

Abstract
AIM: To investigate biological mechanisms underlying pyruvate kinase M2 isoform (PKM2) regulation of cell migration and invasion in hepatocellular carcinoma cells.
METHODS: HepG2 and Huh-7 hepatocellular carcinoma cell lines were stably transfected and cultured in DMEM (HyClone, Logan, UT, United States). To investigate the effects of PKM2 on cellular proliferation, hepatocellular carcinoma cells were subjected to the Cell Counting Kit-8 (Dojindo, Kamimashiki-gun, Kumamoto, Japan). And investigate the effects of PKM2 on cell signal pathway related with migration and invasion, Western immunoblotting were used to find out the differential proteins. All the antibody used was purchaseed from Cell Signal Technology. In order to explore cell motility used Transwell invasion and wound healing assays. The transwell plate with 0.5 mg/mL collagen type I (BD Bioscience, San Jose, CA)-coated filters. The wound-healing assay was performed in 6-well plates. Total RNA was extracted using TRIzol reagent (Invitrogen, CA, United States) and then reverse transcription was conducted. Quantitative reverse transcription-polymerase chain reaction (PCR) analysis was performed with the ABI 7500 real-time PCR system (Applied Biosystems). We further use digital gene expression tag profiling and identification of differentially expressed genes.
RESULTS: The cells seeded in four 96-well plates were measured OD450 by conducted Cell Counting Kit-8. From this conduction we observed that both HepG2 and Huh-7 hepatocellular carcinoma cells with silenced PKM2 turn on a proliferate inhibition; however, cell migration and invasion were enhanced compared with the control upon stimulation with epidermal growth factor (EGF). Our results indicate that the knockdown of PKM2 decreased the expression of E-cadherin and enhanced the activity of the EGF/EGFR signaling pathway, furthermore up-regulate the subsequent signal molecular the PLCγ1 and extracellular signal-regulated kinase 1/2 expression in the hepatocellular carcinoma cell lines HepG2 and Huh-7, which regulates cell motility. These variations we observed were due to the activation of the transforming growth factor beta (TGFβ) signaling pathway after PKM2 knockdown. We also found that the expression of TGFBRI was increased and the phosphorylation of Smad2 was enhanced. Taken together, our findings demonstrate that PKM2 can regulate cell motility through the EGF/EGFR and TGFβ/TGFR signaling pathways in hepatocellular carcinoma cells.
CONCLUSION: PKM2 play different roles in modulating the proliferation and metastasis of hepatocellular carcinoma cells, and this finding could help to guide the future targeted therapies.
Keywords: Pyruvate kinase, Migration, Epidermal growth factor/EGFR signaling pathway, Transforming growth factor beta signaling pathway, Hepatocellular carcinoma
Core tip: The present study is to explore the effects of pyruvate kinase M2 (PKM2) on motility of human hepatocellular carcinoma cells. Here, our results revealed that silenced PKM2 activated the epidermal growth factor (EGF)/EGFR and transforming growth factor beta (TGFβ)/TGFR signaling pathways, in addition, up-regulate the expression of downstream targets, and this is the first research to connect PKM2 with the vital EGF and TGFβ pathways in hepatocellular carcinoma cell motility.
INTRODUCTION
Pyruvate kinase (PK) mediates the final rate-limiting step of glycolysis by catalyzing the dephosphorylation of phosphoenolpyruvate (PEP) to pyruvate to yield one molecule of ATP[1]. Mammalian cells have four pyruvate kinase isoenzymes (M1, M2, L, and R), which are selectively expressed in different types of cells and tissues[2]. In mammals, the M1 isoform (PKM1) is expressed in most adult tissues. The M2 isoform (PKM2), an alternatively spliced variant of M1, is expressed during embryonic development[3]. Studies have shown that cancer cells exclusively express PKM2[4,5], which has been shown to be essential for aerobic glycolysis in tumors (Warburg effect). Over the years, significant advancements have been made in understanding the function and regulation of PKM2 as a pyruvate kinase and protein kinase in cancer cells[6]. A recent study confirmed that the PKM2 induced by epidermal growth factor (EGF) translocates into the nucleus of glioblastoma cells, interacts with β-catenin and leads to cyclinD1 expression, which promotes cell proliferation and tumorigenesis[7]. These findings reveal a novel role for PKM2 as a transcriptional coactivator. However, there are some controversies regarding the specificity and potential of PKM2 as an anti-cancer target in cancer therapy. We previously reported that pyruvate kinase M2 plays a dual role in the regulation of tumor progression[8]; however, the mechanism of this effect is unclear.
Previous studies regarding PKM2 have focused on tumor metabolism and tumor growth; only a few reports have focused on tumor metastasis. E-cadherin plays a critical role in maintaining epithelial integrity, and the loss of E-cadherin affects the adhesive repertoire of a cell[9]. Previous in vitro studies have shown that the loss of E-cadherin in human carcinoma cell lines is associated with poor differentiation and a fibroblastoid morphology[10]. The EGF-dependent activation of the EGFR has been reported to be inhibited in an E-cadherin adhesion-dependent manner, which inhibits the ligand-dependent activation of diverse receptor tyrosine kinases[11].
Transforming growth factor beta (TGFβ) is a cytokine that regulates multiple cellular responses, including inhibition of cell proliferation and induction of differentiation, senescence, and apoptosis[12,13]. Its actions are mediated by binding to the serine/threonine kinase receptor TGFBRII, which recruits and activates TGFBRI. In turn, TGFBRI phosphorylates downstream targets, including the proteins SMAD2 and SMAD3, which translocate to the nucleus in a complex with the common mediator SMAD4 to regulate the transcription of target genes[14,15]. TGFβ1 promotes progression of hepatoma cells by enhancing the (EMT), cell migration, and invasion[16].
Our research demonstrated that the knockdown of PKM2 decreased the expression of E-cadherin and enhanced the EGF/EGFR signaling pathway to promote cell migration and invasion in the hepatocellular carcinoma cell lines HepG2 and Huh-7, which were positive for E-cadherin expression. Meanwhile, the expression levels of TGFBRI and phospho-Smad2 were upregulated when PKM2 was knocked down. The TGFβ/Smad signaling pathway regulates the EMT. Thus, PKM2 may be an important link between EGF and the TGFβ pathway in hepatocellular carcinoma cell migration and invasion. The aim of this study was to elucidate the function and mechanism of PKM2 with regard to cell metastasis in hepatocellular carcinoma cell lines.
MATERIALS AND METHODS
Cell culture conditions and transfection
The human hepatocellular carcinoma cell lines HepG2 and Huh-7 were cultured in DMEM (HyClone, Logan, UT, United States). All cells were cultured in medium containing 10% fetal bovine serum (FBS) (Gibco, Detroit, MI, United States) and 100 IU/mL penicillin-streptomycin at 37 °C in a 5% CO2 humidified atmosphere. The human hepatocellular carcinoma cell lines HepG2 and Huh-7 were obtained from the American Type Culture Collection (ATCC, United States). HepG2 and Huh-7 cells were transfected with the siPKM2 (pcPUR + U6-siPKM2) or the PU6 (pcPUR + U6-siRenilla) plasmid using FuGENE HD (Roche, Indianapolis, IN). Puromycin (0.1 μg/mL) was used to screen for stably transfected clones. The expression of the PKM2 protein was examined via Western blot analysis using an antibody against PKM2 to validate the ability of the constructs to inhibit target gene expression; these experiments were repeated three times. The cell cultures were made quiescent by growing them to confluence, and the medium was replaced with fresh medium containing 0.5% serum for 1 d. EGF (50 ng/mL final concentration) and TGFβ1 (20 ng/mL final concentration) were used for cell stimulation and were obtained from Cell Signaling Technology, Inc.
Stable knockdown of PKM2 and transient transfection
A plasmid containing an RNA interference sequence that targeted the PKM2 gene was constructed. The sense oligo for the siPKM2 sequence was 5′-CACCGCGGCAAGATTTATGTGGAACGTGTGCTGTC-CGTTCCACGTAGATCTTGCTGCTTTTT-3′, and the antisense oligo was 5′-GCATAAAAAGCA-GCAAGATCTACGTGGAACGGACAGCACACGT-TCCACATAAATCTTGCCGC-3′. The HepG2 and Huh-7 cells were transfected with pcPUR + U6-siPKM2 or pcPUR + U6-siRenilla (control) and selected as puromycin-resistant clones. Western blot analysis was performed to confirm the suppression of PKM2.
Protein extraction and Western blot analysis
The cells were re-suspended in lysis buffer containing a protease inhibitor cocktail, and the extracted proteins were separated using 8%-10% SDS-PAGE gels. β-tubulin was used as a loading control. Antibodies against E-cadherin, N-cadherin, RHOG, and MMP1 were obtained from Epitomics. The phospho-EGFR (Tyr1068), phospho-PLCγ1 (Tyr783), phospho-ERK1/2 (Thr202/Tyr204), phospho-Smad2 (Ser465/467), Smad2, TGFBRI and S100P antibodies were obtained from Cell Signaling Technology.
RNA extraction, reverse transcription, and real-time polymerase chain reaction
Total RNA was extracted using TRIzol reagent (Invitrogen, CA, United States). The samples were then treated with DNase for 15 min at room temperature, and the RNA was further purified using an RNA cleanup kit (Qiagen, CA, United States). The reverse transcription (RT) reaction for first-strand cDNA synthesis was performed using reverse transcriptase (Bio-Rad) with 2 μg of total RNA. Quantitative RT- polymerase chain reaction (PCR) analysis was performed with the ABI 7500 real-time PCR system (Applied Biosystems), and the gene expression levels for each individual sample were normalized to GAPDH. The mean relative gene expression was determined, and differences were calculated using the 2-ΔΔCt method. The RT-PCR primer sequences are listed in Table 1.
Table 1.
Sequences of the primers used in quantitative polymerase chain reaction
| Gene | Forward primer sequence (5’-3’) | Reverse primer sequence (5’-3’) |
| PKM2 | ATGGCTGACACATTCCTGGAGC | CCTTCAACGTCTCCACTGATCG |
| GAPDH | GTCTCCTCTGACTTCAACAGCG | ACCACCCTGTTGCTGTAGCCAA |
| CDH2 | CCTCCAGAGTTTACTGCCATGAC | GTAGGATCTCCGCCACTGATTC |
| MMP1 | ATGAAGCAGCCCAGATGTGGAG | TGGTCCACATCTGCTCTTGGCA |
| MMP9 | GCCACTACTGTGCCTTTGAGTC | CCCTCAGAGAATCGCCAGTACT |
| S100P | CTCAAGGTGCTGATGGAGAAGG | GAACTCACTGAAGTCCACCTGG |
| TGFBRI | GACAACGTCAGGTTCTGGCTCA | CCGCCACTTTCCTCTCCAAACT |
| RHOG | CTGCTACACAACTAACGCTTTCC | AGAGTGTACGGAGGCGGTCATA |
| RHOA | TCTGTCCCAACGTGCCCATCAT | CTGCCTTCTTCAGGTTTCACCG |
| RHOB | ACATTGAGGTGGACGGCAAGCA | CTGTCCACCGAGAAGCACATGA |
| NFKB2 | GGCAGACCAGTGTCATTGAGCA | CAGCAGAAAGCTCACCACACTC |
| TIMP2 | ACCCTCTGTGACTTCATCGTGC | GGAGATGTAGCACGGGATCATG |
| VIM | AGGCAAAGCAGGAGTCCACTGA | ATCTGGCGTTCCAGGGACTCAT |
| RAC1 | CGGTGAATCTGGGCTTATGGGA | GGAGGTTATATCCTTACCGTACG |
| RAC2 | CAGCCAATGTGATGGTGGACAG | GGAGAAGCAGATGAGGAAGACG |
| Serpine1 | CTCATCAGCCACTGGAAAGGCA | GACTCGTGAAGTCAGCCTGAAAC |
| KRT19 | AGCTAGAGGTGAAGATCCGCGA | GCAGGACAATCCTGGAGTTCTC |
| CDH1 | GCCTCCTGAAAAGAGAGTGGAAG | TGGCAGTGTCTCTCCAAATCCG |
Cell proliferation assay
Cell proliferation was measured using the Cell Counting Kit-8 (Dojindo, Kamimashiki-gun, Kumamoto, Japan) according to the manufacturer’s instructions. Briefly, the cells were seeded in four 96-well plates at a density of 2 × 103 cells/well. One plate was removed at the same time every day after the cells had adhered to the wells. The absorbance was measured with a microplate reader at a wavelength of 450 nm. All experiments were performed in triplicate.
Transwell invasion and wound healing assays
The transwell invasion assays were performed with 8.0-μm-pore inserts in a 24-well transwell plate. The basement membrane was hydrated with 500 μL of serum-free DMEM 30 min before use. For the invasion assay, the liver cancer cell lines were added to the upper chamber of the transwell plate with 0.5 mg/mL collagen type I (BD Bioscience, San Jose, CA)-coated filters. DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin was added to the lower chamber. The HepG2 and Huh-7 cells were incubated for 36 h. The invading cells were quantified after gentian violet staining. Each experiment was performed in triplicate, and the data are expressed as the mean values. The wound-healing assay was performed in 6-well plates. Tumor cells in medium containing 10% FBS were seeded into 6-well plates (Corning, CA). The cell cultures were made quiescent by growing them to confluence, and the medium was replaced with fresh medium containing 0.5% serum for 1 d. Wounds were made in the monolayer with sterile pipette tips. Then, EGF (50 ng/mL final concentration) was used for cell stimulation. The cells were subsequently washed with PBS and refreshed with medium containing 10% FBS. Photographs were taken at 0 and 24 h. All experiments were performed in triplicate.
Digital gene expression tag profiling and identification of differentially expressed genes
Total RNA was isolated from each sample of HepG2-pu6 and HepG2-siPK cells using TRIzol reagent (Invitrogen). A total of 20 μg of RNA was purified by adsorption onto biotin oligo(dT) magnetic beads. After mRNA binding, cDNA synthesis was performed. Double-stranded cDNA was digested with NlaIII endonuclease to produce fragments containing CATG sites with adjacent poly (A) tails at the 3′ end. After precipitation of the 3′ cDNA fragments, Illumina adaptor 1 was added to the 5′ ends of the fragments. Both adaptor 1 and the CATG site can be recognized by MmeI, which cuts downstream of the CATG site to produce 17-bp fragments tagged with adaptor 1. Adaptor 2 was then added to the 3′ end of these tagged fragments after removal of the poly(A) tail using biotin oligo(dT) beads. Then, these sequences were prepared for Solexa sequencing, and two samples were separately submitted to digital gene expression (DGE) profiling based on Solexa sequencing.
The expression level of each gene was estimated based on the frequency of clean tags and subsequently normalized to the number of transcripts per million clean tags (TPM)[17], which is a standard method and is extensively used in DGE analysis[18]. The threshold of the P value in multiple tests and analyses was determined by the false discovery rate (FDR). A combination of FDR < 0.001 and the absolute value of log2-Ratio ≥ 1 were used as the threshold to determine the significance of differences in gene expression.
Statistical analysis
Statistical analyses were performed using SPSS v13.0 (SPSS, Inc.) software. The independent samples t-test and correlation analysis were used to compare the data. All values are expressed as the mean ± SD. The differences were considered statistically significant at P < 0.05.
RESULTS
Depletion of PKM2 suppressed the proliferation of HepG2 and Huh-7 cells
The expression of the PKM2 protein in the hepatoma cell lines HepG2 and Huh-7 as well as normal human liver L02 cells was evaluated using Western blot and real-time PCR analysis. Hepatoma cells showed a higher level of PKM2 expression than normal human liver L02 cells. Next, stable HepG2 and Huh-7 cells with altered PKM2 expression were established using RNA interference (PKM2 knockdown). As shown in Figure 1A and B, PKM2 knockdown was successfully achieved in the HepG2 and Huh-7 cells. We observed that the proliferation of these cells was decreased after PKM2 was depleted (Figure 1C and D). These results are in agreement with previous studies.
Figure 1.
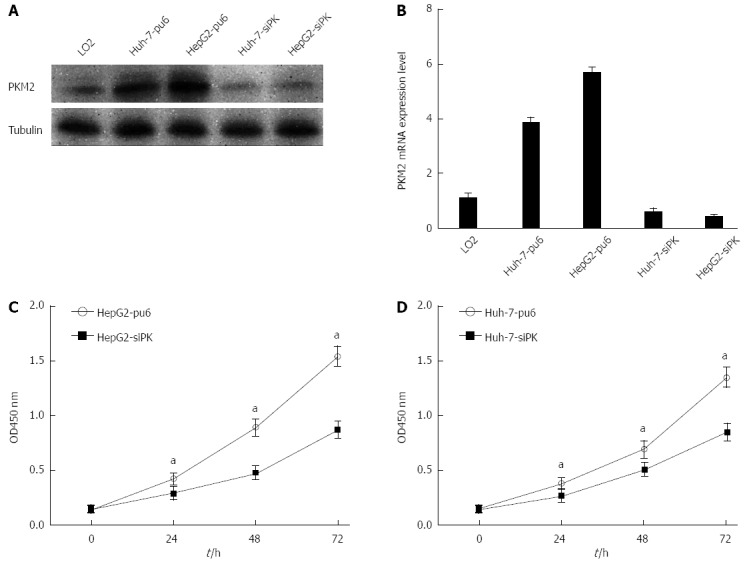
Knockdown of pyruvate kinase M2 isoform suppressed cell proliferation in HepG2 and Huh-7 cells. A: HepG2 and Huh-7 cells were stably transfected with shRNA directed against pyruvate kinase M2 isoform (PKM2). The specific knockdown of PKM2 was monitored by immunoblot (bottom). Cells stably transfected with pcPUR + U6-siPKM2 are referred to as siPK, and those transfected with pcPUR + U6-siRenilla are referred to as pu6; B: PKM2 expression levels were analyzed by quantitative real-time PCR in stable HepG2 and Huh-7 cells. Error bars represent the mean ± SD of triplicate experiments (aP < 0.05 between groups); C and D: Proliferation of the stably transfected cells. The cell number was determined with the CCK-8 assay, and the relative number of cells is shown.
Depletion of PKM2 promoted cell migration and invasion of HepG2 and Huh-7 cells stimulated with EGF
To examine the effect of PKM2 on cell migration and invasion, we employed well-established wound-healing and transwell assays to characterize the cell motility response in HepG2 and Huh-7 cells. A confluent layer of cells was first incubated overnight in medium, and a scratch wound was introduced. Medium containing EGF (50 ng/mL) was then added to stimulate migration, and the percentage of wound sealing was observed after 24 h. The invading cells were quantified 36 h after EGF (50 ng/mL) was added to the lower chamber. The results indicate that the treatment of HepG2-siPK and Huh-7-siPK cells with EGF following a scratch wound and in a transwell assay significantly increased the rate of wound healing (Figure 2A and B) and invasion (Figure 1C and D), respectively, compared with the control cells.
Figure 2.
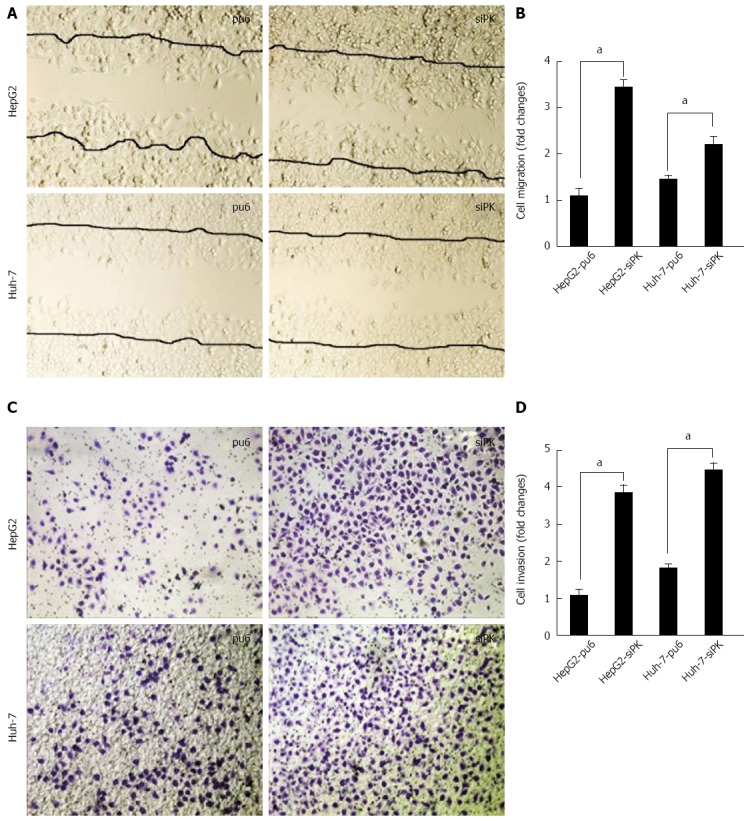
Knockdown of pyruvate kinase M2 isoform promoted the migration and invasion of HepG2 and Huh-7 cells upon epidermal growth factor stimulation. A: A cross-shaped wound was created in the monolayer, and stably transfected HepG2 and Huh-7 cells were cultured for an additional 24 h with epidermal growth factor (EGF) (50 ng/mL). Representative images of the wounded region are shown; B: The results of the migration assay are also shown as graphs (aP < 0.05); C: The invasion potential of the HepG2 and Huh-7 stable cells was assessed using the BD transwell chamber assay with 50 ng/mL EGF in the lower chamber for 36 h. The cells that migrated to the lower side of the filter were stained, photographed, and counted; D: The data are expressed as the mean ± SD from three independent experiments, aP < 0.05 between groups.
Transcriptomic landscape of hepatoma cell lines were revealed and verification
To investigate the mechanisms underlying changes in cell migration and invasion after PKM2 knockdown, we examined the transcriptomic landscape of hepatoma cell lines through mRNA sequencing. To determine the genes and transcripts that were differentially expressed between the HepG2 and HepG2-siPK cells, univariate F-tests were performed. We identified 2100 transcripts that were differentially expressed between the two cell lines. Among the identified transcripts, 450 transcripts were more highly abundant (i.e., were upregulated) in the HepG2-siPK cells, while 1650 showed lower relative abundance (i.e., were downregulated) in the HepG2 cells (Figure 3A). Cell migration- and invasion-related genes were selected for further statistical analysis (Figure 3B). The mRNA sequence analysis was validated by experimental confirmation of the levels of differentially expressed transcripts in HepG2 and HepG2-sipk cells. We used qRT-PCR to validate the expression levels of 16 transcripts, and we observed a high correlation and a similar expression trend between the mRNA-sequence based abundance estimation and the results of qRT-PCR assays (Figure 3C). Protein expression levels were examined by western blot analysis (Figure 3D). Protein expression analysis further confirmed the accuracy of the gene expression levels.
Figure 3.
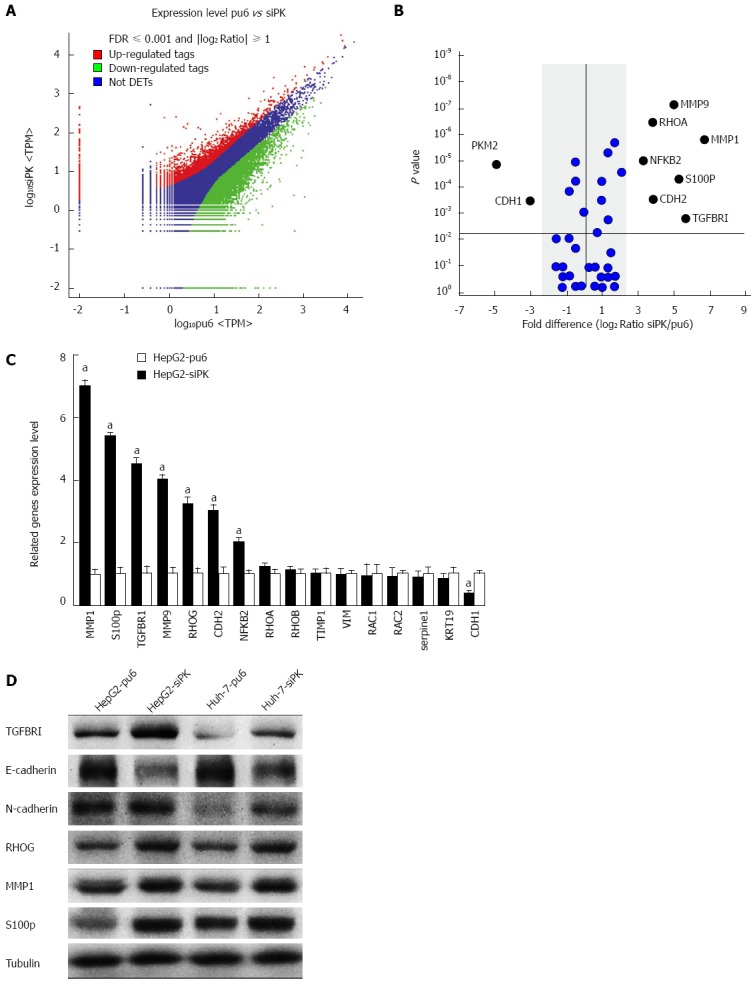
Differential gene expression between HepG2-pu6 and HepG2-siPK cells. A: The number of statistically significant differentially expressed transcripts identified from the F-test. Color plots show the expression levels of significantly differentially expressed transcripts (P < 0.05 between groups and false discovery rate 0.05) between HepG2-pu6 and HepG2-siPK cells. The upregulated genes are indicated in red, and the downregulated genes are indicated in green. Genes that did not show a statistically significant difference are indicated in blue; B: The graph shows gene expression in relation to cell migration and invasion. Fold differences were calculated based on the log2 ratio of siPK/pu6; C: A comparison of the RT-qPCR and RNA sequence expression analysis; D: The expression levels of genes that were determined to have statistically significant differences in expression were analyzed by immunoblot analysis in stable HepG2 and Huh-7 cells.
Depletion of PKM2 enhanced the activities of the EGF/EGFR and TGFβ1/TGFBR signaling pathways
Cell migration and invasion are largely regulated by EGFR activity. To analyze whether the EGFR is involved in the migration and invasion of HepG2 and Huh-7 cells, these cells were treated with EGF, which binds to the EGFR and activates downstream signaling pathways. Treatment with EGF resulted in the phosphorylation of the EGFR and the subsequent activation of the PLCγ1 and ERK1/2 pathways (Figure 4A). We found that PLCγ1 showed a higher level of activity in PKM2-depleted cells than in control cells after either a short or long (24 h) incubation with EGF. PLCγ is a key regulator of cell migration downstream of RTK signaling[19]. Phosphorylation of tyrosine residue 783 of PLCγ1 is critical to its activation[20]. PLCγ1 activation enhanced cell motility, and this effect was observed in both the wound scratch and transwell assays.
Figure 4.
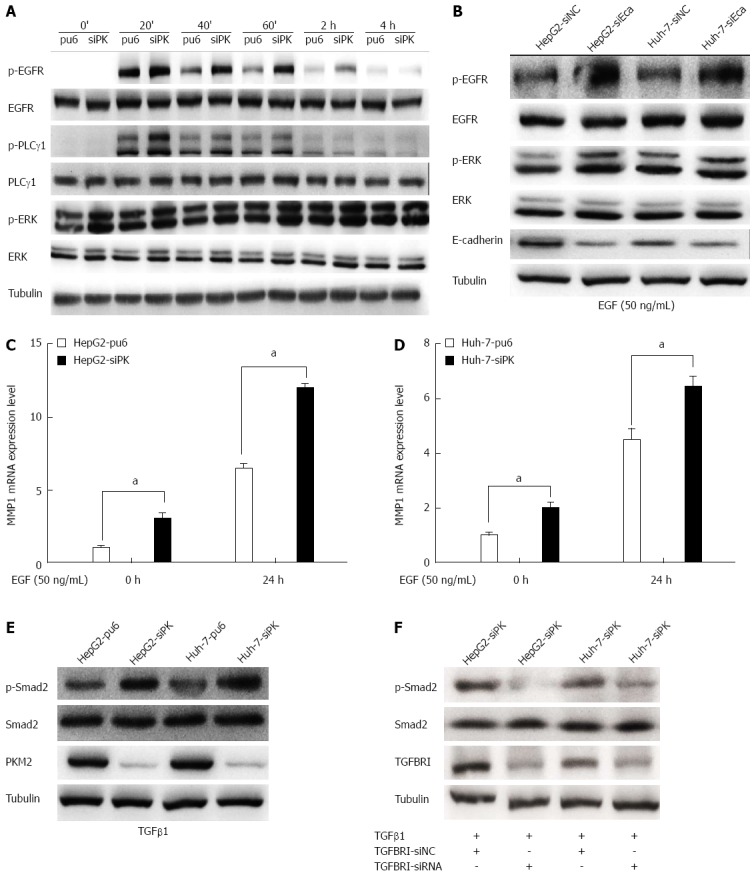
Depletion of pyruvate kinase M2 enhanced the activities of the epidermal growth factor/EGFR and transforming growth factor beta 1/TGFBR downstream signaling pathways. A: Stable HepG2 and Huh-7 cells were exposed to epidermal growth factor (EGF) (50 ng/mL) for different times. Western blots of the cell lysates are shown. The protein levels of phospho-EGFR (Tyr1068), phospho-PLCγ1 (Tyr783), and phospho-ERK1/2 (Thr202/Tyr204) are shown as indicated; B: The expression of E-cadherin was knocked down in HepG2 and Huh-7 cells by transient transfection of siRNA, and after 48 h, these cells were stimulated with EGF (50 ng/mL). The protein levels of phospho-EGFR (Tyr1068) and phospho-ERK1/2 (Thr202/Tyr204) are shown as indicated; C and D: MMP1 expression levels were analyzed by quantitative real-time PCR in HepG2 and Huh-7 stable cells. Error bars represent the mean ± SD of triplicate experiments (aP < 0.05 between groups); E: Phospho-Smad2 (Ser465/467) protein levels are shown as indicated in stable HepG2 and Huh-7 cells stimulated with TGFβ1 (20 ng/mL); F: The expression of TGFBRI was knocked down in HepG2 and Huh-7 cells by transient transfection of siRNA, and after 48 h, these cells were stimulated with transforming growth factor beta 1(TGFβ1) (20 ng/mL). The protein levels of phospho-Smad2 (Ser465/467) are shown as indicated.
The EGF-dependent activation of the EGFR has been reported to be inhibited in an E-cadherin adhesion-dependent manner. Indeed, E-cadherin adhesion inhibits the ligand-dependent activation of diverse receptor tyrosine kinases. To confirm the role of E-cadherin in the activation of EGFR, we used RNA interference to silence E-cadherin gene expression and subsequently observed the level of EGFR phosphorylation in HepG2 cells. We found that the EGFR phosphorylation level was significantly higher when E-cadherin was silenced (Figure 4B). Thus, PKM2 affected the expression level of E-cadherin, thereby regulating the activation of EGFR.
We next investigated the effect of an EGFR ligand on the expression of MMPs using RT-PCR in HepG2-sipk and Huh-7-sipk cells compared with their respective control cells. Treatment with the EGFR ligand EGF enhanced the transcription of MMPs in HepG2 and Huh-7 cells. MMP1 (Figure 4C and D) expression was upregulated in PKM2-depleted cells upon EGF treatment.
In previous experiments, we found that the expression of TGFBRI increased after PKM2 knockdown (Figure 3). We examined the level of Smad2 phosphorylation by immunoblot analysis in PKM2 knockdown cells and the respective control cells (HepG2 and Huh-7). We found that the Smad2/3 phosphorylation level was significantly higher when PKM2 was knocked down (Figure 4E). Next, we used RNA interference to silence TGFBRI gene expression, and we observed that the level of Smad2 phosphorylation was significantly decreased in PKM2 knockdown cells (Figure 4F). These results show that PKM2 is a new modulator of hepatic cell motility and invasiveness that functions through the TGFβ pathway.
DISCUSSION
The invasive and metastatic stage of cancer progression correlates with poor clinical prognosis and represents the most formidable barrier to successful treatment[21]. Cell motility and invasiveness are the defining characteristics of malignant tumors, which enable tumor cells to migrate into adjacent tissues or through limiting basement membranes and extracellular matrices. Cell motility is required for the physiological processes of wound repair and organogenesis and for the pathological process of tumor invasion[22]. Invasive tumor cells are characterized by dysregulated cell motility in response to extracellular signals from growth factors and cytokines[23]. Human tumors express high levels of growth factors and their receptors, and many types of malignant cells appear to exhibit autocrine- or paracrine-stimulated growth. Among the most well-studied growth factor receptor systems is the EGF receptor family[24]. At least two distinct intracellular signaling pathways are required for EGFR-mediated cell motility: the PLCγ and the MAP kinase pathways. PLCγ activity has been proposed to enhance cell motility through the mobilization of actin-modifying proteins from an inactive (membrane-associated) state to an active (sub-membrane cytoskeletal) state[25].
The Erk MAP kinases transmit signals to the nucleus as well as signals that regulate cell-matrix connections[26]. ERK/MAPK pathways play critical roles in EGFR ligand-induced MMP1 expression[27], and sustained ERK phosphorylation is necessary for MMP-9 regulation[28,29]. MMPs have been suggested to serve as key regulators of tumor growth and metastasis based on their enzymatic properties[30].
We observed downregulation of E-cadherin mRNA expression the N-cadherin protein expression level was increased in the HepG2 cell line when PKM2 was depleted. The knockdown of PKM2 promoted cell migration and invasion in HepG2 and Huh-7 cells with EGF stimulation. Increased activity of the EGFR is the critical factor determining the cell motility and invasiveness of HepG2 and Huh-7 cells. The EGF-dependent activation of the EGFR has been reported to be inhibited in an E-cadherin adhesion-dependent manner, which inhibits the ligand-dependent activation of diverse receptor tyrosine kinases. We hypothesized that the downregulation of E-cadherin expression enhanced the phosphorylation of the EGFR and activated downstream signaling pathways, including PLCγ1 and ERK1/2. PLCγ activation enhances cell motility, and ERK1/2 activity plays a critical role in MMP1 and MMP9 expression. In addition, the expression levels of TGFBRI and phospho-Smad2 were upregulated when PKM2 was knocked down. The process of EMT is regulated by the TGFβ/Smad signal pathway. PKM2 may be an important link between EGF and the TGFβ pathway in hepatocellular carcinoma cell migration and invasion. Thus, PKM2 may play a novel role in the reversible inhibition of cell motility and invasion. The biological role of PKM2 in the development of these tumors must be further elucidated.
COMMENTS
Background
Studies have shown that cancer cells exclusively express pyruvate kinase M2 isoform (PKM2). Over the years, significant advancements have been made in understanding the function and regulation of PKM2 as a pyruvate kinase and protein kinase in cancer cells.
Research frontiers
Recently many findings reveal a novel role for PKM2 as a transcriptional coactivator. However, there are more unknown mechanism need to be explored.
Innovations and breakthroughs
There are some controversies regarding the specificity and potential of PKM2 as an anti-cancer target in cancer therapy. The authors found that PKM2 may be an important link between epidermal growth factor (EGF) and the transforming growth factor beta (TGFβ) pathway in hepatocellular carcinoma cell migration and invasion. PKM2 may be act as an inhibitor in cell motility.
Applications
PKM2 may play different roles in modulating the metastasis of hepatocellular carcinoma cells, and this finding could help to guide the development of future targeted therapies. The results of our study reveal that PKM2 may be an important link between the EGF and TGFβ pathways during hepatocellular carcinoma cell migration and invasion.
Terminology
Pyruvate kinase mediates the final rate-limiting step of glycolysis by catalyzing the dephosphorylation of phosphoenolpyruvate to pyruvate to yield one molecule of ATP.
Peer-review
The authors mainly concentrate on to explore the mechanisms of PKM2 inhibits motility in hepatocellular carcinoma cells. Their findings show that PKM2 can regulate cell motility through the EGF/EGFR and TGFβ/TGFR signaling pathways. And this finding could help to guide the future targeted therapies.
Footnotes
Supported by National Natural Science Foundation of China, No. 81370505, No. 81370591, No. 81225025, No. 81100285 and No. 91229201; grants from Ministry of Health Foundation for State Key Clinical Department, 863 and 973 programs in China, No. 2012AA02A201 and No. 2013CB944903.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: September 27, 2014
First decision: December 2, 2014
Article in press: June 10, 2015
P- Reviewer: El Jamali A, Guo ZS, Tanase CP, Yoshitomi H S- Editor: Yu J L- Editor: A E- Editor: Zhang DN
References
- 1.Edwards S, Nguyen BT, Do B, Roberts JKM. Contribution of malic enzyme, pyruvate kinase, phosphoenolpyruvate carboxylase, and the krebs cycle to respiration and biosynthesis and to intracellular pH regulation during hypoxia in maize root tips observed by nuclear magnetic resonance imaging and gas chromatography-mass spectrometry. Plant Physiol. 1998;116:1073–1081. doi: 10.1104/pp.116.3.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43:969–980. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 4.Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005;15:300–308. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 5.Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry. 2005;44:9417–9429. doi: 10.1021/bi0474923. [DOI] [PubMed] [Google Scholar]
- 6.Chaneton B, Gottlieb E. Rocking cell metabolism: revised functions of the key glycolytic regulator PKM2 in cancer. Trends Biochem Sci. 2012;37:309–316. doi: 10.1016/j.tibs.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 7.Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature. 2011;480:118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang LY, Liu YP, Chen LG, Chen YL, Tan L, Liu JJ, Jazag A, Ren JL, Guleng B. Pyruvate kinase M2 plays a dual role on regulation of the EGF/EGFR signaling via E-cadherin-dependent manner in gastric cancer cells. PLoS One. 2013;8:e67542. doi: 10.1371/journal.pone.0067542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tian X, Liu Z, Niu B, Zhang J, Tan TK, Lee SR, Zhao Y, Harris DC, Zheng G. E-cadherin/β-catenin complex and the epithelial barrier. J Biomed Biotechnol. 2011;2011:567305. doi: 10.1155/2011/567305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen HC, Chu RY, Hsu PN, Hsu PI, Lu JY, Lai KH, Tseng HH, Chou NH, Huang MS, Tseng CJ, et al. Loss of E-cadherin expression correlates with poor differentiation and invasion into adjacent organs in gastric adenocarcinomas. Cancer Lett. 2003;201:97–106. doi: 10.1016/j.canlet.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 11.Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 2004;23:1739–1748. doi: 10.1038/sj.emboj.7600136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siegel PM, Massagué J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 13.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 14.Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, Tamaki K, Hanai J, Heldin CH, Miyazono K, et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997;16:5353–5362. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberts AB. TGF-beta signaling from receptors to the nucleus. Microbes Infect. 1999;1:1265–1273. doi: 10.1016/s1286-4579(99)00258-0. [DOI] [PubMed] [Google Scholar]
- 16.Zubeldia IG, Bleau AM, Redrado M, Serrano D, Agliano A, Gil-Puig C, Vidal-Vanaclocha F, Lecanda J, Calvo A. Epithelial to mesenchymal transition and cancer stem cell phenotypes leading to liver metastasis are abrogated by the novel TGFβ1-targeting peptides P17 and P144. Exp Cell Res. 2013;319:12–22. doi: 10.1016/j.yexcr.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 17.‘t Hoen PA, Ariyurek Y, Thygesen HH, Vreugdenhil E, Vossen RH, de Menezes RX, Boer JM, van Ommen GJ, den Dunnen JT. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008;36:e141. doi: 10.1093/nar/gkn705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morrissy AS, Morin RD, Delaney A, Zeng T, McDonald H, Jones S, Zhao Y, Hirst M, Marra MA. Next-generation tag sequencing for cancer gene expression profiling. Genome Res. 2009;19:1825–1835. doi: 10.1101/gr.094482.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Piccolo E, Innominato PF, Mariggio MA, Maffucci T, Iacobelli S, Falasca M. The mechanism involved in the regulation of phospholipase Cgamma1 activity in cell migration. Oncogene. 2002;21:6520–6529. doi: 10.1038/sj.onc.1205821. [DOI] [PubMed] [Google Scholar]
- 20.Poulin B, Sekiya F, Rhee SG. Intramolecular interaction between phosphorylated tyrosine-783 and the C-terminal Src homology 2 domain activates phospholipase C-gamma1. Proc Natl Acad Sci USA. 2005;102:4276–4281. doi: 10.1073/pnas.0409590102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hofmann-Wellenhof R, Woltsche-Kahr I, Smolle J, Kerl H. Clinical and histological features of poor prognosis in cutaneous metastatic melanomas. J Cutan Pathol. 1996;23:199–204. doi: 10.1111/j.1600-0560.1996.tb01467.x. [DOI] [PubMed] [Google Scholar]
- 22.Wang JF, Olson ME, Reno CR, Wright JB, Hart DA. The pig as a model for excisional skin wound healing: characterization of the molecular and cellular biology, and bacteriology of the healing process. Comp Med. 2001;51:341–348. [PubMed] [Google Scholar]
- 23.Han MY, Kosako H, Watanabe T, Hattori S. Extracellular signal-regulated kinase/mitogen-activated protein kinase regulates actin organization and cell motility by phosphorylating the actin cross-linking protein EPLIN. Mol Cell Biol. 2007;27:8190–8204. doi: 10.1128/MCB.00661-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19:6550–6565. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- 25.Arora PD, McCulloch CA. Dependence of fibroblast migration on actin severing activity of gelsolin. J Biol Chem. 1996;271:20516–20523. doi: 10.1074/jbc.271.34.20516. [DOI] [PubMed] [Google Scholar]
- 26.Al-Ayoubi A, Tarcsafalvi A, Zheng H, Sakati W, Eblen ST. ERK activation and nuclear signaling induced by the loss of cell/matrix adhesion stimulates anchorage-independent growth of ovarian cancer cells. J Cell Biochem. 2008;105:875–884. doi: 10.1002/jcb.21889. [DOI] [PubMed] [Google Scholar]
- 27.Park S, Jung HH, Park YH, Ahn JS, Im YH. ERK/MAPK pathways play critical roles in EGFR ligands-induced MMP1 expression. Biochem Biophys Res Commun. 2011;407:680–686. doi: 10.1016/j.bbrc.2011.03.075. [DOI] [PubMed] [Google Scholar]
- 28.Genersch E, Hayess K, Neuenfeld Y, Haller H. Sustained ERK phosphorylation is necessary but not sufficient for MMP-9 regulation in endothelial cells: involvement of Ras-dependent and -independent pathways. J Cell Sci. 2000;113 Pt 23:4319–4330. doi: 10.1242/jcs.113.23.4319. [DOI] [PubMed] [Google Scholar]
- 29.Yu X, Lin SG, Huang XR, Bacher M, Leng L, Bucala R, Lan HY. Macrophage migration inhibitory factor induces MMP-9 expression in macrophages via the MEK-ERK MAP kinase pathway. J Interferon Cytokine Res. 2007;27:103–109. doi: 10.1089/jir.2006.0054. [DOI] [PubMed] [Google Scholar]
- 30.Sato H. [Tumor metastasis and MMP inhibitor] Clin Calcium. 2006;16:621–26. [PubMed] [Google Scholar]