

Abstract
Pituitary adenomas are one of the most common intracranial tumors. Despite their benign nature, dys-regulation of hormone secretion causes systemic metabolic deterioration, resulting in high mortality and an impaired quality of life. Tumorigenic pathogenesis of pituitary adenomas is mainly investigated by performing genetic analyses of somatic mutations in the tumor or germline mutations in patients. Genetically modified mouse models, which develop pituitary adenomas, are also used. Genetic analysis in rare familial pituitary adenomas, including multiple endocrine neoplasia type 1 and type 4, Carney complex, familial isolated pituitary adenomas, and succinate dehydrogenases (SDHs)-mediated paraganglioma syndrome, revealed several causal germline mutations and sporadic somatic mutations in these genes. The analysis of genetically modified mouse models exhibiting pituitary adenomas has revealed the underlying mechanisms, where cell cycle regulatory molecules, tumor suppressors, and growth factor signaling are involved in pituitary tumorigenesis. Furthermore, accumulating evidence suggests that epigenetic changes, including deoxyribonucleic acid (DNA) methylation, histone modification, micro ribonucleic acids (RNAs), and long noncoding RNAs play a pivotal role. The elucidation of precise mechanisms of pituitary tumori-genesis can contribute to the development of novel targeted therapy for pituitary adenomas.
Keywords: pituitary adenoma, tumorigenesis, cyclic adenosine monophosphate, cell cycle, growth factor
Introduction
The pituitary gland is the central mediator for peripheral endocrine homeostasis regulation by secretion of tropic hormones, such as adrenocorticotropic hormone (ACTH), thyroid stimulating hormone (TSH), growth hormone (GH), prolactin, follicle-stimulating hormone (FSH), and luteinizing hormone (LH).1) Pituitary adenomas are common, with 5–10% prevalence rates found at autopsy in postmortem studies,2,3) accounting for 15% of all intra-cranial tumors.4,5) Most common among these are prolactin (PRL)-producing pituitary adenomas (29%) or clinically nonfunctioning pituitary adenomas (NFPAs), derived from all cell types of the adenohypophysis, though mostly gonadotrophs, particularly FSH-producing adenomas. The prevalence of GH-producing pituitary adenomas is 15% and that of ACTH-producing pituitary adenomas is 10%, while TSH-producing pituitary adenomas are rare.6–9) An epidemiological study of 9,519 Japanese patients with pituitary adenoma revealed that 46% were diagnosed with NFPAs, 25% with PRL-producing adenomas, 22% with GH-producing adenomas, and 6% with ACTH-producing adenomas.10) Despite the mostly benign nature of the tumor, hormonal dysregulation and local expansion cause either an excess or impaired secretion of pituitary hormones, causing disturbance in growth, reproductive function, and metabolism, resulting in various morbidities, impaired quality of life, and increased mortality.4)
Surgical resection is the first-line of treatment for pituitary adenomas, except for PRL-producing adenomas. Residual or recurrent tumors require re-operation, medical treatment, or radiation. An understanding of the physiological regulation and molecular mechanisms of pituitary hormone synthesis, secretion, and peripheral action has led to the development of targeted drugs such as dopa-mine agonists, somatostatin analogs, GH receptor antagonists, steroidogenic inhibitors, and glucocorticoid receptor antagonists.11–17) In this regard, a better understanding of pituitary tumorigenesis is crucial for the development of novel targeted drugs for pituitary adenomas. In this review, we discuss human pituitary adenomas and animal models, as well as the involvement of genetic and epigenetic changes in pituitary tumorigenesis.
Pathogenesis of Pituitary Adenomas
Pituitary adenomas are considered to be of monoclonal origin, based on X-chromosome inactivation studies,18–20) suggesting that these tumor cells arise from a single cell. Therefore, it has been hypothesized that a mutation in the cell might cause pituitary adenomas as well as other tumors. Indeed, mutations in the GNAS gene has been reported as a cause of GH-producing pituitary adenomas.21) Furthermore, the analysis of pituitary adenomas related to hereditary syndromes has revealed several causal germline mutations in pituitary adenomas. For example, multiple endocrine neoplasia type 1 (MEN1), Carney complex (CNC), familial isolated pituitary adenomas (FIPAs), and succinate dehydrogenases (SDHs)-related paraganglioma syndrome, shows germline mutations in MEN1, PRKAR1A, CDKN1B, and SDHs genes, respectively,22) and loss of heterozygosity (LOH) at the affected locus in the tumor is generally observed (Table 1).23) However, the frequency of familial pituitary adenomas is less than 5% in patients with pituitary adenomas, demonstrating that the cause of most tumors remains unknown.24) On the other hand, somatic GNAS1 mutations were found in 30–40% of GH-producing pituitary adenomas,25) indicating that mutations contribute to the development of pituitary tumors (Fig. 1).
Table 1.
Genetic changes in human pituitary adenomas and modified mice models with pituitary adenomas
| Locus | Germline mutations |
Somatic mutations |
Syndrome | |||||
|---|---|---|---|---|---|---|---|---|
| Human |
Mice |
Human (pituitary tumors) | Mice (pituitary conditional) | |||||
| Mutations | LOH | Mutations | LOH | |||||
| MEN1 | 11q13 | + | + | + (hetero) | + | + | − | MEN1 |
| PRKAR1A | 17q24.2 | + | ± | − | NA | − | + (GHRH-R) | CNC |
| AIP | 11q13.3 | + | + | + (hetero) | + | − | − | FIPA |
| CDKN1B (p27 kip1 ) | 12p13 | + | ± | + (homo) | NA | Downregulated | + (POMC) | MEN4 |
| SDHs | * | + | + | − | NA | + | − | PGLs |
| GNAS | 20q13.3 | − | NA | − | NA | + | − | MAS |
| Rb | 13q14.2 | − | NA | + (hetero) | + | Downregulated | + (POMC) | |
| CDKN2C (P18 ink4c ) | 1p32 | − | NA | + (homo) | NA | − | − | |
| PTTG1 | 5q35.1 | − | NA | − | NA | Upregulated | + (αGSU) | |
| HMGA1 | 6p21 | − | NA | + | NA | − | − | |
| HMGA2 | 12q15 | − | NA | + | NA | Upregulated | − | |
| Cyclin E | 19q12 | − | NA | − | NA | Upregulated | + (POMC) | |
| TGFα | 2p13 | − | NA | − | NA | − | + (PRL) | |
| FGFR4 | 5q35.2 | − | NA | − | NA | Truncated variant | + (PRL) | |
| D2R | 11q23 | − | NA | + (homo) | NA | − | − | |
| PRLR | 5p13.2 | − | NA | + (homo) | NA | − | − | |
*: SDHA 5p15, SDHB 1p36.1p35, SDHC 1q23.3, SDHD 11q23, CDKN1B: cyclin-dependent kinase inhibitor 1B, CDKN2C: cyclin-dependent kinase inhibitor 2C, CNC: Carney complex, D2R: dopamine receptor type 2, FGFR: fibroblast growth factor receptor, FIPA: familial isolated pituitary adenoma, GHRH-R: growth hormone releasing hormone receptor, GNAS: GNAS complex locus, αGSU: glycoprotein hormone, alpha subunit, hetero: heterozygosity, HMGA: high mobility group A, homo: homozygosity, LOH: loss of heterozygosity, MAS: McCune-Albright syndrome, MEN1: multiple endocrine neoplasia type 1, NA: not applicable, PGL: paraganglioma, PGLs: SDH-related PGL syndrome, POMC: pro-opiomelanocortin, PRLR: prolactin receptor, PRKAR1A: protein kinase, cAMP-dependent, regulatory, type 1, alpha, PTTG1: pituitary tumor transforming 1, Rb: retinoblastoma, SDH: succinate dehydrogenase complex, TGFα: transforming growth factor alfa.
Fig. 1.
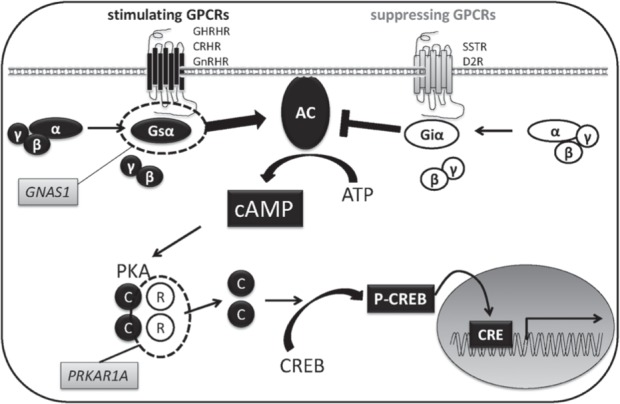
Enhanced cAMP signaling in pituitary adenomas. Activating somatic gain-of function mutations in GNAS1 gene, which encodes α subunit of stimulatory G protein (Gsα), cause GH-producing pituitary adenoma. Loss of expression and/or function mutations in PRKAR1A gene results in Carney complex. PRKAR1A gene encodes type 1 regulatory subunit (R) of protein kinase A that inhibits the catalytic subunits (C) activated by an increase in intracellular cAMP levels. AC: Adenyl cyclase, CRE: cAMP response elements, cAMP: cyclic adenosine monophosphate, CREB: cAMP responsive element binding protein, CRHR: Corticotrophin releasing hormone receptor, D2R: dopamine receptor type 2, GH: growth hormone, GHRHR: growth hormone releasing hormone receptor, Giα: α subunit of inhibitory G protein, GnRHR: gonadotropin releasing hormone receptor, GPCR: G-protein coupled receptor, Gsα: α subunit of stimulatory G protein, p-CREB: phospho-CREB, PKA: protein kinase A, SSTR: somatostatin receptor.
Recently, epigenetic deregulation, including deoxyribonucleic acid (DNA) methylation, histone modification, nucleosomes remodeling, and ribonucleic acid (RNA) mediated targeting, have been shown to play a causative role in pituitary tumori-genesis.26) DNA methylation is a stable modification that leads to chromatin remodeling, resulting in transcriptional silencing without gene mutation.27) It occurs at cytosine residues in CpG islands, frequently located within the promoter region of the gene.28) This mechanism is regulated by DNA methyltransferases (DNMTs), namely DNMT1, DNMT3A, and DNMT3B.29–32) In contrast to DNA methylation, histone modifications are reversible, and can lead to either activation or repression of gene transcription, brought about by specific acetylation or methylation lesions.33) Although several animal models of pituitary tumors have helped to identify potentially causative genes, few mutations of these genes have been detected in human pituitary adenomas. For example, retinoblastoma (Rb)-associated protein gene,34) pituitary tumor transforming gene (PTTG),35) high mobility group A (HMGA),36,37) and cyclin E (CCNE1)38) reportedly play an important role in pituitary tumorigenesis in mice; however, there have been no mutations in these genes in humans, suggesting a possibility of misregulation of expression levels or post-transcriptional regulation of these genes. DNMT3B is highly expressed in human pituitary adenomas, including GH-, PRL-, and ACTH-producing pituitary adenomas, as well as NFPAs, compared to normal pituitary. Knockdown of DNMT3B in AtT20 mouse ACTH-producing pituitary adenoma cell line enhanced Rb expression.39) The promoter region of the Rb gene is frequently hypermethylated in pituitary adenomas.40,41) MicroRNAs (miRNAs) are endogenous small noncoding RNAs that bind to 3′-untranslated regions (3′-UTRs) of target mRNAs, and thus regulate gene expression.42) Deregulated miRNAs have been reported to regulate genes associated with pituitary tumorigenesis.1,43,44) These findings demonstrate a crucial role of epigenetic deregulation in pituitary tumorigenesis.26,45)
I. Genetic changes
Many genetic changes related to pituitary tumor development in humans and mice have been reported. These genes are summarized in Table 1.
1. Evidence in humans
Pituitary adenomas are mostly observed in sporadic conditions, but some also arise in familial tumor syndromes, and both show clonal expansion from a single cell. LOH in the tumor is generally observed in familial syndromes, and somatic mutation occurs in most sporadic tumors.
Germline mutations: MEN1, located on chromosome 11q13, encodes the protein menin.46) Heterozygous mutation in MEN1 is responsible for MEN1, an autosomal dominant syndrome, first identified in 1997.47) Germline mutation of the gene represents tumor development in the parathyroid glands, anterior pituitary, and endocrine pancreas.48) Nonsense or frameshift mutations lead to inactivation of the tumor suppressor function of menin.49) The penetrance of pituitary adenomas in patients with MEN1 varies from 15–50% in different series.50) Estimated prevalence of MEN1-associated pituitary adenomas is 2.7% in all pituitary adenomas.51) All cell types of anterior pituitary adenomas, except the true gonadotropinoma, have been reported in this group.52,53) Pituitary adenomas in patients with MEN1 represent larger size, more aggressive behavior, and reduced response to treatment as compared to non-MEN1.54) Plurihormonal expression is more frequently observed in MEN1-associated pituitary tumors.54,55) No specific histological difference in cellular and nuclear features or proliferative markers is observed between MEN1- and non-MEN1-associated pituitary tumors.55)
PRKAR1A, located on chromosome 17q24.2, encodes type 1 regulatory subunit of protein kinase A.56,57) Heterozygous loss of function mutations in PRKAR1A have been identified in about two-thirds of patients with CNC,58) an autosomal dominant disorder first reported in 1985. CNC is clinically characterized by spotty skin pigmentation, myxomas, endocrine tumors, which include pituitary adenomas, and schwannomas.57,59–61) The incidence of pituitary abnormality in patients with CNC was reported in 12% cases.58) CNC-associated pituitary adenomas can be multi-focal, and plurihormonal staining has identified dysregulation of several hormones, except for ACTH.62–64) GH-producing pituitary adenomas are most common,59,65) while abnormal PRL secretion or PRL-producing pituitary adenomas were also involved in CNC.64,66,67) In somatomammotroph hyperplasia, which appears to predate adenomas, loss of heterozygosity (LOH) of PRKAR1A has not been observed consistently.63)
AIP, located on chromosome 11q13.3, encodes the aryl-hydrocarbon receptor interacting protein (AIP)68). Heterozygous inactivating mutations of AIP were observed in 15–20% of patients with FIPAs.69,70) LOH of AIP was identified in the pituitary adenoma.71) The penetrance of pituitary adenomas in patients with AIP mutations is 40–50% in families with GH-producing adenomas or PRL-producing adenomas.69,70,72) AIP mutation-positive patients have a characteristic clinical phenotype of young-onset and showing GH and/or PRL-producing pituitary adenomas.25,71) In addition, GH-producing pituitary adenomas associated with AIP mutations are generally large and resistant to somatostatin analogs.69)
CDKN1B gene, located on chromosome 12p13, encodes cyclin dependent kinase inhibitor p27kip1, which negatively regulates the Cdk2/Cyclin E and Cdk2/Cyclin A protein complexes and prevents cell cycle progression73) (Fig. 2). Heterozygous loss of function CDKN1B mutations have been identified in patients with MEN4, an autosomal dominant disorder characterized by parathyroid and pituitary tumors.74) Approximately 3% of patients with clinical MEN1 without MEN1 mutations have a mutation in this gene.75,76) CDKN1B mutation was recently identified in AIP mutation-negative patients with FIPA77) (Fig. 2).
Fig. 2.
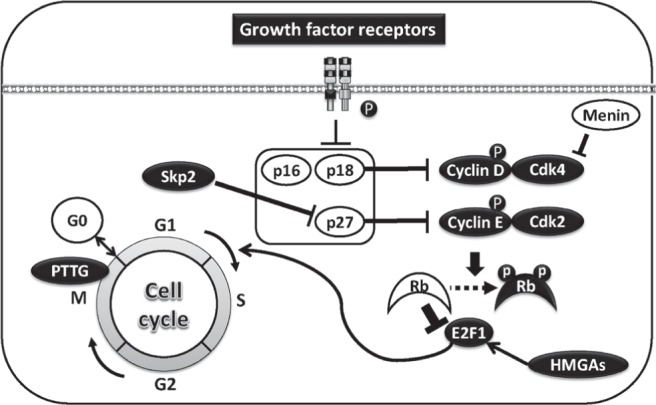
The aberrant regulation of cell cycle in pituitary tumorigenesis. Targeted deletion in Rb gene results in pituitary adenomas depending of a transcription factor E2F, which induces G1 to S phase entry of cell cycle in mice. Cyclin dependent kinases (Cdks), cyclin D and E phosphorylate and inactivate Rb protein. Cyclin D and E are inactivated by Cdk inhibitors (Cdkis) p16, p18, and p27. Skp2 negatively regulates p27 by protein degradation. An activation of growth factor including EGF receptor suppresses Cdks. An overexpression of architectural transcriptional factors HMGAs induces pituitary adenomas in E2F-dependent manner. MEN1 gene encodes menin. This is a tumor suppressor gene, which mediated by Cdk4. Pituitary tumor transforming gene (PTTG) regulates metaphaseanaphase transition as a securin. An overexpression of PTTG induces pituitary adenomas in a downstream of Rb/E2F. E2F1: E2F transcription factor 1, HMGA: high mobility group A.
SDHx genes encode the subunits of SDH or mitochondrial complex II.78) A mutation in this gene is related to familial paraganglioma syndrome78) and several tumors including pituitary adenomas.79) LOH of SDHD has been reported in pituitary tumors,79) though the incidence of SDH mutation in pituitary adenomas may be rare.80)
Somatic mutations: GNAS1, located on chromo-some 20q13, encodes G protein α-subunit (Gsα), which couples numerous hormonal signaling to adenylyl cyclase. Ligands that bind Gsα-coupled receptors stimulates intracellular cyclic adenosine monophosphate (cAMP) production81) (Fig. 1). Activating mutations of the gene are identified as missense mutations, which lead to amino acid substitutions of either residue Arg201 or Gln227, resulting in decreased intrinsic GTPase activity and increased cAMP.21) Somatic mutations of GNAS are identified in 30–40% of GH-producing pituitary adenomas.82) In Japan, it has been reported that 53% of GH-producing adenomas exhibited somatic GNAS mutations.83) Somatic GNAS1 mutations occurring during early prenatal development lead to McCune-Albright syndrome (MAS), characterized by pigmented skin lesions, precocious puberty, fibrous dysplasia of bone, and endocrine hypersecretion.84,85) In pathological analysis, proliferation markers were unaltered in mutated GNAS pituitary tumors and non-mutated tumors, suggesting that GNAS1 mutant affect secretion rather than proliferation.86) In terms of other somatic mutations, MEN1 mutation has been detected in < 5% of pituitary adenomas, indicating that it is rare in sporadic cases.87)
PIK3CA, located on chromosome 3q26.3 encodes the catalytic subunit PIK3CA of class IA PI3-Kinase, which exists as a heterodimer of p110 catalytic-and p85 regulatory-subunits, upstream of the AKT signaling pathway. Activating somatic mutations in PIK3CA at exon 9 and 20 have been identified in pituitary adenomas, including ACTH-producing, PRL-producing, plurihormonal, and NFPAs.88) Interestingly, this mutation was seen in 8.8% of invasive pituitary tumors, while no mutations were detected in noninvasive tumors.88)
2. Animal models
Consistent with human genetic mutation analyses, several mouse models that develop pituitary adenomas and hyperplasia have been generated. Although these models show many phenotypes similar to human pituitary adenomas, several notable difference have also been observed.
Men1± mice develop tumors in the endocrine pancreas and parathyroid within 9 months of age and pituitary tumors within 12 months.89,90) The tumors developed in Men1± mice show LOH and Cdk4 is a critical downstream of Men1-dependent tumor suppression, while Cdk2 is dispensable91) (Fig. 2). Menin, encoded by Men1, interacts with double-stranded DNA and plays a crucial role in regulating cell proliferation by blocking the cell cycle.92,93) Menin reportedly attenuates the effect of activin on PRL and GH suppression in a Pit-1-dependent manner.46,94,95)
PRKAR1A± mice are tumor-prone and tend to develop tumors in cAMP-responsive tissues and sarcomas.96,97) However, these mice do not show any pituitary tumors. Pituitary-specific knockout of PRKAR1A (Prkar1a-pitKO) mice, generated using the rat GHRH receptor promoter to drive Cre expression and crossing them to PRKAR1Aloxp mice, developed pituitary tumors that were multiple and positive for GH-, PRL-, TSH-, and Pit-1-specific strains. Serum GH levels revealed a 3-fold elevation as compared to controls.98) The PKA catalytic subunit has been shown to be downstream of the PKA pathway,99) and its constitutive active mutation in adrenocortical cells results in unilateral cortisol-producing adrenal adenomas, suggesting a common pathway in the tumorigenesis56) (Fig. 1).
Aip± mice are phenotypically normal and fertile.100) The hypomorphic Aip mouse model, expressing 10% normal Aip, shows a patent ductus venosus. Similar phenotypes have been shown in hypomorphic aryl hydrocarbon receptor nuclear translocator (ARNT) mice,101) a well-known interactive partner of AIP,102) indicating the important role of AIP is mediated by its interaction with ARNT.103) No pituitary tumors develop in these animals. In contrast, heterozygous Aip mutations generated by insertion of a gene trap vector construct into an intronic region of genomic DNA between Aip exons 2 and 3 showed 100% of pituitary adenomas, particularly GH-secreting tumors.104) This difference may be due to different sub-strains used for inbreeding or the difference in the placement of the germline mutation to induce the inactivation of Aip.104) Ah receptor nuclear translocator 1 and 2 (ARNT1 and ARNT2) have been shown as possible mediators of AIP function.104) In addition, AIP protein interacts with several proteins including AhR, heat shock protein 90, cAMP, phosphodiesterases (PDE4A5 and PDE2A), epidermal growth factor receptor (EGFR), ret proto-oncogene (RET), and peroxisome proliferator-activated receptor gamma (PPARγ),25,71,102) suggesting a possible role in angiogenesis and cell proliferation.
p27 –/– mice, exhibiting increased body weight and multi-organ hyperplasia, develop intermediate lobe pituitary tumors expressing ACTH.105,106) Deletion of p27 in the pituitary by POMC-Cre generates intermediate lobe pituitary tumors.107) MENX-affected rats, which display diminished expression of p27 due to a Cdkn1b mutation, develop multiple endocrine tumors including pituitary tumors.108) Recently, the deletion of S-phase kinase-associated protein 2 (Skp2),109) the third ubiquitin ligase for p27, has been shown to block co-deletion of Rb and Trp53 and induced pituitary tumorigenesis probably mediated by p27 accumulation in the nucleus.110) This suggests that p27 plays a key role as a tumor suppressor in pituitary tumorigenesis (Fig. 2).
Rb± mice are not predisposed to retinoblastoma, but to high frequency of pituitary adenomas in an E2F-dependent manner.111,112) Rb is a key cell cycle regulator and tumor suppressor, which serves to inhibit the transcription of multiple genes required for entry into the S-phase. Inactivation of Rb function by several Cdks including cyclin D1/Cdk4 induces tumorigenesis.113) Pituitary-specific deletion of Rb by using POMC-Cre results in development of intermediate lobe pituitary tumors, which are completely prevented by Skp2 deletion, demonstrating the essential role of Skp2 in the downstream of Rb pathway114) (Fig. 2).
Two families of Cdk inhibitors exist, namely the INK4a/ARF (p15, p16, p18) and Cip/Kip families (Fig. 2). The INK4a/ARF family inhibits cyclin D1/Cdk4, whereas the Cip/Kip family inhibits cyclin E/Cdk2. Both families of Cdk inhibitors normally act as tumor suppressors by preventing entry into the S phase in an Rb-mediated manner.1,113,115) Genetic deletion of Cdk inhibitors in mice has generated pituitary tumor animal models, demonstrating a pathogenic significance for cell cycle abnormality in pituitary tumorigenesis, at least in animal models. p18ink4c-deficient mice develop GH-producing pituitary adenomas with increasing body size and organomegaly. p27kip1-deficient mice demonstrate similar phenotypes.4,105,116) p18ink4c/p27kip1 double-null mice exhibit more aggressive pituitary tumors than single knockout mice and died within 3.5 months because of the tumors.116) P57kip2-null mice develop pituitary hyperplasia but no adenomas.117) Overexpression of Cyclin E, which is downstream of p27, in pituitary using POMC promoter leads to intermediate lobe pituitary hyperplasia and anterior lobe adenoma, including PRL with few GH cells or non-secreting cells.38) Selective Cyclin E inhibition has been shown to attenuate ACTH-producing tumor growth and hormone secretion118) (Fig. 2).
PTTG encodes pituitary tumor transforming gene 1 (PTTG1), the mammalian protein securin, which is a transcriptional activator119,120) (Fig. 2). PTTG1-null mice exhibit pituitary hypoplasia and when crossbred with Rb± mice, which develop high-penetrance pituitary tumors, showed significant delay in developing pituitary tumors.121) This suggests that PTTG1 is downstream of Rb/E2F in pituitary tumorigenesis.122) Pituitary-specific PTTG1 overexpression in mice by using α-subunit of glycoprotein hormone (αGSU) promoter generates focal pituitary hyperplasia and pituitary tumors, inducing aneuploidy and chromosomal instability.35,123) POMC-Pttg overexpression in zebrafish generates ACTH-producing pituitary tumors and treatment with the Cyclin E inhibitor Roscovitine attenuates tumor development.118)
HMGA encode HMGA proteins that are known as architectural transcriptional factors, namely HMGA1a, HMGA1b, and HMGA1c from HMGA1 gene, and MHGA2 from HMGA2 gene.37) Both HMGA1 and HMGA2 transgenic mice develop mixed GH/PRL-secreting pituitary adenomas.36,37) Absence of E2F1 suppresses these pituitary tumors in HMGA2 transgenic mice, suggesting that the cell cycle is deregulated by HMGA2 in pituitary tumorigenesis37) (Fig. 2).
Overexpression of transforming growth factor (TGF)-α, an EGFR ligand, using PRL promoter in mice, generates PRL-producing pituitary adenomas,124) suggesting the involvement of EGFR in pituitary tumorigenesis. Additionally, inhibition of EGFR and its family of ErbB kinases has been shown to suppress hormone secretion and cell proliferation125–127) (Fig. 2). Fibroblastoma growth factor receptor 4 (FGFR4) is a member of the FGFR family, which includes FGFR-1 through 4. FGFR4 overexpression is associated with chemotherapy resistance and single nucleotide polymorphisms in the gene locus have been identified in breast cancer.128) FGFR4 kinase-containing, N-terminal truncated variant of FGFR4 has been identified as pituitary tumor-derived (ptd)-FGFR-4.129) Overexpression of ptd-FGFR4 in mice generates PRL-producing pituitary adenomas.130)
Dopamine receptor type 2 (D2R), which encodes a predominant dopamine receptor subtype in the anterior pituitary, is the main suppressor of PRL secretion. Knockout of D2R in mice results in development of PRL-producing pituitary adenomas (Fig. 1), especially in females with increasing VEGF-A expression, indicating the physiological importance of dopamine signaling.131) PRL-receptor-deficient mice develop PRL-producing pituitary hyperplasia and adenomas, larger than those developed in D2R knockout mice, suggesting a presence of negative feedback mechanisms.132)
II. Epigenetic changes
Despite aggressive, global, and genetic sequence analyses in human pituitary adenomas, pathogenesis in most tumors remains to be clarified. In this case, epigenetic changes including DNA methylation, histone modification, miRNAs , and long noncoding (Lnc) RNAs have been considered to be related to pathogenesis. The epigenetic changes may also explain some of the discrepancies between observations in humans and animal models.
DNA methylation: Methylation changes occurring within the CpG islands, present in approximately 70% of all mammalian promoters, are the best studied epigenetic alterations in cancer. CpG island methylation plays a key role in regulating transcription and is generally involved in malignant transformation.133)
Low expression levels of Rb in pituitary adenomas have been shown to be due to hypermethylation of the Rb gene promoter.134) Methylation of the Cdkn1b promoter was observed in rat GH3 and mouse GHRH-CL1 pituitary tumor cell lines, but not in primary human pituitary adenomas.135) P16 expression is suppressed in pituitary adenomas, which is ascribed to p16 promoter methylation, especially in NFPAs.136) The FGFR2 promoter is hypermethylated in 45% of human pituitary adenomas and its low expression in tumors is reciprocally correlated to melanoma-associated antigen A3 (MAGE-3) expression, which is hypomethylated in tumors, suggesting that it is epigenetically regulated.137) DNA damage inducible gene 45γ (GADD45γ) is a p53-regulated gene identified as a pituitary-derived growth inhibitor. Promoter CpG island of GADD45γ is hypermethylated in pituitary adenomas, especially NFPAs.138) The smallest member of the Ras-association domain family (RASSF) and a new Ras effector, RASSF3, is a tumor suppressor gene.139) In somatotroph adenomas, hypermethylation of RASSF3 promoter has been identified.140) NNAT encodes Neuronatin, a tumor suppressor, is downregulated in pituitary tumors due to hypermethylation of its promoter.141)
GNAS1 is an imprinting gene that is regulated by DNA methylation. GNAS1 activating mutation in GH-producing pituitary adenomas or MAS is present on the active maternal allele. This is different from normal pituitary, in that Gsα expression has also been observed in the non-mutated paternal allele, demonstrating the impact of GNAS imprinting relaxation on pituitary tumorigenesis.142)
Histone modification: Tail acetylation or methylation of histone lysine residues can lead to either activation or repression of gene transcription. Many of the histone modifications are misregulated in cancer.133)
DNA methyltransferase 3B (DNMT3b), which encodes a protein that produces 5-methylcytosine by adding a methyl group to a cytosine base, was shown to be overexpressed in pituitary adenomas. Down-regulation of DNMT3b in AtT20 mouse corticotroph adenoma cells results in histone 3 acetylation and diminished histone methylation in Rb, p21, and p27.39) IK6 is a dominant-negative isoform of the transcription factor Ikaros, a family of zinc-finger DNA-binding proteins. IK6 has been identified in pituitary adenomas and has been shown to be epigenetically regulated through histone and DNA modifications.143,144)
miRNAs: miRNAs are small, single-stranded, noncoding RNA molecules, which consist of approximately 22 nucleotides. miRNAs bind to sequences at 3′ untranslated regions of mRNAs, resulting in post-transcriptional silencing.145) It has been reported that miRNA dysregulation plays a crucial role in the progression of cancer.146) The analysis of expression profiles and functional properties in pituitary adenomas has revealed that miRNAs play a significant role in pituitary tumorigenesis24,44) (Table 2).
Table 2.
Altered expression of microRNAs related to pituitary adenomas and their target genes
| Target genes of miRNAs | Upregulated miRNAs | Downregulated miRNAs |
|---|---|---|
| AIP | miR-107 | |
| BMI1 | miR-128 | |
| E2F1 | miR-326, miR-603 | |
| HMGA1 and HMGA2 | miR-15, miR-16, miR-26a, miR-34b, miR-548c-3p, miR-196a2, let-7a | |
| HMGA2 | miR-326, miR-432, miR-570 | |
| PRKCD | miR-26a | |
| PTEN | miR-26b | |
| RARS | miR-16-1 | |
| SMAD3 | miR-135a, miR-140-5p, miR-582-3p, miR582-5p, miR-938 | |
| VEGF-R1 | miR24-1 | |
| Wee1 | miR-128a, miR-155, miR-516-3p | |
| ZAC1 | miR-26a |
AIP: aryl hydrocarbon receptor interacting protein, BMI1: BMI1 proto-oncogene, polycomb ring finger, E2F1: E2F transcription factor 1, HMGA: high mobility group A, PRKCD: protein kinase C delta, PTEN: phosphatase and tensin homolog, RARS: arginyl-tRNA synthetase, SMAD3: smad family member 3, VEGF-R1: soluble vascular endothelial growth factor receptor 1, Wee1: WEE1 G2 check point kinase, ZAC1: zinc finger regulator of apoptosis and cell cycle arrest.
AIP was identified as a target for miR-107, which is overexpressed in pituitary adenomas.147) BMI1 polycomb ring finger oncogene 1 is a target for miR-128, which is downregulated in GH-producing pituitary adenomas leading to phosphatase and tensin homolog (PTEN) suppression.148) E2F1 is the target for miR-326 and miR-603, while HMGA1/HMGA2 is the target for miR-15, miR-16, miR-26a, miR-34b, miR-548-3p, miR-196a2, and let-7a, which are downregulated in pituitary adenomas.149,150) miR-326, miR-432, and miR-570 are also downregulated in pituitary adenomas that target HMGA2.149) PRKCD, a serine/threonine kinase involved in proliferation, apoptosis and cell cycle regulation, is a direct target for miR-26a, which is overexpressed in ACTH-producing pituitary adenomas.151) PTEN, a suppressor of the PI3K/AKT signaling pathway, is identified as a direct target for miR-26b, which is overexpressed in GH-secreting pituitary adenomas.148) Arginyl-tRNA synthetase (RARS), a part of the aminoacyl-tRNA synthetase complex, is a target for miR-16-1, whose expression levels are low in pituitary adenomas.152) SMAD3 is a target for miR-135a, miR-140-5p, miR-582-3p, miR-582-5p, and miR-938, which are overexpressed in NFPAs, as compared to normal pituitaries.153) Vascular endothelial growth factor receptor 1 (VEGF-R1) is a target for miR-24-1, which is downregulated in pituitary adenomas.154) Wee1, an inhibitor for Cdk1, is identified as a target for miR-128a, miR-155, and miR-516-3p, which are overexpressed in pituitary adenomas.155) ZAC1, also called as PLAG1, which is a downstream component of a particular signal pathway involving AIP, is a target for miR-26a, which is overexpressed in pituitary adenomas.154) Lnc RNAs: LncRNAs are non-protein coding transcripts, longer than 200 nucleotides. LncRNA are involved in the regulation of molecules related to the cell cycle, including CDK inhibitors, CDKs, Rb, and p53, in addition to functioning as epigenetic regulators, transcription factor regulators, post-transcription regulators, and protein scaffolds.156)
Maternally expressed gene 3 (MEG3), located on chromosome 14q32, belongs to the DLK1-MEG3 imprinting locus, containing multiple maternally and paternally imprinted genes.157,158) MEG3 encodes lncRNA and is downregulated in pituitary adenomas, especially in NFPAs.159) MEG3 stimulates p53-dependent transcription and acts as a tumor suppressor gene.160)
Future Directions and Conclusion
Accumulating evidence suggests that not only genetic changes, but also epigenetic changes play an essential role in the development of pituitary adenomas. Both clinical data and the analysis of animal models are important; however, there are substantial differences between species. In this regard, it is important to establish a human tumor experimental model.
To develop novel therapeutic targeted drugs, it is essential to identify the pathway responsible for pituitary tumorigenesis. Somatostatin analogs are important targeted drugs, that inhibit the pathways essential for GH secretion in GH-producing pituitary adenoma.13) Recent findings suggest that ErbB receptors or Skp2, which is an upstream effector of CDK inhibitors, could be useful as a novel strategy for targeted therapy.114,126,161)
In conclusion, human genetic analysis and establishment of animal models have revealed the mechanisms of pituitary tumorigenesis. Further clarification of underlying mechanisms can contribute to the development of novel targeted drugs for pituitary adenomas.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research from Japanese Ministry of Education Science, Culture, Sports, Science, and Technology 24790945, 23659477, 23591354, and 22591012, Grants-in-Aid for Scientific Research (research on hypothalamic-hypophyseal disorders) from the Ministry of Health, Labor, and Welfare, Japan, Novo Nordisk, Daiichi-Sankyo Fundation of Life Science, and the Naito Foundation.
References
- 1). Melmed S: Pathogenesis of pituitary tumors. Nat Rev Endocrinol 7: 257– 266, 2011. [DOI] [PubMed] [Google Scholar]
- 2). Buurman H, Saeger W: Subclinical adenomas in postmortem pituitaries: classification and correlations to clinical data. Eur J Endocrinol 154: 753– 758, 2006. [DOI] [PubMed] [Google Scholar]
- 3). Teramoto A, Hirakawa K, Sanno N, Osamura Y: Incidental pituitary lesions in 1,000 unselected autopsy specimens. Radiology 193: 161– 164, 1994. [DOI] [PubMed] [Google Scholar]
- 4). Melmed S: Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest 112: 1603– 1618, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5). Sano T: Use of ultrastructural immunohistochemistry in human pituitary pathology. Microsc Res Tech 20: 152– 161, 1992. [DOI] [PubMed] [Google Scholar]
- 6). Asa SL: Diseases of the pituitary. Neurosurg Clin N Am 5: 71– 95, 1994. [PubMed] [Google Scholar]
- 7). Clayton RN: Sporadic pituitary tumours: from epidemiology to use of databases. Baillieres Best Pract Res Clin Endocrinol Metab 13: 451– 460, 1999. [DOI] [PubMed] [Google Scholar]
- 8). Horvath E, Kovacs K: Pathology of prolactin cell adenomas of the human pituitary. Semin Diagn Pathol 3: 4– 17, 1986. [PubMed] [Google Scholar]
- 9). Kovacs K, Horvath E: Pathology of growth hormone-producing tumors of the human pituitary. Semin Diagn Pathol 3: 18– 33, 1986. [PubMed] [Google Scholar]
- 10). Report of Brain Tumor Registry of Japan (1984–2000). Neurol Med Chir (Tokyo) 49( Suppl): PS1– PS96, 2009. [PubMed] [Google Scholar]
- 11). Anthony L, Freda PU: From somatostatin to octreotide LAR: evolution of a somatostatin analogue. Curr Med Res Opin 25: 2989– 2999, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12). Ben-Jonathan N, Hnasko R: Dopamine as a prolactin (PRL) inhibitor. Endocr Rev 22: 724– 763, 2001. [DOI] [PubMed] [Google Scholar]
- 13). Ben-Shlomo A, Melmed S: Pituitary somatostatin receptor signaling. Trends Endocrinol Metab 21: 123– 133, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14). Kohn DT, Kopchick JJ: Growth hormone receptor antagonists. Minerva Endocrinol 27: 287– 298, 2002. [PubMed] [Google Scholar]
- 15). Morris D, Grossman A: The medical management of Cushing's syndrome. Ann N Y Acad Sci 970: 119– 133, 2002. [DOI] [PubMed] [Google Scholar]
- 16). Schteingart DE: Drugs in the medical treatment of Cushing's syndrome. Expert Opin Emerg Drugs 14: 661– 671, 2009. [DOI] [PubMed] [Google Scholar]
- 17). Tritos NA, Biller BM, Swearingen B: Management of Cushing disease. Nat Rev Endocrinol 7: 279– 289, 2011. [DOI] [PubMed] [Google Scholar]
- 18). Alexander JM, Biller BM, Bikkal H, Zervas NT, Arnold A, Klibanski A: Clinically nonfunctioning pituitary tumors are monoclonal in origin. J Clin Invest 86: 336– 340, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19). Herman V, Fagin J, Gonsky R, Kovacs K, Melmed S: Clonal origin of pituitary adenomas. J Clin Endocrinol Metab 71: 1427– 1433, 1990. [DOI] [PubMed] [Google Scholar]
- 20). Jacoby LB, Hedley-Whyte ET, Pulaski K, Seizinger BR, Martuza RL: Clonal origin of pituitary adenomas. J Neurosurg 73: 731– 735, 1990. [DOI] [PubMed] [Google Scholar]
- 21). Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L: GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 340: 692– 696, 1989. [DOI] [PubMed] [Google Scholar]
- 22). Elston MS, McDonald KL, Clifton-Bligh RJ, Robinson BG: Familial pituitary tumor syndromes. Nat Rev Endocrinol 5: 453– 461, 2009. [DOI] [PubMed] [Google Scholar]
- 23). Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjöld M: Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature 332: 85– 87, 1988. [DOI] [PubMed] [Google Scholar]
- 24). Zhou Y, Zhang X, Klibanski A: Genetic and epigenetic mutations of tumor suppressive genes in sporadic pituitary adenoma. Mol Cell Endocrinol 386: 16– 33, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25). Peverelli E, Mantovani G, Lania AG, Spada A: cAMP in the pituitary: an old messenger for multiple signals. J Mol Endocrinol 52: R67– R77, 2014. [DOI] [PubMed] [Google Scholar]
- 26). Yacqub-Usman K, Richardson A, Duong CV, Clayton RN, Farrell WE: The pituitary tumour epigenome: aberrations and prospects for targeted therapy. Nat Rev Endocrinol 8: 486– 494, 2012. [DOI] [PubMed] [Google Scholar]
- 27). Jones PA, Baylin SB: The epigenomics of cancer. Cell 128: 683– 692, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28). Bird AP: CpG-rich islands and the function of DNA methylation. Nature 321: 209– 213, 1986. [DOI] [PubMed] [Google Scholar]
- 29). Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T: DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet 24: 88– 91, 2000. [DOI] [PubMed] [Google Scholar]
- 30). Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T: The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem 278: 4035– 4040, 2003. [DOI] [PubMed] [Google Scholar]
- 31). Okano M, Bell DW, Haber DA, Li E: DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99: 247– 257, 1999. [DOI] [PubMed] [Google Scholar]
- 32). Okano M, Xie S, Li E: Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet 19: 219– 220, 1998. [DOI] [PubMed] [Google Scholar]
- 33). Kouzarides T: Chromatin modifications and their function. Cell 128: 693– 705, 2007. [DOI] [PubMed] [Google Scholar]
- 34). Cryns VL, Alexander JM, Klibanski A, Arnold A: The retinoblastoma gene in human pituitary tumors. J Clin Endocrinol Metab 77: 644– 646, 1993. [DOI] [PubMed] [Google Scholar]
- 35). Vlotides G, Eigler T, Melmed S: Pituitary tumor-transforming gene: physiology and implications for tumorigenesis. Endocr Rev 28: 165– 186, 2007. [DOI] [PubMed] [Google Scholar]
- 36). Fedele M, Pentimalli F, Baldassarre G, Battista S, Klein-Szanto AJ, Kenyon L, Visone R, De Martino I, Ciarmiello A, Arra C, Viglietto G, Croce CM, Fusco A: Transgenic mice overexpressing the wild-type form of the HMGA1 gene develop mixed growth hormone/prolactin cell pituitary adenomas and natural killer cell lymphomas. Oncogene 24: 3427– 3435, 2005. [DOI] [PubMed] [Google Scholar]
- 37). Fedele M, Visone R, De Martino I, Troncone G, Palmieri D, Battista S, Ciarmiello A, Pallante P, Arra C, Melillo RM, Helin K, Croce CM, Fusco A: HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cell 9: 459– 471, 2006. [DOI] [PubMed] [Google Scholar]
- 38). Roussel-Gervais A, Bilodeau S, Vallette S, Berthelet F, Lacroix A, Figarella-Branger D, Brue T, Drouin J: Cooperation between cyclin E and p27(Kip1) in pituitary tumorigenesis. Mol Endocrinol 24: 1835– 1845, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39). Zhu X, Mao X, Hurren R, Schimmer AD, Ezzat S, Asa SL: Deoxyribonucleic acid methyltransferase 3B promotes epigenetic silencing through histone 3 chromatin modifications in pituitary cells. J Clin Endocrinol Metab 93: 3610– 3617, 2008. [DOI] [PubMed] [Google Scholar]
- 40). Ogino A, Yoshino A, Katayama Y, Watanabe T, Ota T, Komine C, Yokoyama T, Fukushima T: The p15(INK4b)/p16(INK4a)/RB1 pathway is frequently deregulated in human pituitary adenomas. J Neuropathol Exp Neurol 64: 398– 403, 2005. [DOI] [PubMed] [Google Scholar]
- 41). Yoshino A, Katayama Y, Ogino A, Watanabe T, Yachi K, Ohta T, Komine C, Yokoyama T, Fukushima T: Promoter hypermethylation profile of cell cycle regulator genes in pituitary adenomas. J Neurooncol 83: 153– 162, 2007. [DOI] [PubMed] [Google Scholar]
- 42). Hatziapostolou M, Polytarchou C, Iliopoulos D: miRNAs link metabolic reprogramming to onco-genesis. Trends Endocrinol Metab 24: 361– 373, 2013. [DOI] [PubMed] [Google Scholar]
- 43). Jiang X, Zhang X: The molecular pathogenesis of pituitary adenomas: an update. Endocrinol Metab (Seoul) 28: 245– 254, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44). Sivapragasam M, Rotondo F, Lloyd RV, Scheithauer BW, Cusimano M, Syro LV, Kovacs K: MicroRNAs in the human pituitary. Endocr Pathol 22: 134– 143, 2011. [DOI] [PubMed] [Google Scholar]
- 45). Tateno T, Zhu X, Asa SL, Ezzat S: Chromatin remodeling and histone modifications in pituitary tumors. Mol Cell Endocrinol 326: 66– 70, 2010. [DOI] [PubMed] [Google Scholar]
- 46). Balogh K, Rácz K, Patócs A, Hunyady L: Menin and its interacting proteins: elucidation of menin function. Trends Endocrinol Metab 17: 357– 364, 2006. [DOI] [PubMed] [Google Scholar]
- 47). Guru SC, Manickam P, Crabtree JS, Olufemi SE, Agarwal SK, Debelenko LV: Identification and characterization of the multiple endocrine neoplasia type 1 (MEN1) gene. J Intern Med 243: 433– 439, 1998. [DOI] [PubMed] [Google Scholar]
- 48). Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG, Libroia A, Lips CJ, Lombardi G, Mannelli M, Pacini F, Ponder BA, Raue F, Skogseid B, Tamburrano G, Thakker RV, Thompson NW, Tomassetti P, Tonelli F, Wells SA, Marx SJ: Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 86: 5658– 5671, 2001. [DOI] [PubMed] [Google Scholar]
- 49). Marx SJ, Simonds WF: Hereditary hormone excess: genes, molecular pathways, and syndromes. Endocr Rev 26: 615– 661, 2005. [DOI] [PubMed] [Google Scholar]
- 50). Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, Melmed S, Sakurai A, Tonelli F, Brandi ML, Endocrine Society : Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 97: 2990– 3011, 2012. [DOI] [PubMed] [Google Scholar]
- 51). Scheithauer BW, Laws ER, Kovacs K, Horvath E, Randall RV, Carney JA: Pituitary adenomas of the multiple endocrine neoplasia type I syndrome. Semin Diagn Pathol 4: 205– 211, 1987. [PubMed] [Google Scholar]
- 52). Corbetta S, Pizzocaro A, Peracchi M, Beck-Peccoz P, Faglia G, Spada A: Multiple endocrine neoplasia type 1 in patients with recognized pituitary tumours of different types. Clin Endocrinol (Oxf) 47: 507– 512, 1997. [DOI] [PubMed] [Google Scholar]
- 53). Tanaka C, Yoshimoto K, Yamada S, Nishioka H, Ii S, Moritani M, Yamaoka T, Itakura M: Absence of germ-line mutations of the multiple endocrine neoplasia type 1 (MEN1) gene in familial pituitary adenoma in contrast to MEN1 in Japanese. J Clin Endocrinol Metab 83: 960– 965, 1998. [DOI] [PubMed] [Google Scholar]
- 54). Vergès B, Boureille F, Goudet P, Murat A, Beckers A, Sassolas G, Cougard P, Chambe B, Montvernay C, Calender A: Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab 87: 457– 465, 2002. [DOI] [PubMed] [Google Scholar]
- 55). Trouillas J, Labat-Moleur F, Sturm N, Kujas M, Heymann MF, Figarella-Branger D, Patey M, Mazucca M, Decullier E, Vergès B, Chabre O, Calender A, Groupe d'études des Tumeurs Endocrines : Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): a case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am J Surg Pathol 32: 534– 543, 2008. [DOI] [PubMed] [Google Scholar]
- 56). Beuschlein F, Fassnacht M, Assié G, Calebiro D, Stratakis CA, Osswald A, Ronchi CL, Wieland T, Sbiera S, Faucz FR, Schaak K, Schmittfull A, Schwarzmayr T, Barreau O, Vezzosi D, Rizk-Rabin M, Zabel U, Szarek E, Salpea P, Forlino A, Vetro A, Zuffardi O, Kisker C, Diener S, Meitinger T, Lohse MJ, Reincke M, Bertherat J, Strom TM, Allolio B: Constitutive activation of PKA catalytic subunit in adrenal Cushing's syndrome. N Engl J Med 370: 1019– 1028, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57). Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, Cho-Chung YS, Stratakis CA: Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet 26: 89– 92, 2000. [DOI] [PubMed] [Google Scholar]
- 58). Bertherat J, Horvath A, Groussin L, Grabar S, Boikos S, Cazabat L, Libe R, René-Corail F, Stergiopoulos S, Bourdeau I, Bei T, Clauser E, Calender A, Kirschner LS, Bertagna X, Carney JA, Stratakis CA: Mutations in regulatory subunit type 1A of cyclic adeno-sine 5'-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab 94: 2085– 2091, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59). Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL: The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 64: 270– 283, 1985. [DOI] [PubMed] [Google Scholar]
- 60). Casey M, Vaughan CJ, He J, Hatcher CJ, Winter JM, Weremowicz S, Montgomery K, Kucherlapati R, Morton CC, Basson CT: Mutations in the protein kinase A R1alpha regulatory subunit cause familial cardiac myxomas and Carney complex. J Clin Invest 106: R31– 38, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61). Michels VV: A new inherited syndrome with cardiac, cutaneous, and endocrine involvement. Mayo Clin Proc 61: 224– 225, 1986. [DOI] [PubMed] [Google Scholar]
- 62). Bossis I, Voutetakis A, Matyakhina L, Pack S, Abu-Asab M, Bourdeau I, Griffin KJ, Courcoutsakis N, Stergiopoulos S, Batista D, Tsokos M, Stratakis CA: A pleiomorphic GH pituitary adenoma from a Carney complex patient displays universal allelic loss at the protein kinase A regulatory subunit 1A (PRKARIA) locus. J Med Genet 41: 596– 600, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63). Kirschner LS: PRKAR1A and the evolution of pituitary tumors. Mol Cell Endocrinol 326: 3– 7, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64). Pack SD, Kirschner LS, Pak E, Zhuang Z, Carney JA, Stratakis CA: Genetic and histologic studies of somatomammotropic pituitary tumors in patients with the “complex of spotty skin pigmentation, myxomas, endocrine overactivity and schwannomas” (Carney complex). J Clin Endocrinol Metab 85: 3860– 3865, 2000. [DOI] [PubMed] [Google Scholar]
- 65). Stratakis CA: Genetics of adrenocortical tumors: Carney complex. Ann Endocrinol (Paris) 62: 180– 184, 2001. [PubMed] [Google Scholar]
- 66). Armstrong DK, Irvine AD, Handley JM, Walsh MY, Hadden DR, Bingham EA: Carney complex: report of a kindred with predominantly cutaneous manifestations. Br J Dermatol 136: 578– 582, 1997. [PubMed] [Google Scholar]
- 67). Raff SB, Carney JA, Krugman D, Doppman JL, Stratakis CA: Prolactin secretion abnormalities in patients with the “syndrome of spotty skin pigmentation, myxomas, endocrine overactivity and schwannomas” (Carney complex). J Pediatr Endocrinol Metab 13: 373– 379, 2000. [PubMed] [Google Scholar]
- 68). Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, Tuppurainen K, Ebeling TM, Salmela PI, Paschke R, Gündogdu S, De Menis E, Mäkinen MJ, Launonen V, Karhu A, Aaltonen LA: Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science 312: 1228– 1230, 2006. [DOI] [PubMed] [Google Scholar]
- 69). Daly AF, Vanbellinghen JF, Khoo SK, Jaffrain-Rea ML, Naves LA, Guitelman MA, Murat A, Emy P, Gimenez-Roqueplo AP, Tamburrano G, Raverot G, Barlier A, De Herder W, Penfornis A, Ciccarelli E, Estour B, Lecomte P, Gatta B, Chabre O, Sabaté MI, Bertagna X, Garcia Basavilbaso N, Stalldecker G, Colao A, Ferolla P, Wémeau JL, Caron P, Sadoul JL, Oneto A, Archambeaud F, Calender A, Sinilnikova O, Montañana CF, Cavagnini F, Hana V, Solano A, Delettieres D, Luccio-Camelo DC, Basso A, Rohmer V, Brue T, Bours V, Teh BT, Beckers A: Aryl hydro-carbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: analysis in 73 families. J Clin Endocrinol Metab 92: 1891– 1896, 2007. [DOI] [PubMed] [Google Scholar]
- 70). Igreja S, Chahal HS, King P, Bolger GB, Srirangalingam U, Guasti L, Chapple JP, Trivellin G, Gueorguiev M, Guegan K, Stals K, Khoo B, Kumar AV, Ellard S, Grossman AB, Korbonits M, International FIPA Consortium : Characterization of aryl hydrocarbon receptor interacting protein (AIP) mutations in familial isolated pituitary adenoma families. Hum Mutat 31: 950– 960, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71). Beckers A, Aaltonen LA, Daly AF, Karhu A: Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev 34: 239– 277, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72). Leontiou CA, Gueorguiev M, van der Spuy J, Quinton R, Lolli F, Hassan S, Chahal HS, Igreja SC, Jordan S, Rowe J, Stolbrink M, Christian HC, Wray J, Bishop-Bailey D, Berney DM, Wass JA, Popovic V, Ribeiro-Oliveira A, Gadelha MR, Monson JP, Akker SA, Davis JR, Clayton RN, Yoshimoto K, Iwata T, Matsuno A, Eguchi K, Musat M, Flanagan D, Peters G, Bolger GB, Chapple JP, Frohman LA, Grossman AB, Korbonits M: The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J Clin Endocrinol Metab 93: 2390– 2401, 2008. [DOI] [PubMed] [Google Scholar]
- 73). Sherr CJ, Roberts JM: CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13: 1501– 1512, 1999. [DOI] [PubMed] [Google Scholar]
- 74). Agarwal SK, Mateo CM, Marx SJ: Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab 94: 1826– 1834, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75). Occhi G, Regazzo D, Trivellin G, Boaretto F, Ciato D, Bobisse S, Ferasin S, Cetani F, Pardi E, Korbonits M, Pellegata NS, Sidarovich V, Quattrone A, Opocher G, Mantero F, Scaroni C: A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS Genet 9: e1003350, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76). Thakker RV: Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol 386: 2– 15, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77). Tichomirowa MA, Lee M, Barlier A, Daly AF, Marinoni I, Jaffrain-Rea ML, Naves LA, Rodien P, Rohmer V, Faucz FR, Caron P, Estour B, Lecomte P, Borson-Chazot F, Penfornis A, Yaneva M, Guitelman M, Castermans E, Verhaege C, Wémeau JL, Tabarin A, Fajardo Montañana C, Delemer B, Kerlan V, Sadoul JL, Cortet Rudelli C, Archambeaud F, Zacharieva S, Theodoropoulou M, Brue T, Enjalbert A, Bours V, Pellegata NS, Beckers A: Cyclin-dependent kinase inhibitor 1B (CDKN1B) gene variants in AIP mutation-negative familial isolated pituitary adenoma kindreds. Endocr Relat Cancer 19: 233– 241, 2012. [DOI] [PubMed] [Google Scholar]
- 78). Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW, Cornelisse CJ, Devilee P, Devlin B: Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287: 848– 851, 2000. [DOI] [PubMed] [Google Scholar]
- 79). Xekouki P, Stratakis CA: Succinate dehydrogenase (SDHx) mutations in pituitary tumors: could this be a new role for mitochondrial complex II and/ or Krebs cycle defects? Endocr Relat Cancer 19: C33– 40, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80). Gill AJ, Toon CW, Clarkson A, Sioson L, Chou A, Winship I, Robinson BG, Benn DE, Clifton-Bligh RJ, Dwight T: Succinate dehydrogenase deficiency is rare in pituitary adenomas. Am J Surg Pathol 38: 560– 566, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81). Weinstein LS, Liu J, Sakamoto A, Xie T, Chen M: Minireview: GNAS: normal and abnormal functions. Endocrinology 145: 5459– 5464, 2004. [DOI] [PubMed] [Google Scholar]
- 82). Landis CA, Harsh G, Lyons J, Davis RL, McCormick F, Bourne HR: Clinical characteristics of acromegalic patients whose pituitary tumors contain mutant Gs protein. J Clin Endocrinol Metab 71: 1416– 1420, 1990. [DOI] [PubMed] [Google Scholar]
- 83). Yasufuku-Takano J, Takano K, Morita K, Takakura K, Teramoto A, Fujita T: Does the prevalence of gsp mutations in GH-secreting pituitary adenomas differ geographically or racially? Prevalence of gsp mutations in Japanese patients revisited. Clin Endocrinol (Oxf) 64: 91– 96, 2006. [DOI] [PubMed] [Google Scholar]
- 84). Schwindinger WF, Francomano CA, Levine MA: Identification of a mutation in the gene encoding the alpha subunit of the stimulatory G protein of adenylyl cyclase in McCune-Albright syndrome. Proc Natl Acad Sci USA 89: 5152– 5156, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85). Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM: Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 325: 1688– 1695, 1991. [DOI] [PubMed] [Google Scholar]
- 86). Freda PU, Chung WK, Matsuoka N, Walsh JE, Kanibir MN, Kleinman G, Wang Y, Bruce JN, Post KD: Analysis of GNAS mutations in 60 growth hormone secreting pituitary tumors: correlation with clinical and pathological characteristics and surgical outcome based on highly sensitive GH and IGF-I criteria for remission. Pituitary 10: 275– 282, 2007. [DOI] [PubMed] [Google Scholar]
- 87). Lemos MC, Thakker RV: Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 29: 22– 32, 2008. [DOI] [PubMed] [Google Scholar]
- 88). Lin Y, Jiang X, Shen Y, Li M, Ma H, Xing M, Lu Y: Frequent mutations and amplifications of the PIK3CA gene in pituitary tumors. Endocr Relat Cancer 16: 301– 310, 2009. [DOI] [PubMed] [Google Scholar]
- 89). Bertolino P, Tong WM, Galendo D, Wang ZQ, Zhang CX: Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol Endocrinol 17: 1880– 1892, 2003. [DOI] [PubMed] [Google Scholar]
- 90). Crabtree JS, Scacheri PC, Ward JM, Garrett-Beal L, Emmert-Buck MR, Edgemon KA, Lorang D, Libutti SK, Chandrasekharappa SC, Marx SJ, Spiegel AM, Collins FS: A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci USA 98: 1118– 1123, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91). Gillam MP, Nimbalkar D, Sun L, Christov K, Ray D, Kaldis P, Liu X, Kiyokawa H: MEN1 tumorigenesis in the pituitary and pancreatic islet requires Cdk4 but not Cdk2. Oncogene 2014. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92). Kaji H, Canaff L, Goltzman D, Hendy GN: Cell cycle regulation of menin expression. Cancer Res 59: 5097– 5101, 1999. [PubMed] [Google Scholar]
- 93). La P, Silva AC, Hou Z, Wang H, Schnepp RW, Yan N, Shi Y, Hua X: Direct binding of DNA by tumor suppressor menin. J Biol Chem 279: 49045– 49054, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94). Lacerte A, Lee EH, Reynaud R, Canaff L, De Guise C, Devost D, Ali S, Hendy GN, Lebrun JJ: Activin inhibits pituitary prolactin expression and cell growth through Smads, Pit-1 and menin. Mol Endocrinol 18: 1558– 1569, 2004. [DOI] [PubMed] [Google Scholar]
- 95). Namihira H, Sato M, Murao K, Cao WM, Matsubara S, Imachi H, Niimi M, Dobashi H, Wong NC, Ishida T: The multiple endocrine neoplasia type 1 gene product, menin, inhibits the human prolactin promoter activity. J Mol Endocrinol 29: 297– 304, 2002. [DOI] [PubMed] [Google Scholar]
- 96). Kirschner LS, Kusewitt DF, Matyakhina L, Towns WH, Carney JA, Westphal H, Stratakis CA: A mouse model for the Carney complex tumor syndrome develops neoplasia in cyclic AMP-responsive tissues. Cancer Res 65: 4506– 4514, 2005. [DOI] [PubMed] [Google Scholar]
- 97). Veugelers M, Wilkes D, Burton K, McDermott DA, Song Y, Goldstein MM, La Perle K, Vaughan CJ, O'Hagan A, Bennett KR, Meyer BJ, Legius E, Karttunen M, Norio R, Kaariainen H, Lavyne M, Neau JP, Richter G, Kirali K, Farnsworth A, Stapleton K, Morelli P, Takanashi Y, Bamforth JS, Eitelberger F, Noszian I, Manfroi W, Powers J, Mochizuki Y, Imai T, Ko GT, Driscoll DA, Goldmuntz E, Edelberg JM, Collins A, Eccles D, Irvine AD, McKnight GS, Basson CT: Comparative PRKAR1A genotype-phenotype analyses in humans with Carney complex and prkar1a haploinsufficient mice. Proc Natl Acad Sci USA 101: 14222– 14227, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98). Yin Z, Williams-Simons L, Parlow AF, Asa S, Kirschner LS: Pituitary-specific knockout of the Carney complex gene Prkar1a leads to pituitary tumorigenesis. Mol Endocrinol 22: 380– 387, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99). Yin Z, Pringle DR, Jones GN, Kelly KM, Kirschner LS: Differential role of PKA catalytic subunits in mediating phenotypes caused by knockout of the Carney complex gene Prkar1a. Mol Endocrinol 25: 1786– 1793, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100). Lin BC, Sullivan R, Lee Y, Moran S, Glover E, Bradfield CA: Deletion of the aryl hydrocarbon receptor-associated protein 9 leads to cardiac malformation and embryonic lethality. J Biol Chem 282: 35924– 35932, 2007. [DOI] [PubMed] [Google Scholar]
- 101). Walisser JA, Bunger MK, Glover E, Harstad EB, Bradfield CA: Patent ductus venosus and dioxin resistance in mice harboring a hypomorphic Arnt allele. J Biol Chem 279: 16326– 16331, 2004. [DOI] [PubMed] [Google Scholar]
- 102). Chahal HS, Chapple JP, Frohman LA, Grossman AB, Korbonits M: Clinical, genetic and molecular characterization of patients with familial isolated pituitary adenomas (FIPA). Trends Endocrinol Metab 21: 419– 427, 2010. [DOI] [PubMed] [Google Scholar]
- 103). Lin BC, Nguyen LP, Walisser JA, Bradfield CA: A hypomorphic allele of aryl hydrocarbon receptor-associated protein-9 produces a phenocopy of the AHR-null mouse. Mol Pharmacol 74: 1367– 1371, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104). Raitila A, Lehtonen HJ, Arola J, Heliövaara E, Ahlsten M, Georgitsi M, Jalanko A, Paetau A, Aaltonen LA, Karhu A: Mice with inactivation of aryl hydrocarbon receptor-interacting protein (Aip) display complete penetrance of pituitary adenomas with aberrant ARNT expression. Am J Pathol 177: 1969– 1976, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105). Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A: Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell 85: 721– 732, 199. [DOI] [PubMed] [Google Scholar]
- 106). Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY, Nakayama K: Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 85: 707– 720, 1996. [DOI] [PubMed] [Google Scholar]
- 107). Chien WM, Rabin S, Macias E, Miliani de Marval PL, Garrison K, Orthel J, Rodriguez-Puebla M, Fero ML: Genetic mosaics reveal both cell-autonomous and cell-nonautonomous function of murine p27Kip1. Proc Natl Acad Sci USA 103: 4122– 4127, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108). Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Höfler H, Fend F, Graw J, Atkinson MJ: Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA 103: 15558– 15563, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109). Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, Chan CH, Yang WL, Wang J, Egia A, Nakayama KI, Cordon-Cardo C, Teruya-Feldstein J, Pandolfi PP: Skp2 targeting suppresses tumorigenesis by Arfp53-independent cellular senescence. Nature 464: 374– 379, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110). Zhao H, Bauzon F, Fu H, Lu Z, Cui J, Nakayama K, Nakayama KI, Locker J, Zhu L: Skp2 deletion unmasks a p27 safeguard that blocks tumorigenesis in the absence of pRb and p53 tumor suppressors. Cancer Cell 24: 645– 659, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111). Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA: Effects of an Rb mutation in the mouse. Nature 359: 295– 300, 1992. [DOI] [PubMed] [Google Scholar]
- 112). Lazzerini Denchi E, Attwooll C, Pasini D, Helin K: Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol Cell Biol 25: 2660– 2672, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113). Knudsen ES, Knudsen KE: Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer 8: 714– 724, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114). Wang H, Bauzon F, Ji P, Xu X, Sun D, Locker J, Sellers RS, Nakayama K, Nakayama KI, Cobrinik D, Zhu L: Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/− mice. Nat Genet 42: 83– 88, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115). Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE: Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev 11: 1464– 1478, 1997. [DOI] [PubMed] [Google Scholar]
- 116). Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, Su L, Xiong Y: CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev 12: 2899– 2911, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117). Bilodeau S, Roussel-Gervais A, Drouin J: Distinct developmental roles of cell cycle inhibitors p57Kip2 and p27Kip1 distinguish pituitary progenitor cell cycle exit from cell cycle reentry of differentiated cells. Mol Cell Biol 29: 1895– 1908, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118). Liu NA, Jiang H, Ben-Shlomo A, Wawrowsky K, Fan XM, Lin S, Melmed S: Targeting zebrafish and murine pituitary corticotroph tumors with a cyclin-dependent kinase (CDK) inhibitor. Proc Natl Acad Sci USA 108: 8414– 8419, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119). Pei L, Melmed S: Isolation and characterization of a pituitary tumor-transforming gene (PTTG). Mol Endocrinol 11: 433– 441, 1997. [DOI] [PubMed] [Google Scholar]
- 120). Zou H, McGarry TJ, Bernal T, Kirschner MW: Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumori-genesis. Science 285: 418– 422, 1999. [DOI] [PubMed] [Google Scholar]
- 121). Chesnokova V, Kovacs K, Castro AV, Zonis S, Melmed S: Pituitary hypoplasia in Pttg−/− mice is protective for Rb+/− pituitary tumorigenesis. Mol Endocrinol 19: 2371– 2379, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122). Zhou C, Wawrowsky K, Bannykh S, Gutman S, Melmed S: E2F1 induces pituitary tumor transforming gene (PTTG1) expression in human pituitary tumors. Mol Endocrinol 23: 2000– 2012, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123). Abbud RA, Takumi I, Barker EM, Ren SG, Chen DY, Wawrowsky K, Melmed S: Early multipotential pituitary focal hyperplasia in the alpha-subunit of glycoprotein hormone-driven pituitary tumor-transforming gene transgenic mice. Mol Endocrinol 19: 1383– 1391, 2005. [DOI] [PubMed] [Google Scholar]
- 124). McAndrew J, Paterson AJ, Asa SL, McCarthy KJ, Kudlow JE: Targeting of transforming growth factor-alpha expression to pituitary lactotrophs in transgenic mice results in selective lactotroph proliferation and adenomas. Endocrinology 136: 4479– 4488, 1995. [DOI] [PubMed] [Google Scholar]
- 125). Cooper O, Vlotides G, Fukuoka H, Greene MI, Melmed S: Expression and function of ErbB receptors and ligands in the pituitary. Endocr Relat Cancer 18: R197– 211, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126). Fukuoka H, Cooper O, Mizutani J, Tong Y, Ren SG, Bannykh S, Melmed S: HER2/ErbB2 receptor signaling in rat and human prolactinoma cells: strategy for targeted prolactinoma therapy. Mol Endocrinol 25: 92– 103, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127). Vlotides G, Cooper O, Chen YH, Ren SG, Greenman Y, Melmed S: Heregulin regulates prolactinoma gene expression. Cancer Res 69: 4209– 4216, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128). Tenhagen M, van Diest PJ, Ivanova IA, van der Wall E, van der Groep P: Fibroblast growth factor receptors in breast cancer: expression, downstream effects, and possible drug targets. Endocr Relat Cancer 19: R115– 129, 2012. [DOI] [PubMed] [Google Scholar]
- 129). Yu S, Asa SL, Weigel RJ, Ezzat S: Pituitary tumor AP-2alpha recognizes a cryptic promoter in intron 4 of fibroblast growth factor receptor 4. J Biol Chem 278: 19597– 19602, 2003. [DOI] [PubMed] [Google Scholar]
- 130). Ezzat S, Zheng L, Zhu XF, Wu GE, Asa SL: Targeted expression of a human pituitary tumor-derived isoform of FGF receptor-4 recapitulates pituitary tumorigenesis. J Clin Invest 109: 69– 78, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 131). Cristina C, García-Tornadú I, Díaz-Torga G, Rubinstein M, Low MJ, Becú-Villalobos D: Dopaminergic D2 receptor knockout mouse: an animal model of prolactinoma. Front Horm Res 35: 50– 63, 2006. [DOI] [PubMed] [Google Scholar]
- 132). Asa SL, Kelly MA, Grandy DK, Low MJ: Pituitary lactotroph adenomas develop after prolonged lactotroph hyperplasia in dopamine D2 receptor-deficient mice. Endocrinology 140: 5348– 5355, 1999. [DOI] [PubMed] [Google Scholar]
- 133). Dawson MA, Kouzarides T: Cancer epigenetics: from mechanism to therapy. Cell 150: 12– 27, 2012. [DOI] [PubMed] [Google Scholar]
- 134). Simpson DJ, Hibberts NA, McNicol AM, Clayton RN, Farrell WE: Loss of pRb expression in pituitary adenomas is associated with methylation of the RB1 CpG island. Cancer Res 60: 1211– 1216, 2000. [PubMed] [Google Scholar]
- 135). Marinoni I, Pellegata NS: p27kip1: a new multiple endocrine neoplasia gene? Neuroendocrinology 93: 19– 28, 2011. [DOI] [PubMed] [Google Scholar]
- 136). Simpson DJ, Bicknell JE, McNicol AM, Clayton RN, Farrell WE: Hypermethylation of the p16/CDKN2A/MTSI gene and loss of protein expression is associated with nonfunctional pituitary adenomas but not somatotrophinomas. Genes Chromosomes Cancer 24: 328– 336, 1999. [PubMed] [Google Scholar]
- 137). Ezzat S: Epigenetic control in pituitary tumors. Endocr J 55: 951– 957, 2008. [DOI] [PubMed] [Google Scholar]
- 138). Zhang X, Sun H, Danila DC, Johnson SR, Zhou Y, Swearingen B, Klibanski A: Loss of expression of GADD45 gamma, a growth inhibitory gene, in human pituitary adenomas: implications for tumorigenesis. J Clin Endocrinol Metab 87: 1262– 1267, 2002. [DOI] [PubMed] [Google Scholar]
- 139). van der Weyden L, Adams DJ: The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim Biophys Acta 1776: 58– 85, 2007.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140). Peng H, Liu H, Zhao S, Wu J, Fan J, Liao J: Silencing of RASSF3 by DNA hypermethylation is associated with tumorigenesis in somatotroph adenomas. PLoS ONE 8: e59024, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141). Revill K, Dudley KJ, Clayton RN, McNicol AM, Farrell WE: Loss of neuronatin expression is associated with promoter hypermethylation in pituitary adenoma. Endocr Relat Cancer 16: 537– 548, 2009. [DOI] [PubMed] [Google Scholar]
- 142). Mantovani G, Lania AG, Spada A: GNAS imprinting and pituitary tumors. Mol Cell Endocrinol 326: 15– 18, 2010. [DOI] [PubMed] [Google Scholar]
- 143). Ezzat S, Zhu X, Loeper S, Fischer S, Asa SL: Tumor-derived Ikaros 6 acetylates the Bcl-XL promoter to up-regulate a survival signal in pituitary cells. Mol Endocrinol 20: 2976– 2986, 2006. [DOI] [PubMed] [Google Scholar]
- 144). Zhu X, Asa SL, Ezzat S: Ikaros is regulated through multiple histone modifications and deoxyribonucleic acid methylation in the pituitary. Mol Endocrinol 21: 1205– 1215, 2007. [DOI] [PubMed] [Google Scholar]
- 145). Bartel DP: MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281– 297, 2004. [DOI] [PubMed] [Google Scholar]
- 146). Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM: Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA 101: 2999– 3004, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147). Trivellin G, Butz H, Delhove J, Igreja S, Chahal HS, Zivkovic V, McKay T, Patócs A, Grossman AB, Korbonits M: MicroRNA miR-107 is overexpressed in pituitary adenomas and inhibits the expression of aryl hydrocarbon receptor-interacting protein in vitro. Am J Physiol Endocrinol Metab 303: E708– E719, 2012. [DOI] [PubMed] [Google Scholar]
- 148). Palumbo T, Faucz FR, Azevedo M, Xekouki P, Iliopoulos D, Stratakis CA: Functional screen analysis reveals miR-26b and miR-128 as central regulators of pituitary somatomammotrophic tumor growth through activation of the PTEN-AKT pathway. Oncogene 32: 1651– 1659, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149). D'Angelo D, Palmieri D, Mussnich P, Roche M, Wierinckx A, Raverot G, Fedele M, Croce CM, Trouillas J, Fusco A: Altered microRNA expression profile in human pituitary GH adenomas: down-regulation of miRNA targeting HMGA1, HMGA2, and E2F1. J Clin Endocrinol Metab 97: E1128– 1138, 2012. [DOI] [PubMed] [Google Scholar]
- 150). Palmieri D, D'Angelo D, Valentino T, De Martino I, Ferraro A, Wierinckx A, Fedele M, Trouillas J, Fusco A: Downregulation of HMGA-targeting microRNAs has a critical role in human pituitary tumorigenesis. Oncogene 31: 3857– 3865, 2012. [DOI] [PubMed] [Google Scholar]
- 151). Gentilin E, Tagliati F, Filieri C, Molè D, Minoia M, Rosaria Ambrosio M, Degli Uberti EC, Zatelli MC: miR-26a plays an important role in cell cycle regulation in ACTH-secreting pituitary adenomas by modulating protein kinase Cδ. Endocrinology 154: 1690– 1700, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152). Bottoni A, Piccin D, Tagliati F, Luchin A, Zatelli MC, degli Uberti EC: miR-15a and miR-16-1 down-regulation in pituitary adenomas. J Cell Physiol 204: 280– 285, 2005. [DOI] [PubMed] [Google Scholar]
- 153). Butz H, Likó I, Czirják S, Igaz P, Korbonits M, Rácz K, Patócs A: MicroRNA profile indicates downregulation of the TGFβ pathway in sporadic non-functioning pituitary adenomas. Pituitary 14: 112– 124, 2011. [DOI] [PubMed] [Google Scholar]
- 154). Bottoni A, Zatelli MC, Ferracin M, Tagliati F, Piccin D, Vignali C, Calin GA, Negrini M, Croce CM, Degli Uberti EC: Identification of differentially expressed microRNAs by microarray: a possible role for microRNA genes in pituitary adenomas. J Cell Physiol 210: 370– 377, 2007. [DOI] [PubMed] [Google Scholar]
- 155). Butz H, Likó I, Czirják S, Igaz P, Khan MM, Zivkovic V, Bálint K, Korbonits M, Rácz K, Patócs A: Down-regulation of Wee1 kinase by a specific subset of microRNA in human sporadic pituitary adenomas. J Clin Endocrinol Metab 95: E181– E191, 2010. [DOI] [PubMed] [Google Scholar]
- 156). Kitagawa M, Kitagawa K, Kotake Y, Niida H, Ohhata T: Cell cycle regulation by long non-coding RNAs. Cell Mol Life Sci 70: 4785– 4794, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157). Benetatos L, Hatzimichael E, Londin E, Vartholomatos G, Loher P, Rigoutsos I, Briasoulis E: The micro-RNAs within the DLK1-DIO3 genomic region: involvement in disease pathogenesis. Cell Mol Life Sci 70: 795– 814, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158). da Rocha ST, Edwards CA, Ito M, Ogata T, Ferguson-Smith AC: Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet 24: 306– 316, 2008. [DOI] [PubMed] [Google Scholar]
- 159). Zhang X, Zhou Y, Mehta KR, Danila DC, Scolavino S, Johnson SR, Klibanski A: A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J Clin Endocrinol Metab 88: 5119– 5126, 2003. [DOI] [PubMed] [Google Scholar]
- 160). Zhou Y, Zhang X, Klibanski A: MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol 48: R45– R53, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161). Fukuoka H, Cooper O, Ben-Shlomo A, Mamelak A, Ren SG, Bruyette D, Melmed S: EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J Clin Invest 121: 4712– 4721, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]