

Abstract
It is counterintuitive that metabolic defects reducing ATP production can cause, rather than protect from, cancer. Yet this is precisely the case for familial paraganglioma, a form of neuroendocrine malignancy caused by loss of succinate dehydrogenase in the tricarboxylic acid cycle. Here we review biochemical, genetic, and epigenetic considerations in succinate dehydrogenase loss and present leading models and mysteries associated with this fascinating and important tumor.
1. Introduction: Overview of Glucose Metabolism
Glucose metabolism is a central source of energy [1]. Glucose also provides metabolic intermediates for biosynthesis of nucleotides, fatty acids, amino acids, and coenzymes. Glucose metabolism to produce carbon skeletons and ATP occurs in three stages: glycolysis, the TCA (tricarboxylic acid) cycle, and oxidative phosphorylation (OXPHOS) (Figure 1). Glycolysis occurs in ten steps in the cytoplasm of cells, producing from each glucose molecule a net of two ATP molecules and two NADH molecules (Figure 1). Under aerobic conditions, NADH produced from glycolysis is oxidized to NAD+ in the electron transport chain (ETC). The resulting high-energy electrons are transferred to oxygen yielding water and additional molecules of ATP by oxidative phosphorylation driven by a proton gradient formed in mitochondria by the electron transfer process. In contrast, under anaerobic conditions, NADH cannot be reoxidized to NAD+. Since NAD+ is required for glycolysis to continue, NADH reduces pyruvate to lactate, resulting in regeneration of NAD+.
Figure 1.

Overview of ATP production in eukaryotic cells. Complete oxidation of glucose to water and carbon dioxide occurs in three processes: glycolysis, the TCA cycle, and oxidative phosphorylation (OXPHOS).
Under aerobic conditions, pyruvate is oxidized by the pyruvate dehydrogenase complex, located in the mitochondria, to the acetyl function of acetyl-coenzyme A (acetyl-CoA) [1]. The TCA cycle then has eight steps whereby citrate, formed from acetyl-CoA and oxaloacetate, is transformed into seven intermediate metabolites releasing two molecules of carbon dioxide and one molecule of each of NADH and FADH2. Per turn of the TCA cycle, there are four oxidation steps where energy is released and stored in the form of three NADH molecules and one FADH2 molecule (Figure 1). One GTP is also produced when succinyl-CoA is converted to succinate.
The TCA cycle is not simply a path for generating ATP by glucose oxidation. The cycle also generates metabolites for many biosynthetic pathways (Figure 2) [1]. Thus, anaplerotic pathways exist to replenish metabolites extracted from the TCA cycle. Examples include production of oxaloacetate from pyruvate or aspartate, production of α-ketoglutarate (α-KG) from glutamate, production of succinyl-CoA from fatty acids, and production of fumarate from adenylosuccinate.
Figure 2.

TCA cycle metabolites in anabolism. TCA cycle metabolites are precursors in a wide spectrum of biosynthetic pathways. In the boxes are final products of anabolism from the four TCA cycle metabolites indicated by red arrows.
The final stage in glucose metabolism is oxidative phosphorylation. This process depends upon five enzyme complexes (Complex I through Complex V) embedded in the inner mitochondrial membrane. Complexes I, III, and IV are proton pumps that generate an electrochemical gradient across the inner mitochondrial membrane, storing the REDOX energy associated with the high-energy electrons extracted from glucose [1]. Complex V then uses this proton-motive force to synthesize ATP. Electrons enter this path from the TCA cycle. Succinate is oxidized to fumarate by succinate dehydrogenase (Complex II). Protons flow passively from the intermembrane space (high concentration) to the matrix (low concentration) through a proton pore in the F o subunit of Complex V (ATP synthase) converting electrochemical gradient energy to protein dynamics required for ATP synthesis catalyzed by the F 1 subunit on Complex V. The overall energy gain from oxidative phosphorylation is 34 ATP per one molecule of glucose (Figure 1).
2. Metabolism and Cancer
Almost a century ago, Warburg made the observation that the metabolism of cancer cells differs from normal cells. Lactate production in slices of normal tissue was only observed upon oxygen deprivation [2]. Remarkably, tumor tissue slices produced large amounts of lactate even when oxygen was available [3]. This led to the hypothesis that cancer cells prefer glycolysis for ATP production even in the presence of oxygen. This is in stark contrast to the observation that normal cells produce ATP from oxidative metabolism of glucose utilizing glycolysis, the TCA cycle, and oxidative phosphorylation.
Energy produced through glycolysis yields two molecules of ATP per one molecule of glucose. This is far less than the 34 molecules of ATP produced per molecule of glucose through oxidative phosphorylation [1]. Why might cancer cells prefer this very poor ATP yield, apparently wasting most of the available energy in the form of partially oxidized carbon skeletons? It remains unclear how aerobic glycolysis causes, correlates with, or facilitates cancer progression. Three hypotheses have been proposed. First, the hypoxic tumor microenvironment selects for cells that have switched to a constitutive glycolytic metabolism. Second, aerobic glycolysis is better suited for providing carbon skeletons for cell proliferation. Third, cancer cells induce a Warburg-like metabolic reprogramming of the microenvironment and exploit these by-products for growth. All three hypotheses imply that efficient energy extraction from glucose is not important.
Tumor vasculature is disorganized, immature, tortuous, and hyperpermeable [4]. Perfusion is ineffective and inefficient [5], imposing nutrient and hypoxic stress on the growing tumor [6–8]. This stress triggers the stabilization of hypoxia-inducible transcription factors (HIFs), which stimulate angiogenesis and increased expression of glycolytic enzymes, glucose transporters, and inhibitors of mitochondrial metabolism [9]. This selective pressure forces tumor cells to decrease their dependence on aerobic metabolism and shift to glycolysis. If this shift becomes irreversible, the Warburg observation that cancer cells produce large amounts of lactate even in room air is explained. Indeed, some cancer cells undergo aerobic glycolysis even upon high oxygen perfusion [10–12].
In contrast to the hypothesis that cancer cells that preferentially rely on glycolysis are selected by hypoxia, the biomass hypothesis is based on the idea that aerobic glycolysis is superior to the TCA cycle for providing necessary carbon skeletons required for cancer cell proliferation [13]. Thus, human physiology provides an abundance of blood glucose and not all glucose should be used to generate ATP [13]. Requirements for biosynthesis of nucleotides and amino acids are particularly important for cancer cell growth [14]. When cancer cells cease aerobic glycolysis upon inhibition of lactate dehydrogenase, tumor cell proliferation is compromised. This suggests that aerobic glycolysis is crucial for cancer growth [15].
Lastly, it is known that interplay with the tumor microenvironment can promote an epithelial-mesenchymal transition in cancer cells, which contributes to cancer progression and metastasis. This interaction is not exclusively one-sided as originally thought. It has been argued that cancer cells can reciprocate by inducing metabolic reprogramming of cancer-associated fibroblasts (CAFs) to increase glucose uptake and lactate production, while simultaneously increasing tumor lactate receptor expression [16]. This reciprocal conditioning of CAFs is proposed to allow cancer cells to exploit their microenvironment for metabolic advantage. Indeed, this has been observed in a neuroblastoma SK-N-AS SDHB-silenced cell culture model [17].
3. Succinate Dehydrogenase: Structure, Regulation, and Assembly
3.1. SDH Structure
This review focuses on the effects of loss of succinate dehydrogenase (SDH). SDH is part of both the TCA cycle and the ETC. In the TCA cycle, SDH catalyzes the oxidation of succinate to fumarate producing one molecule of FADH2. In the ETC, SDH is known as Complex II, the only complex encoded by nuclear DNA. Electrons from succinate oxidation are transferred to ubiquinone in the ETC [18].
Structurally, the SDH complex has four subunits, SDHA, SDHB, SDHC, and SDHD (Figure 3). SDHC and SDHD contain hydrophobic components that anchor the complex to the inner mitochondrial membrane. Protruding into the matrix are SDHA and SDHB. SDHA is covalently attached to a flavin adenine dinucleotide (FAD) prosthetic group. SDHA carries the binding site for succinate. Upon succinate binding, SDHA brings succinate into juxtaposition with the isoalloxazine ring of FAD, where oxidation to fumarate is catalyzed [18]. SDHB connects SDHA to SDHC and SDHD. SDHB contains three Fe-S centers that mediate electron transfer from succinate to ubiquinone [19]. There are two ubiquinone-binding sites on the SDH complex [19, 20]. The high-affinity site is formed by residues from SDHB, SDHC, and SDHD [21, 22], located near the matrix side of the inner mitochondrial membrane. The low-affinity site is formed by SDHC and SDHD, located closer to the intermembrane space of the inner mitochondrial membrane. Lastly, a heme b group is sandwiched between SDHC and SDHD, presumably scavenging free electrons to prevent the formation of reactive oxygen species.
Figure 3.
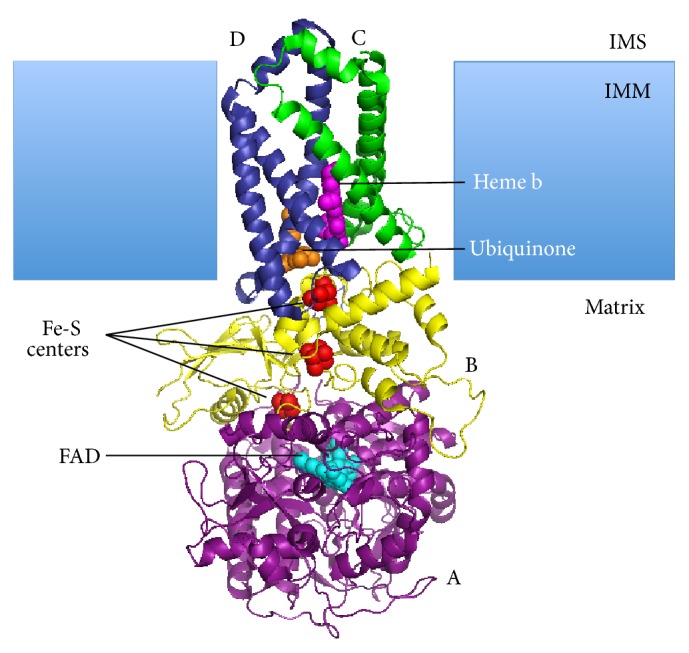
Structure of succinate dehydrogenase [20]. The enzyme is conserved through evolution and has two transmembrane subunits (SDHC and SDHD) and two matrix subunits (SDHA and SDHB). SDHA contains a bound FAD cofactor and the binding site for succinate. SDHB has three Fe-S centers. Heme b and ubiquinone are sandwiched between SDHC and SDHD. Electron pairs move from the substrate succinate to FAD, through the three Fe-S centers, and then to ubiquinone. Heme b prevents reactive oxygen species formation and does not participate in electron transfer. IMS (intermembrane space); IMM (inner mitochondrial membrane).
3.2. Regulation of SDH Activity
SDH activity can be modulated through succinate competitors, ubiquinone inhibitors, or posttranslational modifications. Succinate competitors include malonate, malate, and oxaloacetate [23, 24]. Structurally, these compounds are similar to succinate, which explains their ability to compete for SDH binding. Notably, both malate and oxaloacetate are TCA cycle metabolites. Carboxin and thenoyltrifluoroacetone are synthetic ubiquinone inhibitors that block electron transfer from the SDH complex to ubiquinone [25, 26].
In terms of posttranslational modifications, phosphorylation and acetylation of SDHA lysine residues have been shown to modulate SDH activity [27, 28]. For instance, knockout of SIRT3, a major deacetylase, resulted in accumulation of SDHA lysine acetylation. This decreased the activity of the SDH complex. Upon reexpression of SIRT3, SDHA lysine acetylation was removed and SDH activity was restored [28]. Similarly, phosphorylation of SDHA lysine residues has similar effects on SDH activity [27]. Succinylation of SDHA lysine residues has recently been reported in cells treated with an SDH inhibitor or upon knockdown of succinyl-CoA synthetase [29, 30]. Succinyllysine modification may modulate SDH activity.
3.3. Assembly of SDH
SDH is an intricate complex with four subunits each containing different components that must be properly assembled into the complete enzyme. This presents multiple coordination challenges. In response, protein chaperones facilitate protein folding and factor insertion. These proteins include products of the tcm62, flx1, SDHAF1, sdh5/SDHAF2, SDHAF3, and sdh8/SDHAF4 genes.
Dibrov and colleagues were the first to identify Tcm62 in a genetic screen for mammalian mutations that induced loss of SDH activity [31]. The tcm62 gene encodes a mitochondrial membrane protein that was shown to directly interact with three SDH subunits. Klanner and colleagues confirmed this hypothesis by showing that Tcm62 supported protein folding under heat stress and was required for mitochondrial gene expression [32]. The role of Tcm62 as a general protein folding chaperone was further supported by Heiden and colleagues [33].
During the Sdh1 (yeast ortholog of mammalian SDHA) assembly process, FAD must be imported from the cytosol to the mitochondrial matrix and inserted into the Sdh1 subunit. This is accomplished by Flx1 (flavin exchange) protein. Flx1 is located in the inner mitochondrial membrane and is a member of a superfamily of mitochondrial carriers that exchange substrates between the cytosol and the matrix [34]. The role of Flx1 as a FAD transporter was first described by Tzagoloff and colleagues [35]. They observed that flx1 mutant strain had a decreased mitochondrial flavin adenine dinucleotide/flavin mononucleotide (FAD/FMN) ratio, suggesting that Flx1 is a mitochondrial FAD importer.
The role of Flx1 in FAD insertion into Sdh1 was described by Hao and colleagues [36]. These authors reported complete loss of covalent FAD incorporation into Sdh1 in flx1 mutant cells and inability of the mutant to grow on nonfermentable carbon sources. The flx1 mutant was also hypersensitive to hydrogen peroxide [37]. Interestingly, overexpression of Sdh5 (a protein required for flavination of SDH) partially restored SDH activity in the flx1 mutant.
SDHAF protein family members can be divided into three groups: those that insert Fe-S centers into SDHB, those that insert FAD into SDHA, and those that participate as chaperones. SDHAF1 and SDHAF3 insert Fe-S centers [38–40]. SDHAF1 mutation was reported in an Italian family with progressive infantile leukoencephalopathy and decreased SDH activity [38]. Using a yeast model, a sdhaf1 mutant showed significant loss of SDH activity and inability to grow on acetate as the carbon source. Its role in Fe-S insertion was brought to light by the discovery that Sdhaf1 and Sdhaf3 act together to mediate Fe-S cluster maturation in Sdh2 by protecting the protein from reactive oxygen species [40]. Sdhaf1 or Sdhaf3 deficient yeast and Drosophila specifically showed impaired Sdh2 protein with loss of SDH activity.
Sdh5 and Sdh8 gene products are both SDH assembly factors that facilitate FAD covalent interaction into sdh1. Hao and colleagues showed that mutant sdh5 lacked SDH activity [35]. When Sdh5 was overexpressed, it rescued the FAD incorporation defect previously observed in flx1 mutant cells, which led to the assertion that Sdh5 is required for FAD insertion into the catalytic Sdh1 subunit. This conclusion has been confirmed in multiple studies [41–43].
In a recent study using yeast, Drosophila, and mammalian cells, Sdh8 was shown to interact with Sdh1 in the mitochondrial matrix and mediated interaction with Sdh2 [44]. The authors concluded that Sdh8 interacts with flavo-Sdh1 in the matrix and stabilizes it before Sdh1-Sdh2 is assembled into the SDH complex.
4. Hereditary Paraganglioma
Paraganglioma (PGL) is a rare neuroendocrine neoplasm derived from neural crest cells located between the pelvic floor and the base of the skull. It has an annual incidence of approximately 0.8 per 100,000 [45]. PGL tumors can be derived from cells of the parasympathetic or sympathetic ganglia. These two PGL types occur with similar frequency but have distinct anatomical locations and clinical features [46]. PGLs arising from the parasympathetic ganglia are mainly located around the head and neck (carotid body, glomus jugulare, and glomus typanicum) [46], are typically benign, and are rarely associated with catecholamine secretion [47–49]. Sympathetic PGLs are located in the abdomen and often secrete catecholamines such as epinephrine, norepinephrine, and dopamine. PGL tumors found in the adrenal gland are called pheochromocytomas. Patients with sympathetic PGLs often develop episodic hypertension and are at a higher risk for malignancy [50–54]. Most PGLs are diagnosed between the third to fifth decades of life, with sympathetic PGLs discovered earlier compared to parasympathetic PGLs [55].
There is interesting evidence that PGL development may be associated with chronic hypoxia. The incidence of benign head and neck PGL (carotid body tumors) can approach 1 in 10 for high altitude inhabitants compared to 1 in 500,000 in low altitude dwellers [56–59]. In bovines, the incidence of PGL can be 50% at high altitude [59]. PGLs are also more common in patients with chronic obstructive lung disease and cyanotic congenital heart disease [60–62]. The reason for this association is still unknown and is a subject of this review. We propose here that increased PGL incidence and growth reflect inhibition of α-KG-dependent dioxygenases by hypoxia, an effect that we show to be synergistic with succinate accumulation when SDH function is lost.
Most PGLs appear to be sporadic [46]. About 40% of all PGL cases are inherited [63] involving genes encoding four subunits (A, B, C, and D) of the SDH complex and a gene encoding SDHAF2, the SDHA-specific flavination protein [64]. Several excellent reviews of SDH loss in PGL have recently appeared [65–67]. Other causative genes for familial PGL continue to be discovered [68]. Such genes are therefore tumor suppressor genes whose mutant forms show autosomal dominant inheritance. Carriers are predisposed to PGL with tumorigenesis initiated upon random loss of heterozygosity in a susceptible cell. Because the SDH complex is essential to central metabolism and oxidizes succinate to fumarate, mutations that disrupt the complex will compromise its function. It should be pointed out that it is completely mysterious why various defects in different components of SDH structure and assembly should display different clinical phenotypes. It would be expected that defects in any SDH subunit or assembly factor would display a similar range of effects since all impact performance of the same molecular machine. Classic PGL syndromes are described below.
4.1. SDHA
SDHA-associated mutations are mapped to the SDHA gene on chromosome 5p15.33. Mysteriously, such mutations have very low penetrance in PGL/pheochromocytoma [69]. Thus far, six PGL patients have been reported with SDHA mutations. Five patients developed PGL and one patient had pheochromocytoma [69, 70]. SDHA mutations are also associated with Leigh syndrome [71–73]. Unlike SDH-loss PGL (a disorder displaying autosomal dominant inheritance), Leigh syndrome displays recessive inheritance requiring inherited mutations in both SDHA alleles. These patients develop early onset progressive neurodegenerative disease associated with developmental delay, lactic acidosis, ataxia, and seizures. The peculiar underrepresentation of SDHA mutations in familiar PGL is a mystery.
4.2. SDHB
The PGL syndrome associated with SDHB gene mutations was identified by Astuti and colleagues [74]; see [75, 76]. SDHB mutations were reported to have a 77% penetrance rate by age 50 [77]. SDHB is mapped to chromosome 1p36.1-p35. The mutation types are missense mutations (46%), frameshift mutations (23%), and splicing mutations (12%). This appears to be the most aggressive familial PGL syndrome arising from the sympathetic ganglia of the abdomen. Patients with SDHB-associated mutations are reported to be more likely to become malignant [77, 78]. Some patients with SDHB mutations have also been reported to develop renal cell carcinoma [79].
4.3. SDHC
SDHC mutations leading to head and neck PGLs were first described by Niemann and Muller [80]. SDHC is mapped to chromosome 1q21. These PGLs tend to develop in parasympathetic ganglia with one case of an abdominal norepinephrine-secreting sympathetic paraganglioma [81]. The tumors are reported to be more often benign [82]. SDHC-associated mutations are rare and less penetrant than SDHB and SDHD mutations [46, 80, 82–84]. Reported SDHC mutations include nonsense mutations (47%), splicing mutations (33%), and large deletions (7%) [80].
4.4. SDHD
PGL tumors due to SDHD mutations were first identified by Baysal and colleagues [85]. This is the most common type of familial PGL syndrome. SDHD is located at the gene locus 11q23 and is maternally imprinted [77, 83]. The majority of familial PGL cases with SDHD mutations inherit the mutation from their father and will develop PGL before the age of 50 [46]. Although SDHD-mutant PGLs are reportedly not as aggressive as SDHB mutants, SDHD mutations are considered highly penetrant [77]. SDHD mutations include frameshift mutations (40%), nonsense mutations (25%), and splicing mutations (9%) [86]. SDHD mutations are mainly found in patients with head and neck parasympathetic PGLs occurring in multiple locations [87] and are reported to rarely metastasize [46].
4.5. SDHAF2
The SDHAF2 gene is located on chromosome 11q12.1 [36, 88]. Hao and colleagues discovered that SDHAF2 encodes a protein that inserts the FAD group into SDHA [36]. Mutation of SDHAF2 will result in the lack of SDH activity. Thus far, SDHAF2-associated mutations have been linked to 15 cases of PGL [46]. In this group, the penetrance of PGL is 100% by the age of 45. The disease is confined to only the parasympathetic ganglia, and it may be maternally imprinted [89]. Since the discovery of SDHAF2, there have been multiple SDHAF-related genes that have been implicated in the assembly of the SDH complex [38, 40, 44]. It has yet to be seen whether mutations in the genes will result in PGL [68].
Assuming that PGL mutagenesis is associated with SDH pathology, it is unexplained why there are different tumor phenotypes for different kinds of loss-of-function molecular pathologies in SDH. It might be predicted that any mutation that abolishes SDH function should have the same outcome in susceptible tissues. Such mutations should, in principle, occur in SDH subunit structural genes and in genes encoding proteins that catalyze SDH maturation. It is likely that various hypomorphic alleles among the various tumor suppressor genes complicate interpretation of the genotype-phenotype correlation.
5. Other Tumors Associated with SDH Gene Mutations
Among additional tumors associated with SDH mutations, three autosomal dominant hereditary syndromes are of particular interest: gastrointestinal stromal tumor (GIST), GIST with PGL (Carney-Stratakis dyad), and GIST with PGL and pulmonary chondroma (Carney triad). All of these tumor syndromes are characterized by loss of SDHB in tumor immunohistochemistry staining [90–92].
GISTs are mesenchymal tumors found in the stomach and small intestine of the gastrointestinal tract [93]. The majority of GISTs are caused by KIT or platelet-derived growth factor receptor alpha (PDGFRA) mutations. About 7.5% of GISTs have mutations in the SDH genes. These tumors occur in multiple locations around the gastric wall, often metastasizing to lymph nodes [93]. It is believed that the pathogenesis of SDH-GISTs starts from germline mutations and epigenetic silencing of SDH genes. To date, SDHA mutations are most common, reported in 28% of SDH-deficient GIST [94]. SDHB, C, and D mutations together made up 20–30% of all SDH-deficient GISTs [90, 94].
The Carney-Stratakis dyad characterized by the formation of both GISTs and PGL is associated with germline mutation of SDHB, SDHC, or SDHD [95, 96]. The syndrome has an incomplete penetrance [97].
Finally, the Carney triad characterized by the occurrence of GISTs, PGLs, and pulmonary chondromas is an extremely rare disease. This is typically caused by mutations in KIT or PDGFAR. Thus far, SDHB, C, and D mutations have not been found in patients with Carney triad. The only link is a study showing epigenetic silencing of the SDHC gene locus in a Carney triad patient [98, 99]. The authors showed that the SDHC subunit gene was hypermethylated in some Carney triad patients. This correlated with decreased SDHC mRNA, loss of SDHC protein, and reduced SDH activity.
Importantly, recent integrative genomic studies have confirmed the key roles of established tumor suppressor genes in PGL but suggest that particular combinations of mutations and resulting epigenetic signatures (see below) distinguish different hereditary PGL subtypes [67].
6. Mechanisms of Tumorigenesis in Hereditary PGL
Two models have been proposed to explain how loss of SDH leads to PGL tumorigenesis. The first model hypothesizes that loss of SDH causes mutagenic oxidative stress with tumorigenic consequences. The second model argues that inactivation of SDH produces excess succinate, which can poison α-KG-dependent dioxygenases with tumorigenic consequences that are not yet understood. In this section, we will examine the evidence supporting each model.
Production of reactive oxygen species (ROS) is mapped to two main sources in mitochondria: Complexes I and III [100, 101]. Under normal conditions, Complex II is not known as a ROS producer. However, some evidence suggests that disruption of Complex II, as in SDH subunit gene mutations, can result in defective partial SDH complexes with oxidative stress, genomic instability, tumorigenesis, and decreased lifespan. The first evidence of ROS production in an SDH-deficient model came from studies of a mev-1 mutation in C. elegans [102]. The mev-1 gene is a homolog of the human SDHC gene [103]. Certain mev-1 missense alleles compromised the ability of SDH to interact with ubiquinone, resulting in electron leakage. The mev-1 mutant was hypersensitive to oxygen and had a decreased lifespan [102]. Superoxide anion and lactate levels were higher in the mev-1 mutant than in wild type worms [104]. Since the mev-1 gene is conserved in evolution, the same mutation could be studied in mouse [84] and hamster [105]. Superoxide anion levels were elevated in such animals. There was a higher frequency of DNA damage, and explanation of SDHC mutant cells into immune-compromised mice resulted in benign tumors [84]. In the hamster model, cancer phenotypes were seen in fibroblasts expressing the mutant SDHC protein [105]. Yeast models of complete SDH loss (distinguished from the C. elegans mev-1 missense mutation) have also confirmed increased ROS production [106–108], but no mutagenic DNA damage was detected [106]. Adding to this point, ROS production has not been observed in some recent studies of SDH-deficient models [109–112].
The second model of PGL tumorigenesis proposes that loss of SDH function causes succinate accumulation in mitochondria. Succinate then diffuses into the cytoplasm and competitively inhibits α-KG-dependent dioxygenases (Figure 4) [109]. This fascinating family of iron-dependent enzymes catalyzes the splitting of molecular oxygen to modify target substrates with the simultaneous decarboxylation of α-KG to produce succinate as a by-product [113, 114]. Key features of this enzyme mechanism are (1) the competition between the cosubstrate α-KG and potential inhibitors such as succinate at the active site and (2) the importance of oxygen concentration in determining the reaction rate. The first dioxygenase proposed as a target of succinate accumulation is the prolyl hydroxylase (PHD) that modifies HIF proteins. In normoxia, HIFs are hydroxylated by PHD, which triggers HIF interaction with the von Hippel-Lindau E3 ubiquitin ligase complex. This marks HIF for proteasomal degradation. In contrast, under hypoxic conditions, the PHD reaction rate is decreased by reduction of the oxygen cosubstrate concentration and HIF is stabilized. HIF can then translocate into the nucleus and interact with the HIF1β subunit to activate genes that compensate for the hypoxic condition. According to this model, pseudohypoxic phenotypes are driven by loss of SDH function. It remains completely unknown how pseudohypoxia is tumorigenic and why tumorigenesis is almost completely restricted to neuroendocrine cells.
Figure 4.
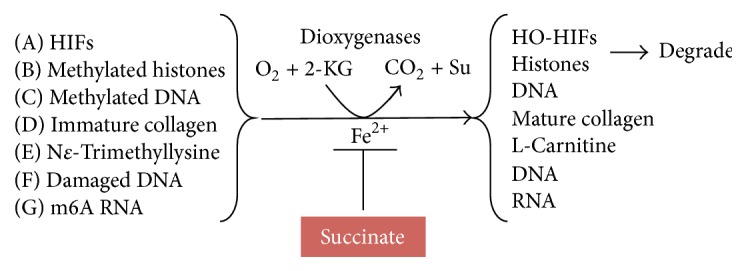
Potential α-KG-dependent dioxygenase targets susceptible to succinate inhibition. In SDH-deficient cells, succinate accumulates in mitochondria and diffuses into the cytosol and nucleus. Succinate inhibits α-KG-dependent dioxygenases resulting in accumulation of (A) HIF1α and HIF2α, (B) hypermethylation of histones, (C) hypermethylation of deoxycytidine, (D) accumulation of immature collagen, (E) accumulation of Nε-trimethyllysine, (F) damaged DNA, and (G) accumulation of N6-methyladenosine on mRNA.
Selak and colleagues were the first to propose succinate inhibition of dioxygenases and show stabilization of HIF in an SDH loss model [109]. In their report, siRNA was used to knock down SDHD expression in HEK293 cells. This resulted in accumulation of succinate and stabilization of HIF. The authors then demonstrated that succinate could inhibit PHD activity in vitro, which led to the conclusion that succinate accumulation in SDH-deficient cells could inhibit PHD activity and induce HIF stabilization. Since α-KG and succinate compete for PHD binding, the authors showed in a separate report that increased intracellular α-KG levels could reverse succinate inhibition of PHD [115]. These studies did not focus on the role of oxygen concentration in these phenomena.
Since the family of α-KG-dependent dioxygenases includes numerous enzymes that participate in a wide range of biological processes, this model is particularly intriguing. Succinate toxicity has been extended to other dioxygenase candidate targets in PGL tumorigenesis. For example, α-KG-dependent Jumonji domain histone demethylases (JMHD) were shown to be susceptible to inhibition. JMHD is responsible for the removal of methyl groups from the tails of histones. Induced global histone hypermethylation could alter epigenetic control of gene expression, with potential tumorigenic consequences. In a yeast Sdh2 loss model, Smith and colleagues confirmed the absence of SDH activity [106]. Succinate accumulated about 10-fold higher in Sdh2 loss cells than in wild type cells, and JMHD activity was inhibited resulting in increased levels of histone methylation. Succinate inhibition could be overcome by increasing α-KG levels in vitro. The authors also showed that mammalian JMHD was susceptible to succinate inhibition. This work suggested for the first time that histone demethylase inhibition by succinate accumulation could alter the gene expression landscape to favor a transformed phenotype. Confirming this hypothesis, Cervera and colleagues showed that knocking down SDHB resulted in 6-fold or more dysregulation of genes that could influence proliferation, adhesion, and the hypoxia pathway [116]. More importantly, SDHB-loss cells displayed some tumor characteristics. In other work, Cervera and colleagues confirmed that loss of SDHB dysregulated histone modifications [117].
Inhibition of α-KG-dependent dioxygenases by succinate in SDH loss cells was subsequently extended to the TET family of DNA demethylases. TET dioxygenases remove methyl groups from 5-methylcytosine residues by first converting these residues to 5-hydroxymethylcytosine. Accumulation of 5-methylcytosine is known to correlate with repressed gene expression. Xiao and colleagues demonstrated that TET dioxygenase could be inhibited by succinate accumulation in vitro and in SDHA and SDHB knockdown cell culture models [118]. In this work, JMHD inhibition by succinate was also confirmed in HEK293 cells models. Cells lacking SDHA or SDHB showed accumulation of succinate and increase global 5-methylcytosine levels. Succinate inhibition of TET dioxygenase was reversible by treating the cells with a cell permeable esterified α-KG. In the same study, collagen synthesis was also shown to be disrupted by succinate inhibition of prolyl-4-hydroxylase, a dioxygenase that hydroxylates proline residues during collagen maturation.
These results have recently been confirmed in a study using SDHB knockout cells. Letouzé and colleagues showed that succinate accumulated in SDHB knockout chromaffin cells led to DNA hypermethylation and established a migratory phenotype [119]. Both DNA and histone hypermethylation were reportedly reversible by increasing intracellular α-KG levels.
The succinate accumulation hypothesis has also been supported by studies of human SDH-loss PGL specimens. Studying SDHB and SDHD mutations in families with PGL, Gimenez-Roqueplo and colleagues showed that PGL tumors accumulated succinate and the SDH complex was inactive [120]. RT-PCR measurements confirmed expression of angiogenic factors. The authors postulated that HIF activation might explain the high vascularization seen in PGL tumors. This was further confirmed by Pollard and coauthors studying SDH-related PGLs and sporadic PGLs [121]. These authors concluded that succinate could cause stabilization of HIF1α. In a study using biopsy material from a patient with a deleterious homozygous SDHA mutation, SDHA mutant fibroblasts showed increased HIF1α translocation into the nucleus [111]. In gene expression studies using micro-array and transcriptional profiling analyses, it was revealed that SDH-mutant PGLs expressed increased levels of hypoxia related genes compared to sporadic and VHL associated PGLs [122–124]. Beyond HIF stabilization, Letouzé and coauthors demonstrated that SDH-related tumors displayed a unique hypermethylation phenotype that is different from the pattern induced by RET, NF1 (neurofibromin 1), and VHL gene mutations [119]. Key genes specifying neuroendocrine differentiation were downregulated, and this might play a factor in PGL tumorigenesis. Recent work from our laboratory employed SDHB knockdown in HEK293 cells and conditional Sdhc knockout mouse embryonic fibroblasts [125]. These studies confirm succinate inhibition of PHD, JMHD, and TET enzymes but also demonstrate that these effects are exquisitely sensitive to oxygen concentration. This is in accord with the dioxygenase reaction mechanism: succinate poisoning can be overcome by increased α-KG and/or increased oxygen. This key observation explains hypoxia as a PGL risk factor and suggests a potential for therapeutic hyperoxia. Together, these findings suggest that succinate is an oncometabolite that can diffuse from the mitochondria to the cytosol and nucleus, perturbing a wide range of biological pathways.
7. Additional α-KG-Dependent Dioxygenase Targets
The initial discovery that PHD, an α-KG-dependent dioxygenase, may play an important role in pseudohypoxic signaling in PGL inspired many groups to examine other α-KG-dependent dioxygenase family members in the setting of SDH loss. After PHD was shown to be inhibited by succinate [109], this effect was extended to Jumonji domain histone demethylases [106], prolyl and lysyl-hydroxylase [118], and TET dioxygenase [118, 119]. The list of α-KG-dependent dioxygenase targets susceptible to inhibition upon SDH loss continues to grow [126–128]. Here we discuss four potential targets (Figure 4). Each has the intriguing potential to play a role in neuroendocrine cell tumorigenesis.
ALKBH5 is a demethylase that removes the methyl group from the exocyclic amine at the 6 position of adenosine in mRNA (m6A). Similar to TET dioxygenase, the enzyme requires α-KG and molecular oxygen generating carbon dioxide and succinate as by-products. Since its substrate requirement is identical to TET dioxygenase, we predict that ALKBH5 will be inhibited by succinate in SDH loss cells. 6-Methyladenine (m6A) is a newly recognized mRNA mark linked to a growing number of functions. In mammalian cells, m6A markers are thought to be important for RNA binding by proteins involved in mRNA export (from the nucleus to the cytosol), RNA metabolism, and mRNA processing [129]. Consistent with ALKBH5 as a dioxygenase that reverses this RNA methylation, depletion of ALKBH5 has been shown to cause accumulation of m6A in mRNA and to be associated with male infertility [129]. m6A in mRNA can also play a role in RNA degradation. For instance, YTHDF2 protein has been shown to bind to m6A in some mRNA positions and relocate RNA to sites of degradation [130]. Thus, it is possible that succinate poisoning of ALKBH5 leads to accumulation of m6A with tumorigenic consequences related to mRNA metabolism.
Another α-KG-dependent dioxygenase family predicted to be inhibited by succinate accumulation is the hABH dioxygenases. These DNA repair proteins remove potentially mutagenic alkylation damage from DNA. These enzymes bind to the methylated base, flip it out from the normal stacked DNA structure while temporarily inserting a hydrophobic amino acid at the repair site [131], and then catalyze oxidative demethylation of the base. Inhibition of hABHs is predicted to result in increased DNA methylation with potentially mutagenic consequences.
Biosynthesis of carnitine involves two α-KG-dependent dioxygenases: 6-N-trimethyllysine-3-hydroxylase (first enzymatic step) and γ-butyrobetaine hydroxylase (final enzymatic step) [128]. L-Carnitine is required to transport fatty acids from the intermembrane space into the matrix for fatty acid metabolism by β-oxidation pathway. Succinate accumulation therefore has the potential to inhibit carnitine biosynthesis and alter fat metabolism.
8. Relationship between Succinate Accumulation and Accumulation of Other Dioxygenase Inhibitory Metabolites in Cancers
Parallel to the succinate accumulation hypothesis, defects in three other TCA cycle enzymes have been linked to competitive inhibition of α-KG-dependent dioxygenases and tumor formation: isocitrate dehydrogenase (IDH), fumarate hydratase (FH), and malate dehydrogenase 2 (MDH2). This section will focus on the defects and the mechanisms that link these enzymes to cancers.
There are three IDH isoforms (IDH1, IDH2, and IDH3). IDH1 is localized to the cytosol and peroxisomes [132]. IDH2 is present in both cytosol and mitochondria, whereas IDH3 is only found in the mitochondria. In the TCA cycle, IDH catalyzes the decarboxylation of isocitrate to α-KG using NADP+ as a cofactor and producing NADPH and CO2 in the process.
Unlike SDH mutations, somatic mutations of IDH result in a gain of function [133, 134]. The majority of these mutations involved amino acid substitution at IDH1 codon 132 (~92% are missense mutations resulting in the R132H substitution). Remarkably, this change alters the enzyme chemistry from a dehydrogenase to a reductase that reduces α-KG to 2-hydroxyglutarate [135]. This leads to depletion of α-KG and accumulation of 2-hydroxyglutarate. These compounds are structurally similar (Figure 5), suggesting that 2-hydroxyglutarate competes unproductively with α-KG for the active site of α-KG-dependent dioxygenases. Indeed, this has been confirmed. In cells with IDH R132H mutations, there is hypermethylation of histones and DNA [136, 137]. Interestingly, even though these metabolites presumably affect the same dioxygenases, mutations in IDH1/2 are linked to glioma and acute myeloid leukemia [138], but not to PGL/pheochromocytoma. Equally mysterious is the fact that no SDH mutations have been found in glioma and practically no IDH mutations have been found in PGL.
Figure 5.
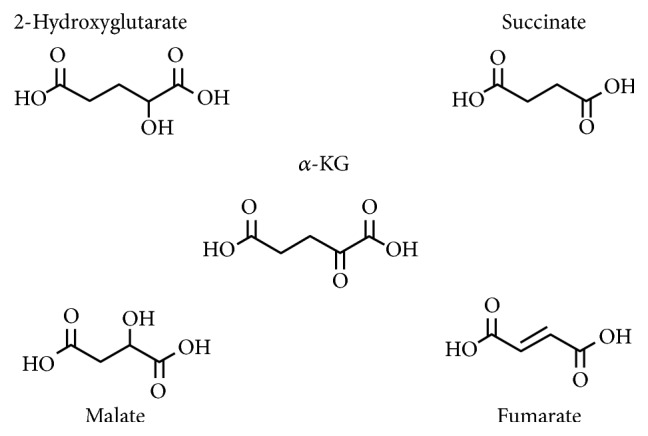
Structural similarity between TCA cycle oncometabolites. IDH gain of function mutations produces 2-hydroxy-α-KG. SDH and FH mutations lead to accumulation of succinate and fumarate, respectively. MDH2 mutations result in increase of malate and fumarate.
FH is a homotetrameric protein in the TCA cycle that catalyzes the hydration of the double bond of fumarate to generate malate [1]. Like SDH, FH is a tumor suppressor, and loss of heterozygosity will predispose FH mutant carriers to develop disorders including renal cell cancer, cutaneous and uterine leiomyomas, and encephalopathies [139]. Interestingly, a subset of patients with FH mutations develop PGL and pheochromocytoma [140]. This could be explained by a shared oncometabolite mechanism of tumorigenesis between SDH-related PGLs and FH-deficient PGLs. It has been reported that FH deficiency results in accumulation of fumarate. Because fumarate has a higher affinity for α-KG-dependent dioxygenases than succinate, it is expected to be a global inhibitor (Figure 5) [141–143]. To date, FH deficiency has been shown to stabilize HIF1α [144] and cause accumulation of histone and DNA methylation [118].
The third TCA cycle enzyme that is linked to inappropriate metabolite accumulation is MDH2 [68]. MDH2 is a mitochondrial enzyme that catalyzes the oxidation of malate to oxaloacetate. There are two isoforms of MDH: MDH1 and MDH2 [1]. MDH1 is found exclusively in the cytosol and is the primary enzyme in the malate-aspartate shuttle. It has been reported that MDH2 mutation can result in 2-fold increase of malate and fumarate levels (Figure 5) [68]. More importantly, MDH2 mutation has recently been linked to PGL. Currently, the mechanism of tumorigenesis is unknown, but there is an obvious and suspicious similarity to the mechanism of SDH and FH mutations.
Together, defects in TCA cycle enzymes with accumulation of dicarboxylic acid oncometabolite variants highlight the importance of continuing research in this field. It is puzzling and mysterious that these defects trigger a common pseudohypoxia pathway and aberrant epigenetic landscape yet have such distinctly different pathological outcomes. Questions regarding tissue specificity and oncogenic pathways will need to be answered in future research. Given the general potential of dicarboxylic acids to inhibit dioxygenases, it seems likely that additional metabolic enzymes will be defined as tumor suppressors.
9. Conclusion
The field of oncometabolite-driven tumorigenesis has gained new interest with discoveries of germline mutations (SDH, isocitrate dehydrogenase, fumarate hydratase, and malate dehydrogenase) linking the TCA cycle with tumorigenesis. It is hypothesized that these mutations result in accumulation of metabolites that competitively inhibit α-KG-dependent dioxygenases. Because there are many enzymes in this family, their inhibition could have profound implications in a wide range of cell regulatory pathways including hypoxic response, collagen maturation, and epigenetic control (histone, DNA, and RNA methylation) of gene expression. Thus, the succinate accumulation hypothesis for familial PGL has been inspirational in proposing part of a mechanism for loss of SDH function leading to tumorigenesis. Though the actual linkage to tumorigenesis remains unknown, this focus places emphasis on the peculiar mechanism of α-KG-dependent dioxygenases and potential approaches to reverse succinate inhibition. In particular, the enzyme reaction mechanism is predicted to be highly sensitive to the balance between succinate and α-KG, encouraging the concept of nontoxic α-KG analog therapy. Likewise, the reaction mechanism is profoundly sensitive to oxygen concentration, a concept not emphasized in work to date. Our recent experiments [125] reexamined the succinate accumulation hypothesis under different oxygen conditions in new SDH-deficient mammalian cell models. We showed that α-KG-dependent dioxygenases are synergistically inhibited by succinate and hypoxia, as predicted from first principles of the enzyme reaction mechanism. Finally, the tissue specificity of tumorigenesis secondary to SDH loss remains a puzzle. We suggest that perhaps neuroendocrine tissue is predisposed to respond uniquely to chronic pseudohypoxia when HIF is perpetually active. Perhaps this chronic signal in neuroendocrine tissue eventually triggers mitogenesis in a failed homeostatic mechanism wherein the tissue expands fruitlessly in an attempt to increase catecholamine section to induce oxygenation.
Acknowledgments
This work was supported by generous funding from the Paradifference Foundation, NIH Grants RO1 CA166025 (L. James Maher) and 1F31CA180698 (Yeng F. Her), benefactor support from the Mayo Clinic Center for Individualized Medicine, the Eagles Cancer Research Fund, and seed funds from the Developmental Therapeutics program of the Mayo Clinic Cancer Center. Additional funding from Mayo Clinic, Mayo Graduate School, and Mayo Clinic MSTP 5T32GM065841 is acknowledged.
Conflict of Interests
The authors declare that there is no conflict of interests that could be perceived as prejudicing the impartiality of the research reported.
References
- 1.Nelson D. L., Cox M. M. Lehninger: Principles of Biochemistry. 4th. New York, NY, USA: W. H. Freeman; 2005. [Google Scholar]
- 2.Warburg O. Tests on surviving carcinoma cultures. Biochemische Zeitschrift. 1923;142:317–333. [Google Scholar]
- 3.Warburg O. On the formation of lactic acid with growth. Biochemische Zeitschrift. 1925;160:307–311. [Google Scholar]
- 4.Konerding M. A., Fait E., Gaumann A. 3D microvascular architecture of pre-cancerous lesions and invasive carcinomas of the colon. British Journal of Cancer. 2001;84(10):1354–1362. doi: 10.1054/bjoc.2001.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gatenby R. A., Gillies R. J. Why do cancers have high aerobic glycolysis? Nature Reviews Cancer. 2004;4(11):891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 6.Carmeliet P., Dor Y., Herbert J. M., et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 7.Bertout J. A., Patel S. A., Simon M. C. The impact of O2 availability on human cancer. Nature Reviews Cancer. 2008;8(12):967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Semenza G. L. HIF-1: upstream and downstream of cancer metabolism. Current Opinion in Genetics & Development. 2010;20(1):51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaelin W. G., Jr., Ratcliffe P. J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Molecular Cell. 2008;30(4):393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Gottschalk S., Anderson N., Hainz C., Eckhardt S. G., Serkova N. J. Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clinical Cancer Research. 2004;10(19):6661–6668. doi: 10.1158/1078-0432.ccr-04-0039. [DOI] [PubMed] [Google Scholar]
- 11.Elstrom R. L., Bauer D. E., Buzzai M., et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Research. 2004;64(11):3892–3899. doi: 10.1158/0008-5472.can-03-2904. [DOI] [PubMed] [Google Scholar]
- 12.Nolop K. B., Rhodes C. G., Brudin L. H., et al. Glucose utilization in vivo by human pulmonary neoplasms. Cancer. 1987;60(11):2682–2689. doi: 10.1002/1097-0142(19871201)60:11<2682::aid-cncr2820601118>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 13.Heiden M. G. V., Cantley L. C., Thompson C. B. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatzivassiliou G., Zhao F., Bauer D. E., et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8(4):311–321. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Fantin V. R., St-Pierre J., Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9(6):425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 16.Fiaschi T., Marini A., Giannoni E., et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Research. 2012;72(19):5130–5140. doi: 10.1158/0008-5472.can-12-1949. [DOI] [PubMed] [Google Scholar]
- 17.Rapizzi E., Fucci R., Giannoni E., et al. Role of microenvironment on neuroblastoma SK-N-AS SDHB-silenced cell metabolism and function. Endocrine Related Cancer. 2015;22(3):409–417. doi: 10.1530/erc-14-0479. [DOI] [PubMed] [Google Scholar]
- 18.Hägerhäll C. Succinate: quinone oxidoreductases: variations on a conserved theme. Biochimica et Biophysica Acta: Bioenergetics. 1997;1320(2):107–141. doi: 10.1016/s0005-2728(97)00019-4. [DOI] [PubMed] [Google Scholar]
- 19.Yankovskaya V., Horsefield R., Törnroth S., et al. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science. 2003;299(5607):700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- 20.Sun F., Huo X., Zhai Y., et al. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell. 2005;121(7):1043–1057. doi: 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 21.Oyedotun K. S., Sit C. S., Lemire B. D. The Saccharomyces cerevisiae succinate dehydrogenase does not require heme for ubiquinone reduction. Biochimica et Biophysica Acta: Bioenergetics. 2007;1767(12):1436–1445. doi: 10.1016/j.bbabio.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 22.Silkin Y., Oyedotun K. S., Lemire B. D. The role of Sdh4p Tyr-89 in ubiquinone reduction by the Saccharomyces cerevisiae succinate dehydrogenase. Biochimica et Biophysica Acta—Bioenergetics. 2007;1767(2):143–150. doi: 10.1016/j.bbabio.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 23.Alston T. A., Mela L., Bright H. J. 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proceedings of the National Academy of Sciences of the United States of America. 1977;74(9):3767–3771. doi: 10.1073/pnas.74.9.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutman M., Silman N. The steady state activity of succinate dehydrogenase in the presence of opposing effectors. II. Reductive activation of succinate dehydrogenase in presence of oxaloacetate. Molecular and Cellular Biochemistry. 1975;7(3):177–185. doi: 10.1007/bf01731407. [DOI] [PubMed] [Google Scholar]
- 25.Gunasekaran P., Tauro P. Mechanism of action of carboxin and the development of resistance in yeast. Journal of Biosciences. 1982;4(2):219–225. doi: 10.1007/bf02702732. [DOI] [Google Scholar]
- 26.Chauveau M., Roussaux J. EPR studies on 6-benzoylaminopurine and thenoyltrifluoroacetone inhibition sites of succinate dehydrogenase in plant mitochondria. Plant and Cell Physiology. 1996;37(7):914–921. doi: 10.1093/oxfordjournals.pcp.a029039. [DOI] [Google Scholar]
- 27.Tomitsuka E., Kita K., Esumi H. Regulation of succinate-ubiquinone reductase and fumarate reductase activities in human complex II by phosphorylation of its flavoprotein subunit. Proceedings of the Japan Academy—Series B: Physical and Biological Sciences. 2009;85(7):258–265. doi: 10.2183/pjab.85.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cimen H., Han M.-J., Yang Y., Tong Q., Koc H., Koc E. C. Regulation of succinate dehydrogenase activity by SIRT3 in mammalian mitochondria. Biochemistry. 2010;49(2):304–311. doi: 10.1021/bi901627u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park J., Chen Y., Tishkoff D. X., et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Molecular Cell. 2013;50(6):919–930. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weinert B. T., Schölz C., Wagner S. A., et al. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Reports. 2013;4(4):842–851. doi: 10.1016/j.celrep.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 31.Dibrov E., Fu S., Lemire B. D. The Saccharomyces cerevisiae TCM62 gene encodes a chaperone necessary for the assembly of the mitochondrial succinate dehydrogenase (complex II) The Journal of Biological Chemistry. 1998;273:32042–32048. doi: 10.1074/jbc.273.48.32042. [DOI] [PubMed] [Google Scholar]
- 32.Klanner C., Neupert W., Langer T. The chaperonin-related protein Tcm62p ensures mitochondrial gene expresssion under heat stress. FEBS Letters. 2000;470(3):365–369. doi: 10.1016/S0014-5793(00)01322-3. [DOI] [PubMed] [Google Scholar]
- 33.Heiden M. G. V., Choy J. S., VanderWeele D. J., et al. Bcl-xL complements Saccharomyces cerevisiae genes that facilitate the switch from glycolytic to oxidative metabolism. The Journal of Biological Chemistry. 2002;277(47):44870–44876. doi: 10.1074/jbc.m204888200. [DOI] [PubMed] [Google Scholar]
- 34.Robinson A. J., Kunji E. R. S. Mitochondrial carriers in the cytoplasmic state have a common substrate binding site. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(8):2617–2622. doi: 10.1073/pnas.0509994103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tzagoloff A., Jang J., Glerum D. M., Wu M. FLX1 codes for a carrier protein involved in maintaining a proper balance of flavin nucleotides in yeast mitochondria. The Journal of Biological Chemistry. 1996;271(13):7392–7397. doi: 10.1074/jbc.271.13.7392. [DOI] [PubMed] [Google Scholar]
- 36.Hao H.-X., Khalimonchuk O., Schraders M., et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325(5944):1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giancaspero T. A., Dipalo E., Miccolis A., Boles E., Caselle M., Barile M. Alteration of ROS homeostasis and decreased lifespan in S. cerevisiae elicited by deletion of the mitochondrial translocator FLX1 . BioMed Research International. 2014;2014:12. doi: 10.1155/2014/101286.101286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghezzi D., Goffrini P., Uziel G., et al. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nature Genetics. 2009;41(6):654–656. doi: 10.1038/ng.378. [DOI] [PubMed] [Google Scholar]
- 39.Bugiani M., Lamantea E., Invernizzi F., et al. Effects of riboflavin in children with complex II deficiency. Brain & Development. 2006;28(9):576–581. doi: 10.1016/j.braindev.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 40.Na U., Yu W., Cox J., et al. The LYR factors sdhaf1 and SDHAF3 mediate maturation of the iron-sulfur subunit of succinate dehydrogenase. Cell Metabolism. 2014;20(2):253–266. doi: 10.1016/j.cmet.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim H. J., Jeong M.-Y., Na U., Winge D. R. Flavinylation and assembly of succinate dehydrogenase are dependent on the C-terminal tail of the flavoprotein subunit. The Journal of Biological Chemistry. 2012;287(48):40670–40679. doi: 10.1074/jbc.m112.405704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zafreen L., Huang L., Walker-Kopp N., Berry E. A. The role of SDH assembly factor 2 and YgfY in flavinylation of succinate dehydrogenase flavoprotein. Biophysical Journal. 2013;104(2, supplement 1):p. 46a. doi: 10.1016/j.bpj.2012.11.296. [DOI] [Google Scholar]
- 43.Huang S., Taylor N. L., Ströher E., Fenske R., Millar A. H. Succinate dehydrogenase assembly factor 2 is needed for assembly and activity of mitochondrial complex II and for normal root elongation in Arabidopsis. The Plant Journal. 2013;73(3):429–441. doi: 10.1111/tpj.12041. [DOI] [PubMed] [Google Scholar]
- 44.van Vranken J. G., Bricker D. K., Dephoure N., et al. SDHAF4 promotes mitochondrial Succinate dehydrogenase activity and prevents neurodegeneration. Cell Metabolism. 2014;20(2):241–252. doi: 10.1016/j.cmet.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beard C. M., Sheps S. G., Kurland L. T., Carney J. A., Lie J. T. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clinic Proceedings. 1983;58(12):802–804. [PubMed] [Google Scholar]
- 46.Welander J., Söderkvist P., Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocrine-Related Cancer. 2011;18(6):R253–R276. doi: 10.1530/erc-11-0170. [DOI] [PubMed] [Google Scholar]
- 47.van Duinen N., Steenvoorden D., Kema I. P., et al. Increased urinary excretion of 3-methoxytyramine in patients with head and neck paragangliomas. The Journal of Clinical Endocrinology & Metabolism. 2010;95(1):209–214. doi: 10.1210/jc.2009-1632. [DOI] [PubMed] [Google Scholar]
- 48.McNicol A. M. Update on tumours of the adrenal cortex, phaeochromocytoma and extra-adrenal paraganglioma. Histopathology. 2011;58(2):155–168. doi: 10.1111/j.1365-2559.2010.03613.x. [DOI] [PubMed] [Google Scholar]
- 49.Erickson D., Kudva Y. C., Ebersold M. J., et al. Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. The Journal of Clinical Endocrinology & Metabolism. 2001;86(11):5210–5216. doi: 10.1210/jc.86.11.5210. [DOI] [PubMed] [Google Scholar]
- 50.Lee J. A., Duh Q.-Y. Sporadic paraganglioma. World Journal of Surgery. 2008;32(5):683–687. doi: 10.1007/s00268-007-9360-4. [DOI] [PubMed] [Google Scholar]
- 51.Dannenberg H., Dinjens W. N. M., Abbou M., et al. Frequent germ-line succinate dehydrogenase subunit D gene mutations in patients with apparently sporadic parasympathetic paraganglioma. Clinical Cancer Research. 2002;8(7):2061–2066. [PubMed] [Google Scholar]
- 52.Ramlawi B., David E. A., Kim M. P., et al. Contemporary surgical management of cardiac paragangliomas. The Annals of Thoracic Surgery. 2012;93(6):1972–1976. doi: 10.1016/j.athoracsur.2012.02.040. [DOI] [PubMed] [Google Scholar]
- 53.Brown M. L., Zayas G. E., Abel M. D., Young W. F., Jr., Schaff H. V. Mediastinal paragangliomas: the mayo clinic experience. The Annals of Thoracic Surgery. 2008;86(3):946–951. doi: 10.1016/j.athoracsur.2008.04.105. [DOI] [PubMed] [Google Scholar]
- 54.Simpson L. N., Hughes B. D., Karikari I. O., et al. Catecholamine-secreting paraganglioma of the thoracic spinal column: report of an unusual case and review of the literature. Neurosurgery. 2012;70(4):E1049–E1052. doi: 10.1227/neu.0b013e31822e5aae. [DOI] [PubMed] [Google Scholar]
- 55.Al-Harthy M., Al-Harthy S., Al-Otieschan A., Velagapudi S., Alzahrani A. S. Comparison of pheochromocytomas and abdominal and pelvic paragangliomas with head and neck paragangliomas. Endocrine Practice. 2009;15(3):194–202. doi: 10.4158/ep.15.3.194. [DOI] [PubMed] [Google Scholar]
- 56.Saldana M. J., Salem L. E., Travezan R. High altitude hypoxia and chemodectomas. Human Pathology. 1973;4(2):251–263. doi: 10.1016/s0046-8177(73)80012-7. [DOI] [PubMed] [Google Scholar]
- 57.Khan Q., Heath D., Smith P., Norboo T. The histology of the carotid bodies in highlanders from Ladakh. International Journal of Biometeorology. 1988;32(4):254–259. doi: 10.1007/bf01080024. [DOI] [PubMed] [Google Scholar]
- 58.Rodríguez-Cuevas S., López-Garza J., Labastida-Almendaro S. Carotid body tumors in inhabitants of altitudes higher than 2000 meters above sea level. Head & Neck. 1998;20(5):374–378. doi: 10.1002/(sici)1097-0347(199808)20:5<374::aid-hed3>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 59.Arias-Stella J., Bustos F. Chronic hypoxia and chemodectomas in bovines at high altitudes. Archives of Pathology & Laboratory Medicine. 1976;100(12):636–639. [PubMed] [Google Scholar]
- 60.Opotowsky A. R., Moko L. E., Ginns J., et al. Pheochromocytoma and paraganglioma in cyanotic congenital heart disease. The Journal of Clinical Endocrinology & Metabolism. 2015;100(4):1325–1334. doi: 10.1210/jc.2014-3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Folger G. M., Jr., Roberts W. C., Mehrizi A., et al. Cyanotic malformations of the heart with pheochromocytoma. A report of five cases. Circulation. 1964;29:750–757. doi: 10.1161/01.cir.29.5.750. [DOI] [PubMed] [Google Scholar]
- 62.Kita T., Imamura T., Date H., et al. Two cases of pheochromocytoma associated with tetralogy of fallot. Hypertension Research. 2003;26(5):433–437. doi: 10.1291/hypres.26.433. [DOI] [PubMed] [Google Scholar]
- 63.Fishbein L., Merrill S., Fraker D. L., Cohen D. L., Nathanson K. L. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. The Annals of Surgical Oncology. 2013;20(5):1444–1450. doi: 10.1245/s10434-013-2942-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bardella C., Pollard P. J., Tomlinson I. SDH mutations in cancer. Biochimica et Biophysica Acta: Bioenergetics. 2011;1807(11):1432–1443. doi: 10.1016/j.bbabio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 65.Dahia P. L. M. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nature Reviews Cancer. 2014;14(2):108–119. doi: 10.1038/nrc3648. [DOI] [PubMed] [Google Scholar]
- 66.Favier J., Amar L., Gimenez-Roqueplo A.-P. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nature Reviews Endocrinology. 2015;11(2):101–111. doi: 10.1038/nrendo.2014.188. [DOI] [PubMed] [Google Scholar]
- 67.Castro-Vega L. J., Lepoutre-Lussey C., Gimenez-Roqueplo A. P., Favier J. Rethinking pheochromocytomas and paragangliomas from a genomic perspective. Oncogene. 2015 doi: 10.1038/onc.2015.172. [DOI] [PubMed] [Google Scholar]
- 68.Cascon A., Comino-Mendez I., Curras-Freixes M., et al. Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. Journal of the National Cancer Institute. 2015;107(5):p. djv053. doi: 10.1093/jnci/djv053. [DOI] [PubMed] [Google Scholar]
- 69.Korpershoek E., Favier J., Gaal J., et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. The Journal of Clinical Endocrinology & Metabolism. 2011;96(9):E1472–E1476. doi: 10.1210/jc.2011-1043. [DOI] [PubMed] [Google Scholar]
- 70.Burnichon N., Brière J.-J., Libé R., et al. SDHA is a tumor suppressor gene causing paraganglioma. Human Molecular Genetics. 2010;19(15):3011–3020. doi: 10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bourgeron T., Rustin P., Chretien D., et al. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nature Genetics. 1995;11(2):144–149. doi: 10.1038/ng1095-144. [DOI] [PubMed] [Google Scholar]
- 72.Parfait B., Chretien D., Rötig A., Marsac C., Munnich A., Rustin P. Compound heterozygous mutations in the flavoprotein gene of the respiratory chain complex II in a patient with Leigh syndrome. Human Genetics. 2000;106(2):236–243. doi: 10.1007/s004390051033. [DOI] [PubMed] [Google Scholar]
- 73.Horváth R., Abicht A., Holinski-Feder E., et al. Leigh syndrome caused by mutations in the flavoprotein (Fp) subunit of succinate dehydrogenase (SDHA) Journal of Neurology, Neurosurgery and Psychiatry. 2006;77(1):74–76. doi: 10.1136/jnnp.2005.067041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Astuti D., Latif F., Dallol A., et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. American Journal of Human Genetics. 2001;69(1):49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gimenez-Roqueplo A.-P., Favier J., Rustin P., et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Research. 2003;63(17):5615–5621. [PubMed] [Google Scholar]
- 76.Gimenez-Roqueplo A.-P., Favier J., Rustin P., et al. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. Journal of Clinical Endocrinology and Metabolism. 2002;87(10):4771–4774. doi: 10.1210/jc.2002-020525. [DOI] [PubMed] [Google Scholar]
- 77.Neumann H. P. H., Pawlu C., Pęczkowska M., et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD and gene mutations. Journal of the American Medical Association. 2004;292(8):943–951. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- 78.Ricketts C. J., Forman J. R., Rattenberry E., et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Human Mutation. 2010;31(1):41–51. doi: 10.1002/humu.21136. [DOI] [PubMed] [Google Scholar]
- 79.Vanharanta S., Buchta M., McWhinney S. R., et al. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. American Journal of Human Genetics. 2004;74(1):153–159. doi: 10.1086/381054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Niemann S., Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nature Genetics. 2000;26(3):268–270. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- 81.Mannelli M., Ercolino T., Giachè V., Simi L., Cirami C., Parenti G. Genetic screening for pheochromocytoma: should SDHC gene analysis be included? Journal of Medical Genetics. 2007;44(9):586–587. doi: 10.1136/jmg.2007.051045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schiavi F., Boedeker C. C., Bausch B., et al. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. Journal of the American Medical Association. 2005;294(16):2057–2063. doi: 10.1001/jama.294.16.2057. [DOI] [PubMed] [Google Scholar]
- 83.Baysal B. E., Willett-Brozick J. E., Lawrence E. C., et al. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. Journal of Medical Genetics. 2002;39(3):178–183. doi: 10.1136/jmg.39.3.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ishii T., Yasuda K., Akatsuka A., Hino O., Hartman P. S., Ishii N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Research. 2005;65(1):203–209. [PubMed] [Google Scholar]
- 85.Baysal B. E., Ferrell R. E., Willett-Brozick J. E., et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287(5454):848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 86.Pasini B., Stratakis C. A. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. Journal of Internal Medicine. 2009;266(1):19–42. doi: 10.1111/j.1365-2796.2009.02111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hensen E. F., Siemers M. D., Jansen J. C., et al. Mutations in SDHD are the major determinants of the clinical characteristics of Dutch head and neck paraganglioma patients. Clinical Endocrinology. 2011;75(5):650–655. doi: 10.1111/j.1365-2265.2011.04097.x. [DOI] [PubMed] [Google Scholar]
- 88.Jiménez C., Cote G., Arnold A., Gagel R. F. Should patients with apparently sporadic pheochromocytomas or paragangliomas be screened for hereditary syndromes? Journal of Clinical Endocrinology and Metabolism. 2006;91(8):2851–2858. doi: 10.1210/jc.2005-2178. [DOI] [PubMed] [Google Scholar]
- 89.Kunst H. P. M., Rutten M. H., de Mönnink J.-P., et al. SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clinical Cancer Research. 2011;17(2):247–254. doi: 10.1158/1078-0432.ccr-10-0420. [DOI] [PubMed] [Google Scholar]
- 90.Miettinen M., Wang Z.-F., Sarlomo-Rikala M., Osuch C., Rutkowski P., Lasota J. Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. American Journal of Surgical Pathology. 2011;35(11):1712–1721. doi: 10.1097/pas.0b013e3182260752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang L., Smyrk T. C., Young W. F., Stratakis C. A., Carney J. A. Gastric stromal tumors in carney triad are different clinically, pathologically, and behaviorally from sporadic gastric gastrointestinal stromal tumors: findings in 104 cases. American Journal of Surgical Pathology. 2010;34(1):53–64. doi: 10.1097/pas.0b013e3181c20f4f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Janeway K. A., Kim S. Y., Lodish M., et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(1):314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Miettinen M., Lasota J. Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs)—a review. The International Journal of Biochemistry & Cell Biology. 2014;53:514–519. doi: 10.1016/j.biocel.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Miettinen M., Killian J. K., Wang Z.-F., et al. Immunohistochemical loss of succinate dehydrogenase subunit a (SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA germline mutation. American Journal of Surgical Pathology. 2013;37(2):234–240. doi: 10.1097/pas.0b013e3182671178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pasini B., McWhinney S. R., Bei T., et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. European Journal of Human Genetics. 2008;16(1):79–88. doi: 10.1038/sj.ejhg.5201904. [DOI] [PubMed] [Google Scholar]
- 96.McWhinney S. R., Pasini B., Stratakis C. A. Familial gastrointestinal stromal tumors and germ-line mutations. The New England Journal of Medicine. 2007;357(10):1054–1056. doi: 10.1056/nejmc071191. [DOI] [PubMed] [Google Scholar]
- 97.Stratakis C. A., Carney J. A. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): molecular genetics and clinical implications. Journal of Internal Medicine. 2009;266(1):43–52. doi: 10.1111/j.1365-2796.2009.02110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Killian J. K., Kim S. Y., Miettinen M., et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discovery. 2013;3(6):648–657. doi: 10.1158/2159-8290.cd-13-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Haller F., Moskalev E. A., Faucz F. R., et al. Aberrant DNA hypermethylation of SDHC: a novel mechanism of tumor development in Carney triad. Endocrine-Related Cancer. 2014;21(4):567–577. doi: 10.1530/erc-14-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Murphy M. P. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/bj20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Raha S., McEachern G. E., Myint A. T., Robinson B. H. Superoxides from mitochondrial complex III: the role of manganese superoxide dismutase. Free Radical Biology and Medicine. 2000;29(2):170–180. doi: 10.1016/s0891-5849(00)00338-5. [DOI] [PubMed] [Google Scholar]
- 102.Adachi H., Fujiwara Y., Ishii N. Effects of oxygen on protein carbonyl and aging in Caenorhabditis elegans mutants with long (age-1) and short (mev-1) life spans. Journals of Gerontology A: Biological Sciences and Medical Sciences. 1998;53(4):B240–B244. doi: 10.1093/gerona/53a.4.b240. [DOI] [PubMed] [Google Scholar]
- 103.Guo J., Lemire B. D. The ubiquinone-binding site of the Saccharomyces cerevisiae succinate-ubiquinone oxidoreductase is a source of superoxide. The Journal of Biological Chemistry. 2003;278(48):47629–47635. doi: 10.1074/jbc.m306312200. [DOI] [PubMed] [Google Scholar]
- 104.Senoo-Matsuda N., Yasuda K., Tsuda M., et al. A defect in the cytochrome b large subunit in complex II causes both superoxide anion overproduction and abnormal energy metabolism in Caenorhabditis elegans. The Journal of Biological Chemistry. 2001;276(45):41553–41558. doi: 10.1074/jbc.m104718200. [DOI] [PubMed] [Google Scholar]
- 105.Slane B. G., Aykin-Burns N., Smith B. J., et al. Mutation of succinate dehydrogenase subunit C results in increased O2.-, oxidative stress, and genomic instability. Cancer Research. 2006;66(15):7615–7620. doi: 10.1158/0008-5472.CAN-06-0833. [DOI] [PubMed] [Google Scholar]
- 106.Smith E. H., Janknecht R., Maher J. L., III Succinate inhibition of α-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Human Molecular Genetics. 2007;16(24):3136–3148. doi: 10.1093/hmg/ddm275. [DOI] [PubMed] [Google Scholar]
- 107.Goffrini P., Ercolino T., Panizza E., et al. Functional study in a yeast model of a novel succinate dehydrogenase subunit B gene germline missense mutation (C191Y) diagnosed in a patient affected by a glomus tumor. Human Molecular Genetics. 2009;18(10):1860–1868. doi: 10.1093/hmg/ddp102. [DOI] [PubMed] [Google Scholar]
- 108.Szeto S. S. W., Reinke S. N., Sykes B. D., Lemire B. D. Ubiquinone-binding site mutations in the Saccharomyces cerevisiae succinate dehydrogenase generate superoxide and lead to the accumulation of succinate. The Journal of Biological Chemistry. 2007;282(37):27518–27526. doi: 10.1074/jbc.m700601200. [DOI] [PubMed] [Google Scholar]
- 109.Selak M. A., Armour S. M., MacKenzie E. D., et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 110.Selak M. A., Durán R. V., Gottlieb E. Redox stress is not essential for the pseudo-hypoxic phenotype of succinate dehydrogenase deficient cells. Biochimica et Biophysica Acta. 2006;1757(5-6):567–572. doi: 10.1016/j.bbabio.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 111.Brière J.-J., Favier J., Bénit P., et al. Mitochondrial succinate is instrumental for HIF1α nuclear translocation in SDHA-mutant fibroblasts under normoxic conditions. Human Molecular Genetics. 2005;14(21):3263–3269. doi: 10.1093/hmg/ddi359. [DOI] [PubMed] [Google Scholar]
- 112.Guzy R. D., Sharma B., Bell E., Chandel N. S., Schumacker P. T. Loss of the SdhB, but not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Molecular and Cellular Biology. 2008;28(2):718–731. doi: 10.1128/mcb.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Myllyla R., Tuderman L., Kivirikko K. I. Mechanism of the prolyl hydroxylase reaction. 2. Kinetic analysis of the reaction sequence. European Journal of Biochemistry. 1977;80(2):349–357. doi: 10.1111/j.1432-1033.1977.tb11889.x. [DOI] [PubMed] [Google Scholar]
- 114.Tuderman L., Myllylä R., Kivirikko K. Mechanism of the prolyl hydroxylase reaction. 1. Role of co-substrates. European Journal of Biochemistry. 1977;80(2):341–348. doi: 10.1111/j.1432-1033.1977.tb11888.x. [DOI] [PubMed] [Google Scholar]
- 115.MacKenzie E. D., Selak M. A., Tennant D. A., et al. Cell-permeating α-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Molecular and Cellular Biology. 2007;27(9):3282–3289. doi: 10.1128/mcb.01927-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cervera A. M., Apostolova N., Crespo F. L., Mata M., McCreath K. J. Cells silenced for SDHB expression display characteristic features of the tumor phenotype. Cancer Research. 2008;68(11):4058–4067. doi: 10.1158/0008-5472.can-07-5580. [DOI] [PubMed] [Google Scholar]
- 117.Cervera A. M., Bayley J.-P., Devilee P., McCreath K. J. Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Molecular Cancer. 2009;8, article 89 doi: 10.1186/1476-4598-8-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xiao M., Yang H., Xu W., et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes & Development. 2012;26(12):1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Letouzé E., Martinelli C., Loriot C., et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23(6):739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 120.Gimenez-Roqueplo A.-P., Favier J., Rustin P., et al. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. The American Journal of Human Genetics. 2001;69(6):1186–1197. doi: 10.1086/324413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pollard P. J., Brière J. J., Alam N. A., et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1α in tumours which result from germline FH and SDH mutations. Human Molecular Genetics. 2005;14(15):2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 122.Dahia P. L. M., Ross K. N., Wright M. E., et al. A HIf1α regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genetics. 2005;1(1):72–80. doi: 10.1371/journal.pgen.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.López-Jiménez E., Gómez-López G., Leandro-García L. J., et al. Research resource: transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Molecular Endocrinology. 2010;24(12):2382–2391. doi: 10.1210/me.2010-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Favier J., Brière J.-J., Burnichon N., et al. The warburg effect is genetically determined in inherited pheochromocytomas. PLoS ONE. 2009;4(9) doi: 10.1371/journal.pone.0007094.e7094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Her Y. F., Nelson-Holte M., Maher L. J., III Oxygen concentration controls epigenetic effects in models of familial paraganglioma. PLoS ONE. 2015;10(2) doi: 10.1371/journal.pone.0127471.e0127471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kaelin W. G., Jr. Cancer and altered metabolism: potential importance of hypoxia-Inducible factor and 2-oxoglutarate-dependent dioxygenases. Cold Spring Harbor Symposia on Quantitative Biology. 2011;76:335–345. doi: 10.1101/sqb.2011.76.010975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kaelin W. G. IDH mutations, 2-oxoglutarate-dependent dioxygenases, and leukemia. Blood. 2014;124(21):p. SCI-6. [Google Scholar]
- 128.Hausinger R. P. FeII/alpha-ketoglutarate-dependent hydroxylases and related enzymes. Critical Reviews in Biochemistry and Molecular Biology. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 129.Zheng G., Dahl J. A., Niu Y., et al. ALKBH5 is a aammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Molecular Cell. 2013;49(1):18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang X., Lu Z., Gomez A., et al. N 6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sundheim O., Talstad V. A., Vågbø C. B., Slupphaug G., Krokan H. E. AlkB demethylases flip out in different ways. DNA Repair. 2008;7(11):1916–1923. doi: 10.1016/j.dnarep.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 132.Geisbrecht B. V., Gould S. J. The human PICD gene encodes a cytoplasmic and peroxisomal NADP+-dependent isocitrate dehydrogenase. The Journal of Biological Chemistry. 1999;274(43):30527–30533. doi: 10.1074/jbc.274.43.30527. [DOI] [PubMed] [Google Scholar]
- 133.Dang L., White D. W., Gross S., et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ward P. S., Patel J., Wise D. R., et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Turkalp Z., Karamchandani J., Das S. IDH mutation in glioma: new insights and promises for the future. JAMA Neurology. 2014;71(10):1319–1325. doi: 10.1001/jamaneurol.2014.1205. [DOI] [PubMed] [Google Scholar]
- 136.Xu W., Yang H., Liu Y., et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chowdhury R., Yeoh K. K., Tian Y.-M., et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Reports. 2011;12(5):463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dang L., Jin S., Su S. M. IDH mutations in glioma and acute myeloid leukemia. Trends in Molecular Medicine. 2010;16(9):387–397. doi: 10.1016/j.molmed.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 139.Alam N. A., Rowan A. J., Wortham N. C., et al. Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Human Molecular Genetics. 2003;12(11):1241–1252. doi: 10.1093/hmg/ddg148. [DOI] [PubMed] [Google Scholar]
- 140.Castro-Vega L. J., Buffet A., De Cubas A. A., et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Human Molecular Genetics. 2014;23(9):2440–2446. doi: 10.1093/hmg/ddt639.ddt639 [DOI] [PubMed] [Google Scholar]
- 141.Koivunen P., Hirsilä M., Remes A. M., Hassinen I. E., Kivirikko K. I., Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. The Journal of Biological Chemistry. 2007;282(7):4524–4532. doi: 10.1074/jbc.m610415200. [DOI] [PubMed] [Google Scholar]
- 142.Hewitson K. S., Liénard B. M. R., McDonough M. A., et al. Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. The Journal of Biological Chemistry. 2007;282(5):3293–3301. doi: 10.1074/jbc.m608337200. [DOI] [PubMed] [Google Scholar]
- 143.Lu H., Dalgard C. L., Mohyeldin A., McFate T., Tait A. S., Verma A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. The Journal of Biological Chemistry. 2005;280(51):41928–41939. doi: 10.1074/jbc.m508718200. [DOI] [PubMed] [Google Scholar]
- 144.Isaacs J. S., Yun Y. J., Mole D. R., et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8(2):143–153. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]