

Abstract
A specialized axonal ending, the basket cell “pinceau,” encapsulates the Purkinje cell axon initial segment (AIS), exerting final inhibitory control over the integrated outflow of the cerebellar cortex. This nonconventional axo-axonic contact extends beyond the perisomatic chemical GABAergic synaptic boutons to the distal AIS, lacks both sodium channels and local exocytotic machinery, and yet contains a dense cluster of voltage-gated potassium channels whose functional contribution is unknown. Here, we show that ADAM11, a transmembrane noncatalytic disintegrin, is the first reported Kv1-interacting protein essential for localizing Kv1.1 and Kv1.2 subunit complexes to the distal terminal. Selective absence of these channels at the pinceau due to mutation of ADAM11 spares spontaneous GABA release from basket cells at the perisomatic synapse yet eliminates ultrarapid ephaptic inhibitory synchronization of Purkinje cell firing. Our findings identify a critical role for presynaptic K+ channels at the pinceau in ephaptic control over the speed and stability of spike rate coding at the Purkinje cell AIS in mice.
SIGNIFICANCE STATEMENT This study identifies ADAM11 as the first essential molecule for the proper localization of potassium ion channels at presynaptic nerve terminals, where they modulate excitability and the release of neural transmitters. Genetic truncation of the transmembrane disintegrin and metalloproteinase protein ADAM11 resulted in the absence of Kv1 channels that are normally densely clustered at the terminals of basket cell axons in the cerebellar cortex. These specialized terminals are responsible for the release of the neurotransmitter GABA onto Purkinje cells and also display electrical signaling. In the ADAM11 mutant, GABAergic release was not altered, but the ultrarapid electrical signal was absent, demonstrating that the dense presynaptic cluster of Kv1 ion channels at these terminals mediate electrical transmission. Therefore, ADAM11 plays a critical role at this central synapse.
Keywords: ataxia, disintegrin, epilepsy, Kv1.1, synchrony, presynaptic terminal
Introduction
Cerebellar basket cell terminals (BCTs) form the first axo-axonic synapses described in the mammalian CNS and possess unique characteristics combining chemical and electrical field inhibitory transmission (Gobel, 1971; Chan-Palay, 1974). Axons converging from three to seven neighboring basket cell interneurons initially establish conventional basal perisomatic GABAergic synaptic contacts at each Purkinje cell axon hillock and then extend distally to ensheath the axon initial segment (AIS) with elaborate ramified microtermini known as septate junctions, which are embedded within a tight glial meshwork (Sotelo, 2008). In these “pinceaux,” the presynaptic contact zone lacks both sodium channels and GABAergic vesicles (Iwakura et al., 2012), but is densely packed with Shaker type Kv1 voltage-gated potassium channels (McNamara et al., 1993; Laube et al., 1996). The postsynaptic AIS membrane lacks detectable GABARa1, bassoon, and neuroligin2 clusters, consistent with an absence of chemical transmission (Iwakura et al., 2012). It has been proposed that the pinceau/glial microcompartment contributes to synchronous basket cell inhibition of Purkinje cell firing by generating a hyperpolarizing electrical field first described at the goldfish Mauthner cell electrical synapse (Furukawa and Furshpan, 1963; Korn and Faber, 1975; Korn and Axelrad, 1980). High-temporal-resolution electrophysiological analysis has demonstrated ultrarapid electrical inhibition at the pinceau, faster than any described in the CNS (Blot and Barbour, 2014). Whether the dense distal cluster of presynaptic potassium channels play a role in the pinceau ephapse has been speculated (Laube et al., 1996; Southan and Robertson, 1998a, 1998b), but this is difficult to isolate experimentally.
The strategic arrangement of ion channels into subcellular compartments is a fundamental organizing principle linking neuronal form and function. In mammalian axons, homomeric and heteromeric Kv1 channels of the Shaker superfamily mediate a range of fast activating, slowly inactivating (delayed rectifier) repolarizing currents. Their precise subcompartmental profiles at the AIS, nodes of Ranvier, branch points, and preterminal axon impart specificity to neuronal signaling dynamics during development, plasticity, and disease (Trimmer, 2015). Although local transport and recycling of these channels at the AIS and nodes rely upon trafficking molecules for site-specific targeting and homeostasis (Gu and Barry, 2011; Rasband, 2011; Galiano et al., 2012; Kole and Stuart, 2012), little is known about Kv1 channel stabilization at central presynaptic terminals. In cerebellar basket cells, Kv1.1/1.2 are expressed in both axons and terminals (McNamara et al., 1993; Laube et al., 1996). These channels coassemble with cytoplasmic Kvβ2 auxiliary subunits that modulate current kinetics but are not required for correct Kv1 trafficking (Pongs and Schwarz, 2010). Similarly, cytoplasmic PSD-95, also present at BCTs, binds to Kv1 heteromers but is not essential for their membrane localization or stabilization (Ogawa et al., 2010). Although functional studies have revealed that molecules such as secretin regulate Kv1.2 endocytosis at BCTs (Williams et al., 2012) and proteomic studies identify other proteins that copurify with Kv1.1, including LGI1 (Schulte et al., 2006), no molecule has yet been shown to be essential for Kv1 targeting and retention at the presynaptic terminal.
ADAM proteins are members of the transmembrane zinc protease superfamily, which have modular design, diverse functions, and region-specific expression in brain (Seals and Courtneidge, 2003). The presence of the ADAM (a disintegrin and metalloprotease) domain define their major activities, whereas the variable length cytoplasmic tail allows for regulation by internal and external cell signaling. Members of the nonenzymatic clade (ADAM11, ADAM22, ADAM23) lack the Zn-finger motif responsible for metalloproteinase catalytic activity and are expressed in the nervous system, where genetic mutation leads to ataxia phenotypes (Mitchell et al., 2001; Sagane et al., 2005; Takahashi et al., 2006). ADAM22 and ADAM23 have been linked to ion channel function in axons and both associate with Kv1.1/Kv1.2 channels at the AIS, juxtaparanodes, and BCTs; however, ADAM22 is not required for K+ channel clustering at the pinceau (Ogawa et al., 2010). The close paralog ADAM11 has strong cerebellar expression and a likely role in neuron–glial relationships during development (Rybnikova et al., 2002). In this study, we examine the role of ADAM11 in Kv channel localization at BCTs and the effects of its loss upon synaptic transmission.
Materials and Methods
Experimental procedures
Animals.
Mutant mice were generated at Rockefeller University and maintained at Baylor College of Medicine, where they were housed at 22°C, fed ad libitum, and submitted to a 12 h light/dark cycle. All procedures were in accordance with the guidelines of the National Institutes of Health and approved by the Animal Care and Use Committees of both institutions.
Construction and confirmation of the Adam11-targeted mutation
The Adam11 gene is composed of up to 27 alternatively spliced exons. A bacterial artificial chromosome (BAC) clone encompassing the Adam11 locus was obtained from a C57BL/6 genomic library (BacPac Resources, RP24-166M9). Using genetic recombination, a targeting vector was constructed from a MC1-TK-containing plasmid with an engineered loxP site within intron 11 and an Frt-Pgk-em7-Neo-Frt-loxP cassette within intron 18 of the Adam11 gene. The Neo allele of the Adam11 gene was generated by homologous recombination in isogenic mouse embryonic stem (ES) cells and correctly targeted ES clones were identified. Once the Adam11 mutant mouse line was established, mRNA was isolated from wild-type (WT) and Adam11Δ12–18 littermates. Using gene-specific primers targeting the region flanking the deletion, the RobusT RT-PCR kit (Finnzyme) was used to produce and subsequently amplify cDNA of the Adam11 transcript. PCR products were size resolved on a 2% gel. The resulting 702-bp (WT) and 128-bp (Adam11Δ12–18) bands were excised, gel purified using the Zymoclean Gel DNA Recovery kit (Zymogen), and sequenced in both directions. The resulting sequences were aligned against the reference Adam11 sequence (NM_001110778) to verify the deletion and resulting frameshift leading to a premature stop codon and truncated ADAM11 protein. Homozygous Adam11Δ12–18 mice were also crossed with transgenic mice expressing a BAC construct containing channelrhodopsin-2 (CHR-2) under control of the nNOS promoter (Kim et al., 2014) to allow selective optogenetic activation of interneurons.
Genotyping
Genomic DNA was isolated from tail clips using DirectPCR Lysis Reagent (Viagen Biotech). The genotypes of Adam11 mutant mice of either sex were determined using PCR amplification of specific alleles using three primers: (1) a mutant-specific primer (ggttgccatggaaacgtgggca), (2) a WT-specific primer (gcggtgaatgagtgtgacattg), and (3) a common reverse primer (accgtccagcttgtgaaggttag). The PCR yields a 620-bp band corresponding to the WT allele and a 700-bp band corresponding to the mutant allele.
mRNA isolation and RT-PCR
Mice of either sex were killed by cervical dislocation. Whole brains were promptly removed under sterile, RNAase-free conditions, dissected, and frozen immediately before use. Samples were homogenized in TRIzol reagent with a Tissue-Tearor (BioSpec Products), followed by phenol-chloroform extraction. mRNA was resuspended in sterile, RNase-free water and resolved on a nondenaturing agarose gel to verify a successful extraction. The RT-PCR to generate cDNA was performed with poly-dT primers using the Phusion RT-PCR Kit (Finnzyme) for transcript detection.
Primer design and PCR amplification
Gene-specific primers were designed against mouse nucleotide sequences using the Primer3Plus webserver. For specific amplification of Adam11 transcripts, primers were designed targeting both the N and C termini of the protein, as well as spanning the targeted deletion from intron 11 to intron 18 [ENSMUST00000103081 (short isoform); ENSMUST00000068150 (long isoform)], with at least one primer designed to cross exon–exon boundaries. Primers against Tuba1 (β-actin), Kcna4 (Kv1.4), Kcnb1 (Kv2.1), Hcn1 (HCN1), Adam22 (ADAM22), and Adam23 (ADAM23) were similarly designed. Primers for Kcna1 (Kv1.1) and Kcna2 (Kv1.2), which contain a single coding exon, were each designed within a single exon. In brief, PCR amplification of cDNA from WT and Adam11Δ12–18 mice was performed using the gene-specific primers and appropriate kit controls according to recommended protocol. All reactions were performed using the HotStar Master Mix (QIAGEN) with an initial 10 min 94°C denaturation, followed by 35 cycles of 30 s denaturation (94°C), 1 min of annealing (56°C), and 1 min of elongation (72°C), followed by a final 10 min elongation step (72°C).
Immunofluorescence studies
Age-matched adult WT and mutant mice (2–4 months old) of either sex were killed with isofluorane and perfused by left ventricular cannulation with cold PBS. Whole brains were extracted, postfixed in 4% PFA for 40 min, and cryoprotected 4°C in a 30% sucrose in PBS solution overnight. Brain tissue was blocked and frozen in OCT medium (Tissue-Tek) for 2 h. Then, 20 μm parasagittal and horizontal sections were cut using a cryostat maintained at −20°C (Microm, HM505E) and then mounted on gel-coated slides for processing. (Superfrost Plus; Fisher Scientific).
After thawing, brain sections were washed three times in PBS and then incubated in blocking solution containing 10% BSA and 0.3% Triton X-100 in PBS for 1 h at room temperature. Primary antibodies [Kv1.1 (K20/78), Kv1.2 (K14/16), Kv1.4 (K13/31), Kvβ2 (K17/70), PSD-95 (K28/43), ADAM22 (N46/30) and HCN1 (N70/28); all from Neuromab]; LGI1 (ab30868; Abcam); and ADAM11 (ab56101; Abcam; N terminus) were diluted in blocking solution and tissue sections were incubated for 2 h at room temperature. Sections were washed three times in antibody vehicle and incubated with secondary Alexa Fluor (Invitrogen) antibodies for 1 h in the dark. Finally, sections were washed in blocking solution followed by PBS and then dried at room temperature in the dark. Coverslips were applied using ProLong Gold Antifade containing DAPI (Invitrogen, P36931). Images were captured using an Olympus IX71 inverted microscope and a Zeiss Axioplan 2 confocal microscope equipped with LSM 510 acquisition software. Whole images were colored to reflect the fluorophore used and were adjusted equally for brightness and contrast using Adobe Photoshop (version 12.0.2). For each comparison, immunohistochemical staining of all sections was performed identically in a single batch, mounted, and then imaged. Each image was batch adjusted automatically in an identical manner so that all images are fully comparable in light intensity and contrast between genotypes. All scaling of brightness and contrast were applied to every pixel in the image. No nonlinear adjustments were performed.
Immunoprecipitation and Western blot
Whole mouse brains from WT or Adam11 mutant mice of either sex were extracted and homogenized with a Tissue-Tearor, followed by sonication in ice-cold RIPA buffer containing protease and phosphatase inhibitors (Santa Cruz Biotechnology) and centrifugation to pellet cell debris. The supernatant protein lysates were separated on 12% Tris-HEPES-SDS polyacrylamide gels and analyzed by Western immunoblot using anti-Kv1.1 antibody diluted in vehicle (1:400), anti-Kv1.2 antibody (1:400), anti-Kvβ2 antibody (1:200), anti- PSD-95 antibody (1:400), anti-ADAM22 antibody (1:200), and anti-HCN1 antibody (1:200) (all from Neuromab), followed by HRP-tagged donkey anti-mouse secondary antibody (1:5000; Santa Cruz Biotechnology), and detected with a commercial chemiluminescent substrate (SuperSignal; Pierce).
For immunoprecipitation experiments, cell lysates were diluted to 1 mg/ml and each 1 ml sample was precleared for 1 h with 30 μl of protein A-Sepharose (Pharmacia) and incubated overnight with 5 μg of anti-ADAM11 antibody (H-19; Santa Cruz Biotechnology; “internal region” per datasheet). All incubations were performed at 4°C with constant rotation. Antibody-bound protein complexes were captured by the addition of 30 μl of protein A-Sepharose and incubated for another 2 h. Protein A-Sepharose was pelleted by centrifugation and the immunoprecipitated protein complexes were eluted using Laemmli sample buffer before SDS-PAGE and Western blotting as described above.
Electrophysiology
Purkinje cell recordings.
Parasagittal cerebellar slices (300 μm thickness) were prepared from mutant and WT of either sex cerebella at postnatal day 55 (P55)–P75. Whole-cell voltage-clamp recordings of spontaneous IPSCs were made from visually identified Purkinje cell somas. Electrodes were pulled from borosilicate glass capillary pipettes and fire-polished to yield a resistance of 3∼4 MΩ before seal formation,
Solutions and drugs.
Experiments were performed at 29–30°C using an extracellular bath solution composed of the following (in mm): NaCl 120, KCl 3, NaHCO3 25, MgCl2 1.0, CaCl2 2, NaH2PO4 1.25, and glucose 10. The solution was maintained during an experiment at pH 7.4 by bubbling with 5% CO2 + 95% O2. The pipette for whole-cell Purkinje cell recording was filled with a solution containing the following (in mm): Cs-Methanesulfonate 140, HEPES 5, TEA-Cl 10, EGTA 1, Mg-ATP 1, Na2-phosphocreatine 10, 0.2% biocytin, pH 7.3, with CsOH. For cell-attached recordings, the recording pipette was filled with the extracellular bath solution. For cell-attached recordings of PC action potential firing at the soma, action currents were detected with the holding potential set at 0 mV.
Light stimulation and recordings.
A custom-made array of 3 × 7 LEDs with the center emitting wavelength of 469 nm was used as a light source to illuminate the field centered parallel to and immediately above the Purkinje cell layer. Experiments were performed in a darkened room. Changes in the light intensity reflected from the slice surface were monitored with a photodiode positioned adjacent to and oriented toward the slice surface. Microelectrode patch Purkinje cell recordings were sampled at a frequency of 40 kHz. The stimulation protocol consisted of a train of 5 light pulses (each 3 ms) at an interval of 1 s. The protocol was repeated 200 times (1000 photic stimuli total) at a rate of every 10 s for each cell. The LED light signal was used as a time marker to align each recording electrode trace. The number of action potentials falling into each discrete 1 ms interval was counted and accumulated over traces. The counting of Purkinje cell action potentials started 47 ms before the onset of light stimulation and ended 100 ms after the offset of the light-charging current. To pool the results from different cells, data from each cell is first normalized to its mean value before the light onset and then grouped.
Behavior
All experiments were performed on mice of either sex 4–8 weeks of age between 9:00 h and 18:00 h during the normal light cycle time for the mice.
Footprint test.
To assess gait, paws were painted with black ink and the mouse placed in a 28 × 9 inch (L × W × H) path floored with lined paper. Stride length was measured as the distance connecting two consecutive paw prints from the same paw. Gait width was measured as the distance between contralateral paw prints.
Rotarod test.
Performance was scored on a 3-cm-diameter accelerating rotating rod (Ugo Basile, 47600). On the first day of training, mice were trained for 5 min with the apparatus at a constant 4 RPM. Mice were then placed on the apparatus with a 5 min protocol set to accelerate from 4 to 20 RPM and again from 4 to 40 RPM. All subsequent testing was performed with a 4–80 RPM accelerating protocol and time to fall was measured. Three trials were performed on days 1, 2, and 7. No training was provided after the first day. Mice rested a minimum of 30 min between each trial.
Cold swim test.
A plastic cage was filled with water (17°C) to a depth of 10 cm. Mice were placed in the center of the tank to swim for a period of 2 min and then placed on a dry platform at room temperature and video-monitored.
Video-EEG monitoring.
Mice of either sex were briefly anesthetized with avertin and implanted with silver wire cortical EEG electrodes positioned through bilateral cranial burr holes and connected to a microconnector. Several days after recovery, video-EEG activity was acquired during daily 4 h monitoring sessions using a Stellate Systems Harmonie system.
Statistical analysis
Spike frequency data are expressed as mean ± SD. Data in the gait analysis were analyzed using unpaired t test and error bars in the figures show mean ± SEM. Data in the rotating rod test were analyzed by two-way ANOVA with repeated-measures and Bonferroni post hoc testing. Statistical significance was considered to be p < 0.05. Statistical analysis was conducted using GraphPad Prism 5.0 software.
Results
To explore the role of ADAM11 in potassium channel localization, we examined a mutant (C57BL/6−Adam11Δ12–18) mouse line in which targeted disruption of the Adam11 gene removed exons 12–18, resulting in a truncated ADAM11 protein lacking the disintegrin and transmembrane domains (Fig. 1).
Figure 1.
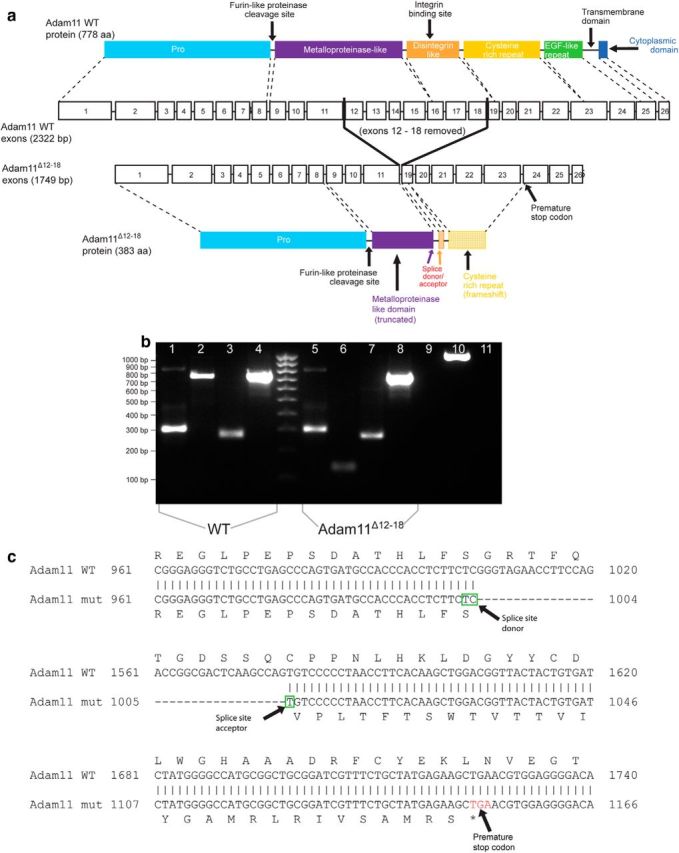
Generation of Adam11Δ12-18 mutant mice. a, Schematic of Adam11 gene structure and location of targeted deletion. b, PCR showing that the N- (lanes 1, 5) and C termini (lanes 3, 7) of the transcript are the same size in WT and mutant brains. The amplicon of the mutant deletion (lanes 2, 6) is ∼570 bp shorter than WT. Controls shown: 4, 8 = β-actin; 9 = (−)cDNA control; 10 = (+)RT control; 11 = (−)RT control. c, Sanger sequencing of the final transcript reveals the deletion of exons 12–18 results in a frameshift giving rise to a premature stop codon and truncated protein. Only the portion of the ADAM11 protein upstream of the mutation is expected to be translated in the mutant mouse. This mutation shortens the metalloproteinase-like domain, removing the entire disintegrin-like domain including the integrin-binding site. The final protein also lacks the cysteine-rich and EGF-like repeats and the transmembrane and cytoplasmic domains.
ADAM11 is expressed in basket cell pinceaux and is essential for Kv1 channel subunit localization
We evaluated the expression of Kv1 potassium channels reported to interact with ADAM family members in brain sections of WT and Adam11Δ12–18 mutant mice using subunit-specific antibodies. Both the Kv1.1 and Kv1.2 subunits are highly expressed in cerebellar BCTs and intense staining surrounding the Purkinje cell soma and the pinceaux expansions encircling the AIS is evident in confocal images of WT cerebellum (Fig. 2a,g,e). In contrast, Adam11Δ12–18 mutants (n = 5) retained perisomatic axon Kv1 staining, but all lacked immunofluorescence for both Kv1 subunits in the pinceaux (Fig. 2b,h,f). ADAM11 is expressed at low levels in PCs, but highly expressed in the BCT and pinceaux of WT mice (Fig. 2c,i) and spatially overlaps with Kv1.1/Kv1.2. ADAM11 is absent in the BCTs of mutants (Fig. 2d,j,f; n = 2), suggesting a critical role in heteromeric Kv1 localization. We next investiated whether the Kv1-associated proteins Kvβ2 and PSD-95, also normally present at BCTs, were mislocalized. Staining for Kvβ2 (Fig. 3a) and PSD-95 (Fig. 3c) was also absent from the BCT pinceaux (Fig. 3b,d; n = 3). ADAM22, normally prominent in WT BCTs, was also missing in the mutant pinceaux (n = 2; Fig. 3e,f).
Figure 2.
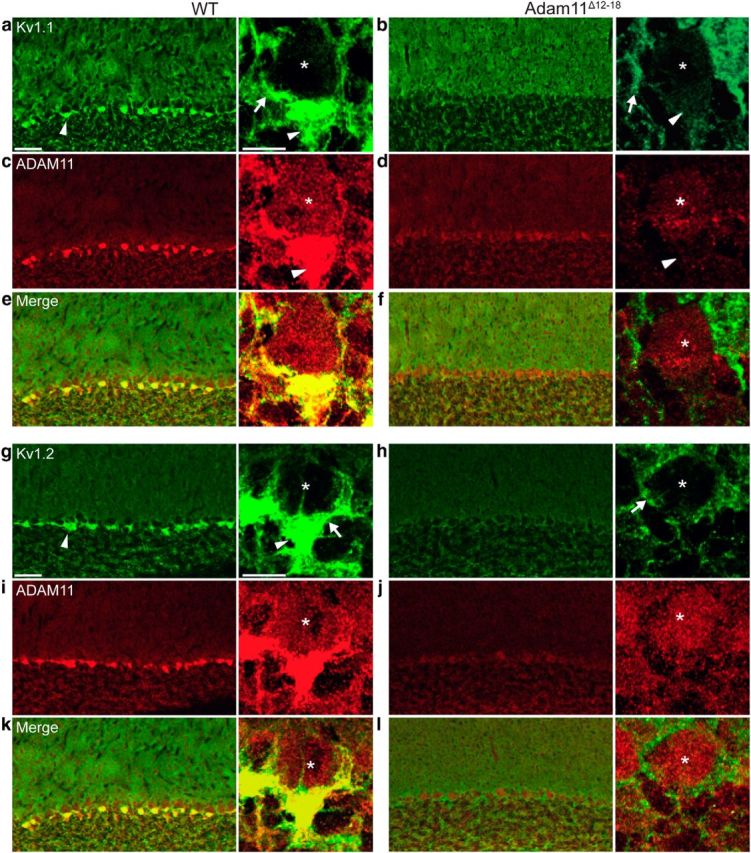
Absence of Kv1.1 and Kv1.2 channels in Adam11Δ12-18 mutant BCT pinceaux. Epifluorescence detection of specific antibodies to Kv1.1/Kv1.2 channel subunits reveals their high density at BCT's in cerebellar cortex of WT mouse (a, g, arrowheads). High-magnification inset shows BCT terminal axon (arrow) surrounding Purkinje cell soma (*), whereas the terminal pinceau surrounds the AIS (arrowhead). Kv1.1/Kv1.2 staining in Adam11Δ12-18 BCTs is detectable at perisomatic axon membrane (arrow), but absent at the pinceau (b, h, arrowheads). ADAM11 is detected faintly in PC soma (*) and densely at BCTs of WT (c, i, arrowheads), but truncated protein is reduced in mutant BCTs (d, j, arrowheads). Merged images verify ADAM11, Kv1 colocalization (e, f, and k, l; n = 5 mutant mice). Scale bars in a, 50 μm/10 μm (inset).
Figure 3.

Mutant BCTs lack Kvβ2 subunit, PSD-95, and ADAM22, but accurately target HCN1 channels. Kvβ2 expression in WT pinceaux (a, arrowhead) is lost in Adam11Δ12-18 BCTs (b). PSD-95 and ADAM22 are present in WT pinceaux (c, e), but missing from pinceaux in the mutant (d, f). HCN1 localization at WT BCT pinceaux (g) is unaffected by ADAM11 truncation (h). i, Coimmunoprecipitation using an ADAM11-specific antibody revealed native interactions with Kv1.1, Kv1.2, PSD-95, and ADAM22, but not HCN1, from WT and Adam11Δ12-18 whole-brain lysates. Scale bars in a, 50 μm/10 μm (inset).
ADAM11 associates with Kv1.1, Kv1.2, PSD-95, and ADAM22
To further characterize the pivotal localizing role of Adam11, we investigated whether ADAM11 and Kv1 subunits associate in vivo. A specific antibody was used to immunoprecipitate ADAM11 from WT whole-brain protein lysates, followed by Western immunoblotting with subunit-specific antibodies to Kv1.1 and Kv1.2, revealing a native association of ADAM11 with both subunits (n = 3; Fig. 3i). Similar experiments revealed an association between the cytosolic scaffolding protein PSD-95 (n = 3) and ADAM11. We found that ADAM22 also coimmunoprecipitates with ADAM11 (n = 2), suggesting that it may also participate in stabilizing the Kv1 cluster at the terminal. These data identify ADAM11 as an essential targeting factor for multiple interacting proteins at the pinceau, consisting of Kv1.1, Kv1.2, Kvβ2, PSD-95, and ADAM22.
HCN1 and LGI1 demonstrate unaltered lamination of ADAM11-deficient pinceaux
Because the extracellular disintegrin-like domain of ADAM11 could participate in cell adhesion, axonal guidance, and synaptic stabilization (Rybnikova et al., 2002) and because Kv1 channels themselves may share a role in axon terminal differentiation (Budnik et al., 1990), we considered whether their absence at BCTs might simply reflect a defect in terminal development. We found that specific antibody staining for HCN1, a channel present at WT pinceaux (Luján et al., 2005) that does not associate with ADAM11 (Fig. 3i), confirmed both a normal BCT morphology and lamination in the mutants (n = 4; Fig. 3g,h). Furthermore, transcription of all Kv1 channel complex genes was detected in both WT and mutant brains (data not shown). These data indicate that, in contrast to an adhesion factor such as neurofascin (Buttermore et al., 2012), neither the missing ADAM11 and ADAM22 proteins nor the presynaptic Kv1.x complex is essential for BCT site-specific synaptogenesis. Furthermore, the HCN1 expression indicates that the general integrity of axonal ion channel trafficking to the pinceaux is retained despite their absence. We also examined the localization of LGI1, a known ADAM11/ADAM22- and Kv1-interacting protein (Sagane et al., 2008). Two models currently describe how LGI1 may function as a ligand packaged into vesicles and secreted into the cleft, where it may link presynaptic ADAM22 and postsynaptic ADAM23 (Senechal et al., 2005; Fukata et al., 2010), and/or as an integral presynaptic auxiliary subunit of Kv1-containing channels capable of altering channel kinetics (Schulte et al., 2006). In agreement with the first model, we found a punctate intracellular pattern of LGI1 antibody staining in BCTs distinct from that displayed by the membrane-linked Kv1.2 subunit complex in WT BCTs (Fig. 4a,c,e). Furthermore, despite the loss of the Kv1-associated cluster, presynaptic LGI1 localization was not altered in Adam11Δ12–18 BCTs (Fig. 4b,d,f) or in Kv1.1-null mice (data not shown). These data suggest that LGI1 is not an exclusively integral membrane Kv1.1 complex subunit at these presynaptic terminals.
Figure 4.

LGI1 localization is retained in BCTs of WT, Adam11Ä12-18, and Kv1.1-null mutants. Kv1.2 and LGI1 staining in WT (a, c, merged image e) and mutant (b, d, merged image d) BCTs. Unlike Kv1 channels (Kv1.2 shown here), immunofluorescence for LGI1, a known interactor with ADAM11, ADAM22, ADAM23, and Kv1.2, is present in punctate granules in perisomatic axons surrounding PCs and in pinceaux in both WT and mutant BCTs (c, d, arrowheads). Scale bars, 50 μm/10 μm.
Absence of ephaptic inhibition of Purkinje cell firing in Adam11 mutants
Transmission at the basket cell/Purkinje cell synapse combines chemical and ephaptic signaling. To evaluate both components, we crossed Adam11Δ12–18 mutants with transgenic mice expressing a BAC construct containing CHR-2 under control of the nNOS promoter to allow selective optogenetic activation of interneurons. In the cerebellar cortex of these mice, only basket and stellate cells express CHR-2, which is intensely expressed throughout their cellular compartments, including the terminal pinceaux. The intrinsic properties of these interneurons are unaltered and photostimulation of cerebellar slices inhibits spontaneous firing of Purkinje cells (Kim et al., 2014). Due to the presence of CHR-2 within the pinceaux, it is likely that all BCT terminals contacting the patched PC are activated directly by the stimulus. Figure 5a shows a sample trace of IPSCs recorded from a Purkinje cell in WT CHR-2 labeled mice in response to a train of light stimulation of the cerebellar cortex slice (3 ms current charging pulse to LED light source at 1 s intervals). Under this protocol, basket/stellate cells recover well from light simulation, as evidenced by the stable generation of evoked IPSCs in Purkinje cells. Figure 5b shows the effect of photostimulation on Purkinje cell spontaneous firing. As expected, light- evoked IPSCs briefly silence Purkinje cells, followed by recovery of spontaneous firing. Figure 5c quantifies the pooled results from experiments (n = 6) shown in Figure 5b. For each cell, 1000 photic stimuli were delivered (200 traces, each sweep containing five light pulses). Traces of Purkinje cell firing during light stimulation were acquired and the number of action potentials during 1 ms intervals was plotted. As shown, light stimulation induces a fast onset (<1 ms) inhibition of Purkinje cell firing, followed by a silent period (10–20 ms), with variable recovery of the baseline firing rate that is complete by 50–70 ms after the stimulus. The inset of Figure 5c expands the analysis 10-fold to a time window of 10 ms with a shorter discrete spike counting interval (0.1 ms) for higher temporal resolution. As shown, ultrarapid inhibition of Purkinje cell firing by light stimulation starts at least 1 ms before the onset of the IPSC, indicating that the earliest phase of inhibition at the AIS spike initiation site cannot be explained by distant GABAergic synapses arising at dendritic (stellate cell) and perisomatic (basket cell) membranes and is likely caused by ephaptic transmission directly at the AIS.
Figure 5.

Illumination suppresses PC firing in mice expressing nNOS-driven CHR-2 in interneurons. a, Light-evoked IPSC recorded from a PC in cerebellar slice from WT mice expressing CHR-2. Inset shows graded IPSCs in response to different current-charging LED protocols. b, Sample trace demonstrating inhibition of spontaneous PC firing by a single light stimulus. c, Grouped responses of firing in PC cells (n = 6) reveals time course of PC inhibition in response to light stimulation. Inset shows at 10× expanded time resolution that the onset of firing inhibition (black arrow) starts before the onset of IPSC (red arrow).
To exclude the involvement of GABAergic transmission and to confirm this model, we examined the effect of the GABAA receptor blocker bicuculline. As shown in Figure 6a, application of 10 μm bicuculline spared the initial ultrarapid arrest of spike electrogenesis in Purkinje cells, but completely abolished the late phase of inhibition. Surprisingly, without the influence of the GABAergic IPSC, the Purkinje cell firing rate immediately rebounds and overshoots from its initial inhibited state, as also seen in extracellular recordings of Purkinje cells exposed to gabazine (Blot and Barbour, 2014), implying that the ephaptic mechanism not only blocks spike electrogenesis, but also allows a biphasic rebound at the AIS that is normally stabilized by the arrival of GABAergic hyperpolarization from somatodendritic synapses, producing a longer lasting interval of synchronized firing inhibition among Purkinje cells. Given the fact that the onset and duration of the early inhibition observed here is extremely rapid, brief, and independent of GABAA receptor activation, and in agreement with the conclusions of detailed mapping of induced extracellular fields (Blot and Barbour, 2014), we concur that this initial phase of the inhibition is indeed the ephaptic effect of the pinceau field potential.
Figure 6.

Absence of ephaptic inhibition of PC firing in Adam11Δ12-18 mutants expressing CHR-2. a, Top, Grouped time course of PC firing from WT Purkinje cells (n = 4) in response to light stimulation before application of bicuculline (10 μm). Bottom, Grouped time course of PC cell firing from WT in response to light stimulation after application of bicuculline. GABA receptor blockade abolishes the late phase of the inhibition but reveals the ultrarapid ephaptic inhibition and rebound of PC firing. b, Top, Grouped time course of PC firing from Adam11Δ12-18 mutant mice (n = 5) in response to light stimulation revealing persistence of BCT GABAergic synaptic inhibition before application of bicuculline (10 μm). Bottom, GABA receptor blockade revealing the absence of the early ephaptic inhibition and firing rebound in mutant mice.
Next, we performed similar experiments using cerebellar slices from Adam11Δ12–18 mutants expressing nNos-CHR-2. As shown in Figure 6b, light stimulation evokes a long-lasting phase of inhibition in Purkinje cell firing, similar to that seen in +/+ mice and this spike inhibition was completely abolished by GABAA receptor blockade. However, the early spike-firing inhibition and rebound excitation seen in +/+ mice was entirely absent, suggesting that the ephaptic field within the pinceau is strongly diminished. The ultrarapid effect of basket cell activation on the inhibition of Purkinje cell firing is selectively absent in Adam11 mutants lacking the high density of presynaptic K+ channels in their pinceaux.
Specificity of ADAM11 compartmental control over presynaptic Kv1 channel expression
Despite the absence of K+ channel clusters at the pinceaux, perisomatic regions of basket cell axons were visibly stained by Kv1 and Kv1.2 antibodies (Fig. 2b,h, arrows) and perinodal immunostaining of Kv1 and Kv1.2 was preserved and easily visualized in peripheral myelinated axons of ADAM mutants (Fig. 7). To better define the proximal functional boundary of the axon terminal compartment controlled by ADAM11, we investigated whether the mutation might also diminish Kv1 activity at the preterminal axon regions immediately proximal to the pinceau that overlap the perisomatic GABAergic release sites. Kv1 reduction at this site should enhance GABA release, as demonstrated in genomic Kv1 knock-out mice that show enhanced spontaneous GABA release onto Purkinje cells (Zhang et al., 1999).
Figure 7.

Kv1 channels are present at juxtaparanodes of Adam11Δ12-18 mutant mice. Sciatic nerve colabeled with either Kv1.1 or Kv1.2 and KCNQ2 shows the normal arrangement of Kv1 channels at juxtaparanodes in both WT and mutant mice. KCNQ2 localization is also unaltered in mutant mice.
To assess this possibility, we analyzed whole-cell patch-clamp records of spontaneous IPSCs in Purkinje cells. Figure 8a shows representative IPSC sample traces of IPSC activity in WT and Adam11Δ12–18 cells. On average, we found that the mutant cells (n = 17; 5 mice) received a very slightly larger amplitude (∼10%) inhibitory input, measured as the integral of the cumulative IPSC, than the +/+ group (n = 20, 5 mice); however, the difference was not significant (Fig. 8b, t test, p = 0.36). To ensure that spontaneous GABA release onto PCs was indeed dependent on potassium channels in the mutant, we examined IPSCs in response to bath application of 4-AP, a voltage-gated potassium channel blocker. On average, the depolarizing effects of 4-AP increased the mean integrated IPSC amplitude to ∼250% of that measured in physiological saline and had a similar effect on IPSC amplitude in both groups (Fig. 8c,d), These results are consistent with the persistent presence of functional Kv1 heteromeric channels at perisomatic boutons (Fig. 2b,h) and indicate that GABAergic transmission at perisomatic release sites is unaltered in Adam11Δ12–18 mutants despite the loss of Kv1 at the terminal pinceaux, demonstrating that ADAM11 is selectively responsible for K+ channel clustering in the pinceau and not other axonal compartments.
Figure 8.

Spontaneous IPSC activity is similar in both WT and mutant genotypes. a, Sample traces of spontaneous IPSCs from WT (top trace) and mutant (bottom trace) PCs. b, Summary data for group IPSC measurements. For each cell, the integral value of the IPSCs is normalized to the average value among WT cells (WT: 100 ± 27%, n = 20; mutant: 110 ± 39%, n = 17). There is no significant difference in the basal inhibitory synaptic input onto Purkinje cells between the two genotypes (t test p = 0.36). c, Sample traces of IPSCs after application of 4-AP (100 μm) in WT PCs. d, 4-AP enhances IPSCs equally in both genotypes (WT: 265 ± 58% of baseline, n = 7; mutant: 272 ± 62% of baseline, n = 7).
Adam11Δ12–18 neurological phenotype
Because ADAM11 is widely expressed in the nervous system, any mutant phenotype present in the Adam11Δ12–18 cannot be attributed solely to the loss of BCT potassium channel clusters, which would require conditional mutation of only ADAM11 in basket cells to elucidate. Nevertheless we compared the neurological phenotype with Kv1.1 and Kv1.2 genomic knock-out mouse models. Tests of vestibulocerebellar function in adult Adam11Δ12–18 mice revealed a normal spontaneous gait and only minimal impairment of rotarod performance (Fig. 9a–c), which emerged at a late stage after repeated trials; both are consistent with behavioral observations in homozygous Adam11-null mutants (Takahashi et al., 2006). Because Kv1.1 mutations in both mouse and human (Tomlinson et al., 2013) are linked to stress-induced episodic ataxia as well as seizures, we first examined mutant mice after a brief cold-water swim. Upon exit, Adam11Δ12–18 mutants remain stationary and within 1–2 min develop an abnormal, high amplitude coarse truncal tremor with a markedly ataxic gait lasting 15–30 min. Conversely, WT mice subjected to the same cold-water swim show no tremor and promptly resume normal exploratory behavior. Interestingly, adult Kv1.1-, Kv1.2-, and Kvβ2-null mutant mice also show cold-induced tremor and ataxia; however, in Kv1.1-null mutants, these are accompanied by rapid and incapacitating myoclonic limb movements (Zhou et al., 1998; McCormack et al., 2002; Xie et al., 2010). The latter are not accompanied by abnormal EEG activity and are due to hyperexcitability and bursting in peripheral axons due to pervasive loss of the channel throughout the nervous system, including the nodes of Ranvier (Zhou et al., 1998). In Adam11Δ12–18 mice, axonal Kv channels are preserved and no myoclonic jerking was seen after the cold-water swim. Examination of peripheral nodes of Ranvier in Adam11Δ12–18 mutants showed the normal presence of Kv1.1 and Kv1.2 channels (Fig. 7). Although these findings are consistent with a central origin of the cold-induced tremor in all of these models, the ability to fully ascribe the phenotype to defective signaling at the pinceaux will require cell-type-specific expression of the ADAM11 trafficking mutation. We also discovered neocortical seizures in adult homozygotes (data not shown), as seen in Kv1.1- and Kv1.2-null mice (Robbins and Tempel, 2012), further suggesting that extracerebellar circuits are also affected by the ADAM11 mutation; however, no specific mislocation of Kv1 channels could be discerned at presynaptic terminals by confocal microscopy of forebrain networks.
Figure 9.

Neurological phenotype of adult Adam11Δ12-18 mutant mice. Gait and rotarod performance of Adam11Δ12-18 mutant mice are similar to WT mice. Gait analysis was performed by measuring stride length and width of inked footprints. a, Comparison revealed no significant difference between genotypes. b, Daily trials of mice on an accelerating rotarod test showed no significant difference in latency to fall.
Discussion
Our findings provide the first direct evidence that ADAM11 is instrumental in targeting voltage-gated K+ channels to the distal termini of basket cell pinceaux membranes and that this densely localized K+ channel cluster contributes directly to fast electrical inhibition of the Purkinje cell AIS and neuronal cerebellar cortical outflow. Inhibition at this axo-axonic junction is due to extracellular hyperpolarizing electrical field effects arising from the depolarization of tightly arranged septate terminals within the pinceau (Korn and Axelrad, 1980; Blot and Barbour, 2014). Mutation of ADAM11 leads to mistrafficking of Kv1.1 and 1.2 channels, along with their Kvβ2 regulatory subunits that normally cluster at this site, and absence of this cluster eliminates a path for K+ efflux that would normally dissipate the electrical field across the postsynaptic membrane within this confined space.
The mechanism underlying ephaptic hyperpolarization at the pinceau was first described at the classical electrical inhibitory synapse in goldfish, in which stimulation of the eighth nerve projecting to the Mauthner cell lateral dendrite produced an extracellular field at the axon cap termed the extrinsic hyperpolarizing potential (Furukawa and Furshpan, 1963). The axon cap describes a tight extracellular compartment containing multiple nerve terminals enveloped by glia, an arrangement that spatially directs the passive spread of the field. Similarly, the basket cell pinceau contains an astroglial meshwork of septate junctions arising from distal axon terminals that adhere to the Purkinje cell AIS, which may insulate and amplify the electrotonic spread of current. Korn and Axelrad (1980) first demonstrated that the brief hyperpolarization of the PC AIS after BC stimulation is not chemically mediated, but rather reflects a local electrical modulation of the firing rate. Blot and Barbour (2014) reexamined transmission at the BCT and found that the ephaptic fields, although ∼100× smaller in amplitude than those reported earlier, inhibit action potential electrogenesis directly at the Purkinje cell AIS. In their study, the contribution of Kv1 channels was excluded using local application of 4-AP, which blocked all Kv1.1 and Kv1.2 channels of basket cells rather than those solely located at the pinceau. Our finding that genetic elimination of low-threshold K+ channels only at the pinceau spares GABA release at preterminal synaptic boutons yet abolishes the fast ephaptic phase of basket cell inhibition at the PC AIS definitively identifies a critical physiological role for the high-density membrane K+ channel cluster as an amplifier of this signal. The significance of the very brief electrical hyperpolarization that precedes a prolonged GABAergic inhibition remains unclear. Because it is evolutionarily conserved and strategically located, it is likely to enhance the security and synchrony of cerebellar cortical output.
The microanatomical arrangement of K channels in central axons has been subject to considerable analysis. At the large, well visualized calyx of Held, several subtypes reside in the terminal calyx (Kv1.1, Kv1.2) and unmyelinated distal preterminal zone (Kv1.3; Forsythe, 1994; Gazula et al., 2010). In hippocampal mossy fibers, axonal Kv1.1/1.4 subunits are located near, but not at, the giant terminal boutons (Cooper et al., 1998). Interaction with Kvβ2, KIF3, and EB1 is important for the dynamics of kinesin-mediated axonal transport of Kv1 channels (Gu and Gu, 2010) and heteromeric interaction of Kv1.1 with Kv1.4 alters axonal distribution patterns (Jenkins et al., 2011). At BCTs, physiological studies of potassium currents have identified distinct functional contributions for Kv1 channels in perisomatic GABAergic chemotransmission. Southan and Robertson (1998a, 1998b, 2000) showed that α-dendrotoxin (a specific blocker of Kv1.1, Kv1.2, and Kv1.6) abolished K+ currents at BCTs. Tan and Llano (1999) used two-photon imaging of basket cell axons to show that α-dendrotoxin sensitive K+ currents did not affect calcium entry into axons or action potential waveform, but accelerated PC firing, and concluded that a major role of these channels may be to set the local resting potential at strategic points along the axon. Blockade of charybdotoxin- and TEA-sensitive K+ currents showed only a minor role in spike repolarization and therefore alter inhibition through a mechanism distinct from spike-broadening at axon branch points. Patch-clamp recordings of PCs in Kv1.1-null mutants reveal increased tonic GABAergic inhibition (a two-fold increase in sIPSC frequency), which was attributed to prolonged depolarization at axonal preterminal branch points (Zhang et al., 1999). Mice engineered with a dominant-negative Kv1.1 v408A/+ human EA1 mutation for episodic ataxia (Herson et al., 2003), as well as a Kv1.2 missense mutation that reduces Kv1.1 and Kv1.2 channels at BCTs in the ataxic pingu mouse (Xie et al., 2010), also show a greater frequency and amplitude of sIPSCs at Purkinje cells. In the current study, K+ channel cluster loss restricted to the basket cell ephapse did not alter GABAergic release at more proximal sites of the terminal. However, due to the complex architecture of the pinceau, additional understanding of the ultrastructure and biophysics of the presynaptic membranes juxtaposed with the Purkinje cell AIS will be needed to fully model the dynamic contributions of capacitative and ionic currents at this ephapse.
The selectivity of ADAM11-mediated Kv1.1/Kv1.2 channel trafficking to BCT pinceaux adds a GABA-independent mechanism for fast synchronization by basket cells, allowing PC output to be integrated without exocytotic synaptic delay. In addition, physical interaction between ADAM11 and the ion channel complex may provide a further level of regulation and offer biophysical insight into the fidelity of the ephapse. ADAM proteins contain multiple ectodomains implicated in extracellular disintegrin signal transduction and metalloproteinase activity linked to cell adhesion, membrane remodeling, and cytokine/growth factor activation. A physical link between ion channels and extracellular integrin signaling has been analyzed in numerous tissues, where they form macromolecular complexes capable of reciprocal regulation (Arcangeli and Becchetti, 2006). The recurrence of this biological motif, although lacking in enzymatic activity, in the ADAM11/Kv1.x association at presynaptic terminals raises the possibility of external regulation that may couple extracellular ligands to membrane electrogenesis at synapses, as well as the physical distance separating the apposition of presynaptic and postsynaptic membranes within the pinceau. In addition, ADAM11 mutant mice also display stress-induced tremor and seizures, implicating a novel candidate gene for these disorders. Given that other ADAM family members associate with axonal membrane at the AIS and nodal compartments, further unraveling of ADAMs and their interactions with membrane ion channels will enrich our understanding of the molecular assembly and plasticity of axon terminal excitability.
Footnotes
This work was supported by the HHMI (Medical Student Research Training Fellowship to M.J.K.) and the National Institute of Neurological Disorders and Stroke–National Institutes of Health (Grant NS 29709 to J.L.N.). We thank Matthew Rasband and Ed Cooper for helpful discussions during preparation of the manuscript.
The authors declare no competing financial interests.
References
- Arcangeli A, Becchetti A. Complex functional interaction between integrin receptors and ion channels. Trends Cell Biol. 2006;16:631–639. doi: 10.1016/j.tcb.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Blot A, Barbour B. Ultra-rapid axon-axon ephaptic inhibition of cerebellar Purkinje cells by the pinceau. Nat Neurosci. 2014;17:289–295. doi: 10.1038/nn.3624. [DOI] [PubMed] [Google Scholar]
- Budnik V, Zhong Y, Wu CF. Morphological plasticity of motor axons in Drosophila mutants with altered excitability. J Neurosci. 1990;10:3754–3768. doi: 10.1523/JNEUROSCI.10-11-03754.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttermore ED, Piochon C, Wallace ML, Philpot BD, Hansel C, Bhat MA. Pinceau organization in the cerebellum requires distinct functions of neurofascin in Purkinje and basket neurons during postnatal development. J Neurosci. 2012;32:4724–4742. doi: 10.1523/JNEUROSCI.5602-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Palay V. A new synaptic specialization: filamentous braids. Brain Res. 1974;79:280–284. doi: 10.1016/0006-8993(74)90417-X. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Milroy A, Jan YN, Jan LY, Lowenstein DH. Presynaptic localization of Kv1.4-containing A-type potassium channels near excitatory synapses in the hippocampus. J Neurosci. 1998;18:965–974. doi: 10.1523/JNEUROSCI.18-03-00965.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ID. Direct patch recording from identified presynaptic terminals mediating glutamatergic EPSCs in the rat CNS, in vitro. J Physiol. 1994;479:381–387. doi: 10.1113/jphysiol.1994.sp020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Lovero KL, Iwanaga T, Watanabe A, Yokoi N, Tabuchi K, Shigemoto R, Nicoll RA, Fukata M. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc Natl Acad Sci U S A. 2010;107:3799–3804. doi: 10.1073/pnas.0914537107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Furshpan EJ. Two inhibitory mechanisms in the Mauthner neurons of goldfish. J Neurophysiol. 1963;26:140–176. doi: 10.1152/jn.1963.26.1.140. [DOI] [PubMed] [Google Scholar]
- Galiano MR, Jha S, Ho TS, Zhang C, Ogawa Y, Chang KJ, Stankewich MC, Mohler PJ, Rasband MN. A distal axonal cytoskeleton forms an intra-axonal boundary that controls axon initial segment assembly. Cell. 2012;149:1125–1139. doi: 10.1016/j.cell.2012.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazula VR, Strumbos JG, Mei X, Chen H, Rahner C, Kaczmarek LK. Localization of Kv1.3 channels in presynaptic terminals of brainstem auditory neurons. J Comp Neurol. 2010;518:3205–3220. doi: 10.1002/cne.22393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobel S. Axo-axonic septate junctions in the basket formations of the cat cerebellar cortex. J Cell Biol. 1971;51:328–333. doi: 10.1083/jcb.51.1.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Barry J. Function and mechanism of axonal targeting of voltage-sensitive potassium channels. Prog Neurobiol. 2011;94:115–132. doi: 10.1016/j.pneurobio.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Gu C. Dynamics of Kv1 channel transport in axons. PloS one. 2010;5:e11931. doi: 10.1371/journal.pone.0011931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herson PS, Virk M, Rustay NR, Bond CT, Crabbe JC, Adelman JP, Maylie J. A mouse model of episodic ataxia type-1. Nat Neurosci. 2003;6:378–383. doi: 10.1038/nn1025. [DOI] [PubMed] [Google Scholar]
- Iwakura A, Uchigashima M, Miyazaki T, Yamasaki M, Watanabe M. Lack of molecular-anatomical evidence for GABAergic influence on axon initial segment of cerebellar Purkinje cells by the pinceau formation. J Neurosci. 2012;32:9438–9448. doi: 10.1523/JNEUROSCI.1651-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins PM, McIntyre JC, Zhang L, Anantharam A, Vesely ED, Arendt KL, Carruthers CJ, Kerppola TK, Iniguez-Lluhi JA, Holz RW, Sutton MA, Martens JR. Subunit-dependent axonal trafficking of distinct alpha heteromeric potassium channel complexes. J Neurosci. 2011;31:13224–13235. doi: 10.1523/JNEUROSCI.0976-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee S, Tsuda S, Zhang X, Asrican B, Gloss B, Feng G, Augustine GJ. Optogenetic mapping of cerebellar inhibitory circuitry reveals spatially biased coordination of interneurons via electrical synapses. Cell Rep. 2014;7:1601–1613. doi: 10.1016/j.celrep.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Stuart GJ. Signal processing in the axon initial segment. Neuron. 2012;73:235–247. doi: 10.1016/j.neuron.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Korn H, Axelrad H. Electrical inhibition of Purkinje cells in the cerebellum of the rat. Proc Natl Acad Sci U S A. 1980;77:6244–6247. doi: 10.1073/pnas.77.10.6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn H, Faber DS. An electrically mediated inhibition in goldfish medulla. J Neurophysiol. 1975;38:452–471. doi: 10.1152/jn.1975.38.2.452. [DOI] [PubMed] [Google Scholar]
- Laube G, Röper J, Pitt JC, Sewing S, Kistner U, Garner CC, Pongs O, Veh RW. Ultrastructural localization of Shaker-related potassium channel subunits and synapse-associated protein 90 to septate-like junctions in rat cerebellar Pinceaux. Brain Res Mol Brain Res. 1996;42:51–61. doi: 10.1016/S0169-328X(96)00120-9. [DOI] [PubMed] [Google Scholar]
- Luján R, Albasanz JL, Shigemoto R, Juiz JM. Preferential localization of the hyperpolarization-activated cyclic nucleotide-gated cation channel subunit HCN1 in basket cell terminals of the rat cerebellum. Eur J Neurosci. 2005;21:2073–2082. doi: 10.1111/j.1460-9568.2005.04043.x. [DOI] [PubMed] [Google Scholar]
- McCormack K, Connor JX, Zhou L, Ho LL, Ganetzky B, Chiu SY, Messing A. Genetic analysis of the mammalian K+ channel beta subunit Kvbeta 2 (Kcnab2) J Biol Chem. 2002;277:13219–13228. doi: 10.1074/jbc.M111465200. [DOI] [PubMed] [Google Scholar]
- McNamara NM, Muniz ZM, Wilkin GP, Dolly JO. Prominent location of a K+ channel containing the alpha subunit Kv 1.2 in the basket cell nerve terminals of rat cerebellum. Neuroscience. 1993;57:1039–1045. doi: 10.1016/0306-4522(93)90047-J. [DOI] [PubMed] [Google Scholar]
- Mitchell KJ, Pinson KI, Kelly OG, Brennan J, Zupicich J, Scherz P, Leighton PA, Goodrich LV, Lu X, Avery BJ, Tate P, Dill K, Pangilinan E, Wakenight P, Tessier-Lavigne M, Skarnes WC. Functional analysis of secreted and transmembrane proteins critical to mouse development. Nat Genet. 2001;28:241–249. doi: 10.1038/90074. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Oses-Prieto J, Kim MY, Horresh I, Peles E, Burlingame AL, Trimmer JS, Meijer D, Rasband MN. ADAM22, a Kv1 channel-interacting protein, recruits membrane-associated guanylate kinases to juxtaparanodes of myelinated axons. J Neurosci. 2010;30:1038–1048. doi: 10.1523/JNEUROSCI.4661-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongs O, Schwarz JR. Ancillary subunits associated with voltage-dependent K+ channels. Physiol Rev. 2010;90:755–796. doi: 10.1152/physrev.00020.2009. [DOI] [PubMed] [Google Scholar]
- Rasband MN. Composition, assembly, and maintenance of excitable membrane domains in myelinated axons. Semin Cell Dev Biol. 2011;22:178–184. doi: 10.1016/j.semcdb.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins CA, Tempel BL. Kv1.1 and Kv1.2: similar channels, different seizure models. Epilepsia. 2012;53:134–141. doi: 10.1111/j.1528-1167.2012.03484.x. [DOI] [PubMed] [Google Scholar]
- Rybnikova E, Kärkkäinen I, Pelto-Huikko M, Huovila AP. Developmental regulation and neuronal expression of the cellular disintegrin ADAM11 gene in mouse nervous system. Neuroscience. 2002;112:921–934. doi: 10.1016/S0306-4522(02)00124-0. [DOI] [PubMed] [Google Scholar]
- Sagane K, Hayakawa K, Kai J, Hirohashi T, Takahashi E, Miyamoto N, Ino M, Oki T, Yamazaki K, Nagasu T. Ataxia and peripheral nerve hypomyelination in ADAM22-deficient mice. BMC Neurosci. 2005;6:33. doi: 10.1186/1471-2202-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagane K, Ishihama Y, Sugimoto H. LGI1 and LGI4 bind to ADAM22, ADAM23 and ADAM11. Int J Biol Sci. 2008;4:387–396. doi: 10.7150/ijbs.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte U, Thumfart JO, Klöcker N, Sailer CA, Bildl W, Biniossek M, Dehn D, Deller T, Eble S, Abbass K, Wangler T, Knaus HG, Fakler B. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron. 2006;49:697–706. doi: 10.1016/j.neuron.2006.01.033. [DOI] [PubMed] [Google Scholar]
- Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 2003;17:7–30. doi: 10.1101/gad.1039703. [DOI] [PubMed] [Google Scholar]
- Senechal KR, Thaller C, Noebels JL. ADPEAF mutations reduce levels of secreted LGI1, a putative tumor suppressor protein linked to epilepsy. Hum Mol Genet. 2005;14:1613–1620. doi: 10.1093/hmg/ddi169. [DOI] [PubMed] [Google Scholar]
- Sotelo C. Development of “Pinceaux” formations and dendritic translocation of climbing fibers during the acquisition of the balance between glutamatergic and gamma-aminobutyric acidergic inputs in developing Purkinje cells. J Comp Neurol. 2008;506:240–262. doi: 10.1002/cne.21501. [DOI] [PubMed] [Google Scholar]
- Southan AP, Robertson B. Patch-clamp recordings from cerebellar basket cell bodies and their presynaptic terminals reveal an asymmetric distribution of voltage-gated potassium channels. J Neurosci. 1998a;18:948–955. doi: 10.1523/JNEUROSCI.18-03-00948.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan AP, Robertson B. Modulation of inhibitory post-synaptic currents (IPSCs) in mouse cerebellar Purkinje and basket cells by snake and scorpion toxin K+ channel blockers. Br J Pharmacol. 1998b;125:1375–1381. doi: 10.1038/sj.bjp.0702218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan AP, Robertson B. Electrophysiological characterization of voltage-gated K(+) currents in cerebellar basket and purkinje cells: Kv1 and Kv3 channel subfamilies are present in basket cell nerve terminals. J Neurosci. 2000;20:114–122. doi: 10.1523/JNEUROSCI.20-01-00114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi E, Sagane K, Oki T, Yamazaki K, Nagasu T, Kuromitsu J. Deficits in spatial learning and motor coordination in ADAM11-deficient mice. BMC Neurosci. 2006;7:19. doi: 10.1186/1471-2202-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan YP, Llano I. Modulation by K+ channels of action potential-evoked intracellular Ca2+ concentration rises in rat cerebellar basket cell axons. J Physiol. 1999;520:65–78. doi: 10.1111/j.1469-7793.1999.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson SE, Rajakulendran S, Tan SV, Graves TD, Bamiou DE, Labrum RW, Burke D, Sue CM, Giunti P, Schorge S, Kullmann DM, Hanna MG. Clinical, genetic, neurophysiological and functional study of new mutations in episodic ataxia type 1. J Neurol Neurosurg Psychiatry. 2013;84:1107–1112. doi: 10.1136/jnnp-2012-304131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS. Subcellular localization of K channels in mammalian brain neurons: remarkable precision in the midst of extraordinary complexity. Neuron. 2015;85:238–256. doi: 10.1016/j.neuron.2014.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MR, Fuchs JR, Green JT, Morielli AD. Cellular mechanisms and behavioral consequences of Kv1.2 regulation in the rat cerebellum. J Neurosci. 2012;32:9228–9237. doi: 10.1523/JNEUROSCI.6504-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Harrison J, Clapcote SJ, Huang Y, Zhang JY, Wang LY, Roder JC. A new Kv1.2 channelopathy underlying cerebellar ataxia. J Biol Chem. 2010;285:32160–32173. doi: 10.1074/jbc.M110.153676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, Messing A, Chiu SY. Specific alteration of spontaneous GABAergic inhibition in cerebellar purkinje cells in mice lacking the potassium channel Kv1. J Neurosci. 1999;19:2852–2864. doi: 10.1523/JNEUROSCI.19-08-02852.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Zhang CL, Messing A, Chiu SY. Temperature-sensitive neuromuscular transmission in Kv1.1 null mice: role of potassium channels under the myelin sheath in young nerves. J Neurosci. 1998;18:7200–7215. doi: 10.1523/JNEUROSCI.18-18-07200.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]