

Abstract
This is a case report of a 40-year-old man with an adrenal mass that was found incidentally on routine check-up examination. MRI showed a 30×51×57 mm cystic-semisolid heterogeneous mass; hormonal functions were within normal limits. Operative removal was planned because of the large size of the mass. Histopathological and immunohistochemical findings were consistent with adenomatoid tumour. The patient was disease-free at 1 year follow-up. We present this case with its radiological and histological characteristics under the review of the literature.
Background
The adrenal gland, with its two derivative composition, mesodermal cortex and neuroectodermal medulla, is an important endocrine organ with secretory function for corticosteroids. In addition to its more usual non-functioning and functioning lesions, there are less common lesions under the title of ‘other adrenal lesions’ in the adrenal gland chapters of our books.1 Adenomatoid tumour is an extremely rare primary tumour of the adrenal gland with its peculiar morphology for pathologists and unexpected diagnosis for clinicians. The aim of this report is to highlight the histological features of this tumour with its potential clinical and histopathological mimickers, under the review of the literature.
Case presentation
A 40-year-old man presented to the hospital for a routine check-up procedure and a mass lesion was incidentally detected in the right adrenal gland by ultrasonography. Physical examination revealed nothing unusual; he neither had a specific illnessnor any clinical symptom of hormonal hypersecretion. Abdominal MRI revealed a 30×51×57 mm well-marginated right adrenal mass with mostly hyperintense content on axial short τ inversion recovery (STIR) image (figure 1A). There were no signal changes between in phase (figure 1B) and out of phase (figure 1C) axial T1-weighted images. The mass contained a few internal nodular and thin septal components with hypointense concentration on axial STIR image (figure 1A). Axial VIBE image obtained after administration of gadolinium-based contrast material showed enhancement of internal components (figure 1D).
Figures 1.

Abdominal MRI examination showing well-marginated right adrenal mass (arrow) with mostly hyperintense content on axial short τ inversion recovery (STIR) image (A). There is no signal change between in phase (B) and out of phase (C) axial T1-weighted images. The mass contains a few internal nodular and thin septal components (arrowhead) with hypointense intensity on axial STIR image (A). Axial volumetric interpolated breath-hold examination (VIBE) image (D) obtained after administration of gadolinium-based contrast material showing enhancement of internal components (arrowhead).
No abnormality of hormonal function was detected; the basal plasma levels of cortisol and androgens, and urine catecholamines, were within normal limits, and dexamethasone suppression test was positive. Even though the mass was non-functioning, the patient underwent surgery because of its large size. Laparoscopic transperitoneal right adrenalectomy was performed.
On gross examination, the adrenal gland was 7.7×4.6×2.7 cm. Cut sections revealed a 5.5 cm yellow solid and cystic spongy tumoural lesion (figure 2). Histopathology showed a neoplasm composed of loose, dishesive papillary and channel-like structures floating in the cystic lumen, forming microcysts and tubules at the periphery (figure 3A, B). These structures were lined by flat to low cuboidal endothelial-like cells with cytoplasmic vacuoles (figure 3C). No cellular atypia or mitosis was present. Aggregates of macrophages and lymphoid follicles with germinal centres were scattered in or around the lesion (figure 3D). The tumour was surrounded by the adrenal cortex and limited to the gland; capsular or periadrenal invasion was not detected. Immunohistochemically, the tumour cells were positive with panCK, calretinin and negative with CD34 and CD31 (figure 4). These findings were consistent with adenomatoid tumour.
Figure 2.

Gross examination: (A) The adrenal gland showing a well-defined enlargement. (B) Cut surface showing a yellow solid and cystic spongy tumoural lesion.
Figure 3.

Histology: (A) Tumoural lesion composed of papillary and channel-like structures (H&E, ×40). (B) Papillary structures and adrenal gland in the left (H&E, ×100). (C) Vacuolated cells and gland-like structures (H&E, ×100). (D) Lymphoid follicles with germinal centres in the lesion (H&E, ×100).
Figure 4.
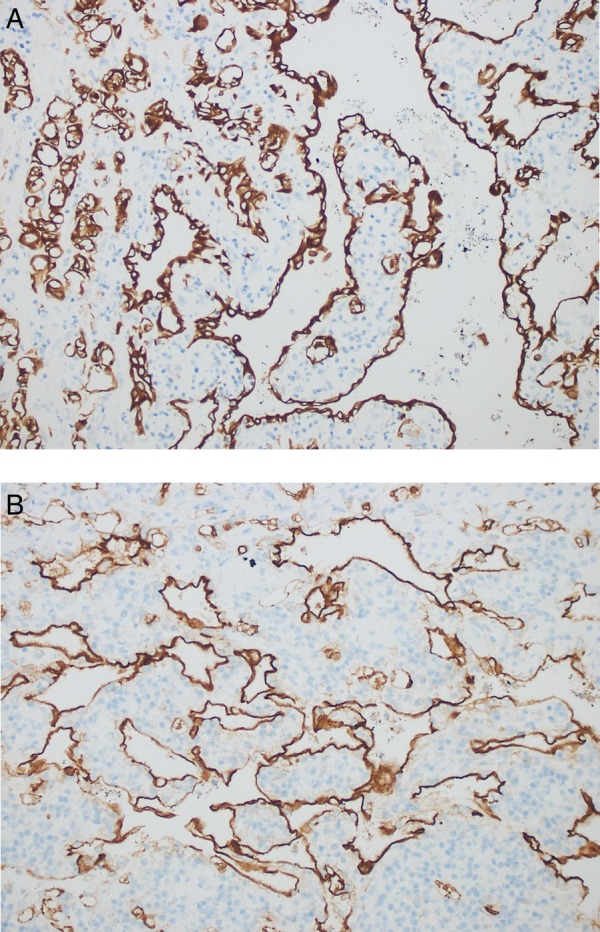
Immnunohistochemical staining showing diffuse and strong positivity for tumour cells; (A) cytokeratin, (B) calretinin (IHC, ×200).
Outcome and follow-up
After 1 year follow-up with abdominal MRI, our patient had no clinical evidence of disease.
Discussion
Adenomatoid tumours of the adrenal gland usually occur in middle-aged men, with a mean age of 41 years, and are generally regarded as benign neoplasms.2 They are either detected incidentally by a routine screening procedure for non-specific symptoms such as vague abdominal pain, hypertension and occasional haematuria or at autopsy.2
From the first description of adenomatoid tumour in the literature, their histogenesis has been argued about for years. The proposed candidates for the cell of origin are mesothelial, mesonephric, Müllerian and endothelial, such as originally proposed by Masson et al.3 Today, the evidence obtained from the ultrastructural and immunohistochemical findings and the sporadic detection of a continuity between the peritoneal lining and the cells lining the channel-like/tubular structures indicate that adenomatoid tumours are of a mesothelial nature.1 Although the adrenal gland is devoid of a mesothelial lining, the most accepted hypothesis for an adenomatoid tumour originating in the adrenal gland is derivation from mesothelial rests.4
Limbach et al reported on a patient with adrenal adenomatoid tumour associated with a personal and family-specific germline mutation in succinate dehydrogenase complex subunit D (SDHD). Some authors have paid special attention to SDHx mutations, and these mutations were reported in association with increased risk of renal tumours, thyroid tumours and gastrointestinal stromal tumours.5 There are also two reported cases of adrenal adenomatoid tumour with HIV. Roy et al suggest the possibility of antiretroviral therapies or chronic immunosuppression increasing this association.6 7
Imaging techniques such as CT/MRI/positron emission tomography cannot always reliably distinguish adrenal adenomatoid tumours from other adrenal tumours.6
In addition to adrenal adenoma, pheochromocytoma and adrenocortical carcinoma, differential diagnoses of adenomatoid tumour also include haemangioma, angiosarcoma, lymphangioma, metastatic adenocarcinoma, malignant mesothelioma and cystic adenoma. Exact diagnosis should be made using immunohistochemical markers such as cytokeratin, calretinin, D2–40, CD34, CD31 and factor VIII. Haemangioma, angiosarcoma and lymphangioma are positive for endothelial markers (CD34, CD31, factor VIII) and negative for keratin and mesothelial markers. Strong immunoreactivity for keratin and calretinin is helpful to confirm the diagnosis of adenomatoid tumour.2 8–10
Given that pleura, epididymis, ovary, uterus and fallopian tube are the most common reported sites for adenomatoid tumour, we suggest that in cases with an unusual histopathological pattern on microscopy, adenomatoid tumour should be included in the differential diagnosis. Clinicopathological features with immunohistochemical markers can substantiate the diagnosis. If available, molecular genetic analysis can be used for research purposes.
Adenomatoid tumours are benign in behaviour and without metastatic potential.
Patient's perspective.
The discovery of the tumour was quite coincidental. In fact, it was during my annual check-up. We went through the previous check-up records and pursued advanced analysis. Positive news was it was there previously and did not grow, but the negative news was it had to be taken out. I waited a couple of weeks for the birth of my second boy, and then had the operation. After the operation, I went back home the following day and did not go to work for two weeks. The result of the pathology test was, “nothing to worry about, continue with regular check-ups”. Currently, the year after the operation, there is not much difference in my daily routine.
Learning points.
Adenomatoid tumour should be taken into consideration in the differential diagnosis of clinically undefined adrenal masses.
It can lead to diagnostic difficulty for unexperienced pathologists due to its wide differential diagnosis.
Keeping in mind the histological features of adenomatoid tumours will allow us to reach an accurate diagnosis.
Immunohistochemical markers and, if available, molecular genetic analysis, can be used for diagnosis.
Footnotes
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Desmet VJ, Rosai J. Adrenal gland and other paraganglia; Other adrenal lesions. In: Rosai J, ed. Rosai and Ackerman's surgical pathology. New York, USA: Elsevier Mosby, 2011:1080. [Google Scholar]
- 2.Cheng L, Ulbright TM. Adenomatoid tumor. In: DeLellis RA, Llyod RV, Heitz PU, Eng C, eds. World Health Organization classification of tumors pathology and genetics of tumours of endocrine organs. Lyon, France: IARC Press, 2004:167. [Google Scholar]
- 3.Masson P, Riopelle JL, Simard LC. Le mésothéliomé bénin de la sphéregénitale. Rev Can Biol 1942;1:720–51. [Google Scholar]
- 4.El-Daly H, Rao P, Palazzo F et al. . A rare entity of an unusual site: adenomatoid tumour of the adrenal gland: a case report and review of the literature. Pathology Res Int 2010;2010:702472 10.4061/2010/702472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lİmbach AL, Ni Y, Huang J et al. . Adenomatoid tumour of the adrenal gland in a patient with germline SDHD mutation: a case report and review of the literature. Pathology 2011;43:495–8. 10.1097/PAT.0b013e3283486bb9 [DOI] [PubMed] [Google Scholar]
- 6.Phitayakom R, MacLennan G, Sadow P et al. . Adrenal adenomatoid tumor in a patient with human immunodeficiency virus. Rare Tumors 2011;3:e21 10.4081/rt.2011.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glasgow BJ, Steinsapir KD, Anders K et al. . Adrenal pathology in the acquired immune deficiency syndrome. Am J ClinPathol 1985;84:594–7. [DOI] [PubMed] [Google Scholar]
- 8.Bialas M, Szczepanski W, Szpor J et al. . Adenomatoid tumour of the adrenal gland: a case report and literature review. Pol J Pathol 2010;2:97–102. [PubMed] [Google Scholar]
- 9.Li SY, Wang X, Zhang S. Adenomatoid tumor of adrenal gland: a rare case report. Indian J Pathol Microbiol 2013;56:319–21. 10.4103/0377-4929.120413 [DOI] [PubMed] [Google Scholar]
- 10.Zhao M, Li C, Zheng J et al. . Cystic lymphangioma-like adenomatoid tumor of the adrenal gland: report of a case and review of the literature. Int J Clin Exp Pathol 2013;6:943–50. [PMC free article] [PubMed] [Google Scholar]