macone A, Mariottini P, Persichini T. A role for spermine oxidase as a mediator of reactive oxygen species production in HIV-Tat-induced neuronal toxicity. Free Radic Biol Med. 2013;63:99–107. doi: 10.1016/j.freeradbiomed.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Carey AN, Sypek EI, Singh HD, Kaufman MJ, McLaughlin JP. Expression of HIV-Tat protein is associated with learning and memory deficits in the mouse. Behav Brain Res. 2012;229:48–56. doi: 10.1016/j.bbr.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaliere F, Urra O, Alberdi E, Matute C. Oligodendrocyte differentiation from adult multipotent stem cells is modulated by glutamate. Cell Death Dis. 2012;3:e268. doi: 10.1038/cddis.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras X, Bennasser Y, Chazal N, Moreau M, Leclerc C, Tkaczuk J, Bahraoui E. Human immunodeficiency virus type 1 Tat protein induces an intracellular calcium increase in human monocytes that requires DHP receptors: involvement in TNF-alpha production. Virology. 2005;332:316–328. doi: 10.1016/j.virol.2004.11.032. [DOI] [PubMed] [Google Scholar]
- De Biase LM, Nishiyama A, Bergles DE. Excitability and synaptic communication within the oligodendrocyte lineage. J Neurosci. 2010;30:3600–3611. doi: 10.1523/JNEUROSCI.6000-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Rosenberg PA, Volpe JJ, Jensen FE. Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc Natl Acad Sci U S A. 2003;100:6801–6806. doi: 10.1073/pnas.1136624100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis J, White MA, Forrest AD, Yuelling LM, Nogaroli L, Afshari FS, Fox MA, Fuss B. Phosphodiesterase-Ialpha/autotaxin's MORFO domain regulates oligodendroglial process network formation and focal adhesion organization. Mol Cell Neurosci. 2008;37:412–424. doi: 10.1016/j.mcn.2007.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach V. Zooming in on AMPA receptor regulation by CaMKII. Nat Neurosci. 2011;14:674–675. doi: 10.1038/nn.2852. [DOI] [PubMed] [Google Scholar]
- Derkach V, Barria A, Soderling TR. Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:3269–3274. doi: 10.1073/pnas.96.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois-Dalcq M, Behar T, Hudson L, Lazzarini RA. Emergence of three myelin proteins in oligodendrocytes cultured without neurons. J Cell Biol. 1986;102:384–392. doi: 10.1083/jcb.102.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF. Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia. 2005;50:91–106. doi: 10.1002/glia.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Wu G, Wang J, Ambati J, Knapp PE, Reed JL, Bruce-Keller AJ, Hauser KF. HIV-1 Tat and opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia. 2006;53:132–146. doi: 10.1002/glia.20262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Bruce-Keller AJ, Knapp PE, Hauser KF. CCL5/RANTES gene deletion attenuates opioid-induced increases in glial CCL2/MCP-1 immunoreactivity and activation in HIV-1 Tat-exposed mice. J Neuroimmune Pharmacol. 2008a;3:275–285. doi: 10.1007/s11481-008-9127-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Bruce-Keller AJ, Yakovleva T, Bazov I, Bakalkin G, Knapp PE, Hauser KF. Morphine exacerbates HIV-1 Tat-induced cytokine production in astrocytes through convergent effects on [Ca2+]i, NF-κB trafficking and transcription. PLoS One. 2008b;3:e4093. doi: 10.1371/journal.pone.0004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Ignatowska-Jankowska BM, Bull C, Skoff RP, Lichtman AH, Wise LE, Fox MA, Su J, Medina AE, Krahe TE, Knapp PE, Guido W, Hauser KF. Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol Psychiatry. 2013;73:443–453. doi: 10.1016/j.biopsych.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Knapp PE, Zou S, Marks WD, Bowers MS, Akbarali HI, Hauser KF. Interactive HIV-1 Tat and morphine-induced synaptodendritic injury is triggered through focal disruptions in Na+ influx, mitochondrial instability, and Ca2+ overload. J Neurosci. 2014;34:12850–12864. doi: 10.1523/JNEUROSCI.5351-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follett PL, Deng W, Dai W, Talos DM, Massillon LJ, Rosenberg PA, Volpe JJ, Jensen FE. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: a protective role for topiramate. J Neurosci. 2004;24:4412–4420. doi: 10.1523/JNEUROSCI.0477-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Ghiani CA. Glutamate receptors in glia: new cells, new inputs and new functions. Trends Pharmacol Sci. 2000;21:252–258. doi: 10.1016/S0165-6147(00)01494-2. [DOI] [PubMed] [Google Scholar]
- Hahn YK, Podhaizer EM, Hauser KF, Knapp PE. HIV-1 alters neural and glial progenitor cell dynamics in the central nervous system: coordinated response to opiates during maturation. Glia. 2012;60:1871–1887. doi: 10.1002/glia.22403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn YK, Podhaizer EM, Farris SP, Miles MF, Hauser KF, Knapp PE. Effects of chronic HIV-1 Tat exposure in the CNS: heightened vulnerability of males versus females to changes in cell numbers, synaptic integrity, and behavior. Brain Struct Funct. 2015;220:605–623. doi: 10.1007/s00429-013-0676-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughey NJ, Mattson MP. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S55–S61. doi: 10.1097/00126334-200210012-00005. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem. 2001;78:457–467. doi: 10.1046/j.1471-4159.2001.00396.x. [DOI] [PubMed] [Google Scholar]
- Hauser KF, McLaughlin PJ, Zagon IS. Endogenous opioid systems and the regulation of dendritic growth and spine formation. J Comp Neurol. 1989;281:13–22. doi: 10.1002/cne.902810103. [DOI] [PubMed] [Google Scholar]
- Hauser KF, Hahn YK, Adjan VV, Zou S, Buch SK, Nath A, Bruce-Keller AJ, Knapp PE. HIV-1 Tat and morphine have interactive effects on oligodendrocyte survival and morphology. Glia. 2009;57:194–206. doi: 10.1002/glia.20746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J. 2002;364:593–611. doi: 10.1042/BJ20020228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imgrund S, Hartmann D, Farwanah H, Eckhardt M, Sandhoff R, Degen J, Gieselmann V, Sandhoff K, Willecke K. Adult ceramide synthase 2 (CERS2)-deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J Biol Chem. 2009;284:33549–33560. doi: 10.1074/jbc.M109.031971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Beesley J, Itoh A, Cohen AS, Kavanaugh B, Coulter DA, Grinspan JB, Pleasure D. AMPA glutamate receptor-mediated calcium signaling is transiently enhanced during development of oligodendrocytes. J Neurochem. 2002;81:390–402. doi: 10.1046/j.1471-4159.2002.00866.x. [DOI] [PubMed] [Google Scholar]
- Jayadev S, Yun B, Nguyen H, Yokoo H, Morrison RS, Garden GA. The glial response to CNS HIV infection includes p53 activation and increased expression of p53 target genes. J Neuroimmune Pharmacol. 2007;2:359–370. doi: 10.1007/s11481-007-9095-x. [DOI] [PubMed] [Google Scholar]
- Johnson TP, Patel K, Johnson KR, Maric D, Calabresi PA, Hasbun R, Nath A. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc Natl Acad Sci U S A. 2013;110:13588–13593. doi: 10.1073/pnas.1308673110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Káradóttir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. 2005;438:1162–1166. doi: 10.1038/nature04302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh B, Beesley J, Itoh T, Itoh A, Grinspan J, Pleasure D. Neurotrophin-3 (NT-3) diminishes susceptibility of the oligodendroglial lineage to AMPA glutamate receptor-mediated excitotoxicity. J Neurosci Res. 2000;60:725–732. doi: 10.1002/1097-4547(20000615)60:6<725::AID-JNR4>3.3.CO%3B2-M. [DOI] [PubMed] [Google Scholar]
- Kim BO, Liu Y, Ruan Y, Xu ZC, Schantz L, He JJ. Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol. 2003;162:1693–1707. doi: 10.1016/S0002-9440(10)64304-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Martemyanov KA, Thayer SA. Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci. 2008;28:12604–12613. doi: 10.1523/JNEUROSCI.2958-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JE, Eugenin EA, Buckner CM, Berman JW. HIV tat and neurotoxicity. Microbes Infect. 2006;8:1347–1357. doi: 10.1016/j.micinf.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Scherer J, Möller T, Verkhratsky A, Kettenmann H. Subcellular heterogeneity of voltage-gated Ca2+ channels in cells of the oligodendrocyte lineage. Glia. 1995;13:1–12. doi: 10.1002/glia.440130102. [DOI] [PubMed] [Google Scholar]
- Knapp PE, Hauser KF. mu-Opioid receptor activation enhances DNA synthesis in immature oligodendrocytes. Brain Res. 1996;743:341–345. doi: 10.1016/S0006-8993(96)01097-9. [DOI] [PubMed] [Google Scholar]
- Knapp PE, Bartlett WP, Skoff RP. Cultured oligodendrocytes mimic in vivo phenotypic characteristics: cell shape, expression of myelin-specific antigens, and membrane production. Dev Biol. 1987;120:356–365. doi: 10.1016/0012-1606(87)90238-7. [DOI] [PubMed] [Google Scholar]
- Kristensen AS, Jenkins MA, Banke TG, Schousboe A, Makino Y, Johnson RC, Huganir R, Traynelis SF. Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat Neurosci. 2011;14:727–735. doi: 10.1038/nn.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Huang Y, Reid R, Steiner J, Malpica-Llanos T, Darden TA, Shankar SK, Mahadevan A, Satishchandra P, Nath A. NMDA receptor activation by HIV-Tat protein is clade dependent. J Neurosci. 2008;28:12190–12198. doi: 10.1523/JNEUROSCI.3019-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ. Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med. 2000;6:1380–1387. doi: 10.1038/82199. [DOI] [PubMed] [Google Scholar]
- Lokensgard JR, Hu S, Hegg CC, Thayer SA, Gekker G, Peterson PK. Diazepam inhibits HIV-1 Tat-induced migration of human microglia. J Neurovirol. 2001;7:481–486. doi: 10.1080/135502801753170345. [DOI] [PubMed] [Google Scholar]
- Matute C. Glutamate and ATP signalling in white matter pathology. J Anat. 2011;219:53–64. doi: 10.1111/j.1469-7580.2010.01339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute C, Alberdi E, Ibarretxe G, Sánchez-Gómez MV. Excitotoxicity in glial cells. Eur J Pharmacol. 2002;447:239–246. doi: 10.1016/S0014-2999(02)01847-2. [DOI] [PubMed] [Google Scholar]
- Matute C, Domercq M, Sánchez-Gómez MV. Glutamate-mediated glial injury: mechanisms and clinical importance. Glia. 2006;53:212–224. doi: 10.1002/glia.20275. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Levine JM, Qu Y. Multiple classes of the oligodendrocyte lineage are highly vulnerable to excitotoxicity. Neuroreport. 1998;9:2757–2762. doi: 10.1097/00001756-199808240-00014. [DOI] [PubMed] [Google Scholar]
- Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X, Trapp BD, McRory JE, Rehak R, Zamponi GW, Wang W, Stys PK. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature. 2006;439:988–992. doi: 10.1038/nature04474. [DOI] [PubMed] [Google Scholar]
- Milstein AD, Nicoll RA. Regulation of AMPA receptor gating and pharmacology by TARP auxiliary subunits. Trends Pharmacol Sci. 2008;29:333–339. doi: 10.1016/j.tips.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montag D, Giese KP, Bartsch U, Martini R, Lang Y, Blüthmann H, Karthigasan J, Kirschner DA, Wintergerst ES, Nave KA, et al. Mice deficient for the myelin-associated glycoprotein show subtle abnormalities in myelin. Neuron. 1994;13:229–246. doi: 10.1016/0896-6273(94)90472-3. [DOI] [PubMed] [Google Scholar]
- Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD. Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann Neurol. 2000;47:186–194. doi: 10.1002/1531-8249(200002)47:2<186::AID-ANA8>3.3.CO%3B2-V. [DOI] [PubMed] [Google Scholar]
- Perry SW, Barbieri J, Tong N, Polesskaya O, Pudasaini S, Stout A, Lu R, Kiebala M, Maggirwar SB, Gelbard HA. Human immunodeficiency virus-1 Tat activates calpain proteases via the ryanodine receptor to enhance surface dopamine transporter levels and increase transporter-specific uptake and Vmax. J Neurosci. 2010;30:14153–14164. doi: 10.1523/JNEUROSCI.1042-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popko B, Puckett C, Lai E, Shine HD, Readhead C, Takahashi N, Hunt SW, 3rd, Sidman RL, Hood L. Myelin deficient mice: expression of myelin basic protein and generation of mice with varying levels of myelin. Cell. 1987;48:713–721. doi: 10.1016/0092-8674(87)90249-2. [DOI] [PubMed] [Google Scholar]
- Power C, Hui E, Vivithanaporn P, Acharjee S, Polyak M. Delineating HIV-associated neurocognitive disorders using transgenic models: the neuropathogenic actions of Vpr. J Neuroimmune Pharmacol. 2012;7:319–331. doi: 10.1007/s11481-011-9310-7. [DOI] [PubMed] [Google Scholar]
- Reynolds ES. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol. 1963;17:208–212. doi: 10.1083/jcb.17.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh JA, Bachani M, Li W, Butler TR, Smith KJ, Bianchet MA, Wang T, Prendergast MA, Sacktor N, Nath A. HIV immune complexes prevent excitotoxicity by interaction with NMDA receptors. Neurobiol Dis. 2013;49:169–176. doi: 10.1016/j.nbd.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MG, Fern R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature. 2005;438:1167–1171. doi: 10.1038/nature04301. [DOI] [PubMed] [Google Scholar]
- Sánchez-Gómez MV, Alberdi E, Ibarretxe G, Torre I, Matute C. Caspase-dependent and caspase-independent oligodendrocyte death mediated by AMPA and kainate receptors. J Neurosci. 2003;23:9519–9528. doi: 10.1523/JNEUROSCI.23-29-09519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattentau QJ, Dalgleish AG, Weiss RA, Beverley PC. Epitopes of the CD4 antigen and HIV infection. Science. 1986;234:1120–1123. doi: 10.1126/science.2430333. [DOI] [PubMed] [Google Scholar]
- Sheng WS, Hu S, Hegg CC, Thayer SA, Peterson PK. Activation of human microglial cells by HIV-1 gp41 and Tat proteins. Clin Immunol. 2000;96:243–251. doi: 10.1006/clim.2000.4905. [DOI] [PubMed] [Google Scholar]
- Staprans S, Marlowe N, Glidden D, Novakovic-Agopian T, Grant RM, Heyes M, Aweeka F, Deeks S, Price RW. Time course of cerebrospinal fluid responses to antiretroviral therapy: evidence for variable compartmentalization of infection. AIDS. 1999;13:1051–1061. doi: 10.1097/00002030-199906180-00008. [DOI] [PubMed] [Google Scholar]
- Tan IL, McArthur JC. HIV-associated neurological disorders: a guide to pharmacotherapy. CNS Drugs. 2012;26:123–134. doi: 10.2165/11597770-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Quarles RH, Suzuki K. Immunocytochemical studies of quaking mice support a role for the myelin-associated glycoprotein in forming and maintaining the periaxonal space and periaxonal cytoplasmic collar of myelinating Schwann cells. J Cell Biol. 1984;99:594–606. doi: 10.1083/jcb.99.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbinati C, Bugatti A, Giacca M, Schlaepfer D, Presta M, Rusnati M. alpha(v)beta3-integrin-dependent activation of focal adhesion kinase mediates NF-kappaB activation and motogenic activity by HIV-1 Tat in endothelial cells. J Cell Sci. 2005a;118:3949–3958. doi: 10.1242/jcs.02518. [DOI] [PubMed] [Google Scholar]
- Urbinati C, Mitola S, Tanghetti E, Kumar C, Waltenberger J, Ribatti D, Presta M, Rusnati M. Integrin alphavbeta3 as a target for blocking HIV-1 Tat-induced endothelial cell activation in vitro and angiogenesis in vivo. Arterioscler Thromb Vasc Biol. 2005b;25:2315–2320. doi: 10.1161/01.ATV.0000186182.14908.7b. [DOI] [PubMed] [Google Scholar]
- Urbinati C, Ravelli C, Tanghetti E, Belleri M, Giacopuzzi E, Monti E, Presta M, Rusnati M. Substrate-immobilized HIV-1 Tat drives VEGFR2/alpha(v)beta(3)-integrin complex formation and polarization in endothelial cells. Arterioscler Thromb Vasc Biol. 2012;32:e25–e34. doi: 10.1161/ATVBAHA.111.242396. [DOI] [PubMed] [Google Scholar]
- Waggener CT, Dupree JL, Elgersma Y, Fuss B. CaMKIIβ regulates oligodendrocyte maturation and CNS myelination. J Neurosci. 2013;33:10453–10458. doi: 10.1523/JNEUROSCI.5875-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Jiang Z, Hou W, Li Z, Cheng S, Green LA, Wang Y, Wen X, Cai L, Clauss M, Wang Z. HIV Tat protein affects circadian rhythmicity by interfering with the circadian system. HIV Med. 2014;15:565–570. doi: 10.1111/hiv.12154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R, Yao H, Dhillon NK, Buch SJ. HIV-1 Tat co-operates with IFN-gamma and TNF-alpha to increase CXCL10 in human astrocytes. PLoS One. 2009;4:e5709. doi: 10.1371/journal.pone.0005709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, Jeang KT. Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc Natl Acad Sci U S A. 2000;97:11466–11471. doi: 10.1073/pnas.97.21.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz A, Svennerholm B, Hagberg L, Gisslén M. Cerebrospinal fluid viral loads reach <2 copies/ml in HIV-1-infected patients with effective antiretroviral therapy. Antivir Ther. 2006;11:833–837. [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Yao H, Peng F, Callen S, Buch S. PDGF-mediated protection of SH-SY5Y cells against Tat toxin involves regulation of extracellular glutamate and intracellular calcium. Toxicol Appl Pharmacol. 2009;240:286–291. doi: 10.1016/j.taap.2009.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou S, Fitting S, Hahn YK, Welch SP, El-Hage N, Hauser KF, Knapp PE. Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at μ-opioid receptor-expressing glia. Brain. 2011;134:3616–3631. doi: 10.1093/brain/awr281. [DOI] [PMC free article] [PubMed] [Google Scholar]