

Abstract
Ras homolog enriched in brain (Rheb) is a key regulator of mammalian target of rapamycin complex 1 (mTORC1). The Rheb-mTORC1 axis is a pivotal pathway that mediates cell growth. It was previously reported that upon energy-stress stimulation, the phosphorylation of Rheb at serine 130 by p38-regulated/activated protein kinase (PRAK) results in the impaired nucleotide binding ability of Rheb and inhibits Rheb-mediated mTORC1 activation. However, the role of Rheb phosphorylation in cancer development remains to be elucidated. The aim of the present study was to determine the effect of Rheb phosphorylation on tumor growth in vitro and in vivo. In addition, tissue samples were obtained from 70 hepatocellular carcinoma (HCC) patients in order to determine any associations between Rheb phosphorylation and the clinicopathological characteristics of patients. In vitro and ex vivo kinase assays were performed to determine the phosphorylation of Rheb by PRAK. A xenograft assay was performed to assess tumorigenicity of MEF cell lines. In addition, western blot and immunohistochemical analyses were performed to detect Rheb protein expression and phosphorylation. The results of the present study revealed that Rheb phosphorylation may be induced through Ras overexpression. In addition, kinase-dead PRAK and dominant-negative PRAK mutation were demonstrated to abolish the Rheb phosphorylation induced by Ras overexpression. Xenograft assays in nude mice revealed that Rheb phosphorylation was involved in PRAK-mediated tumor suppression. Of note, the clinicopathological analysis of 70 HCC samples determined that Rheb phosphorylation was associated with poor proliferation and the progression of HCC. In conclusion, the results of the present study suggested that Rheb phosphorylation may have an important role as an intracellular barrier to cancer development.
Keywords: Ras, p38-regulated/activated protein kinase, Rheb phosphorylation, liver cancer
Introduction
Tumor formation is driven by the activation of oncogenes; however, cells harbor several barriers to tumor development, including cell growth inhibition, premature senescence and cell death (1,2). Ras-induced cellular stress is a widely explored ex vivo model employed in order to study oncogene-induced cellular barriers (3–6). Overexpression of Ras activates p38-regulated/activated protein kinase (PRAK) (7), which phosphorylates p53 at serine 37, leading to premature cellular senescence (8).
Ras homolog enriched in brain (Rheb) is a member of Ras family (9), serving as the upstream activator of mammalian target of rapamycin complex 1 (mTORC1) (10). Previous studies have suggested that Rheb-mTORC1 may be the pivotal axis in regulating cell growth in coordination with nutrient availability in the environment (11,12). Rheb is overexpressed in numerous types of cancer (13) and may be a critical target for farnesyltransferase inhibitors (FTIs) therapy in non-small-cell lung cancer cells (14).
A previous study reported that PRAK phosphorylated Rheb at serine 130 and that this post-translational modification of Rheb attenuated its guanine nucleotide binding activities, leading to its inactivation and subsequent cell growth inhibition (15). However, the role of Rheb phosphorylation in cancer development remains to be elucidated. Therefore, the aim of the present study was to determine the effect of Rheb phosphorylation on tumor growth in vitro and in vivo. In addition, tissue samples were obtained from 70 hepatocellular carcinoma (HCC) patients in order to determine any associations between Rheb phosphorylation and the clinicopathological characteristics of patients.
Materials and methods
Materials
Mouse monoclonal antibodies against hemagglutinin (cat. no. sc-7392), c-Myc (cat. no. sc-40) and HRas (cat. no. sc-29) were obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Anti-phospho-Rheb (S130) antibodies were raised against an S130 phospho-modified peptide of Rheb by Abmart Co., Ltd. (Shanghai, China). Rabbit anti-Rheb polyclonal antibody was from Proteintech (Wuhan, Hubei, China). Rabbit polyclonal antibody against p-PRAK(T182) was a gift from Professor Jiahuai Han. Mouse anti-ribosomal protein S6 monoclonal antibody (cat. no. 2317), rabbit anti-p-S6 (S235/S236) polyclonal antibody (cat. no. 2211), rabbit anti-Erk polyclonal antibody (cat. no. 9102) and rabbit anti-Erk (T202/Y204) polyclonal antibody (cat. no. 4370) were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA). For western blot analysis, antibodies were used at a dilution of 1:3,000 for exogenous proteins and 1:1,000 for endogenous proteins (if not otherwise indicated).
All mutant constructs of PRAK were created through polymerase chain reaction (PCR) mutagenesis and were verified by DNA sequencing. The sense primers used were as follows: for PRAK-KM mutation, 5′-gaacggtttgcgctgaugattcttcttgatcg-3′; for PRAK-DN, 5′-caaggtgacttgatggcaccccagttcac-3′. The antisense primers used were the exact reverse complementary sequence of the sense primers. DNA sequencing was performed by Sangon Biotech (Shanghai, China).
Cell culture and transfection
Human embryonic kidney 293 (HEK293) cells and mouse embryonic fibroblast (MEF) were purchased from ATCC (Manassas, VA, USA). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Inc., Waltham, MA, USA), as previously described (15). Calcium phosphorylate transfection was used to transfect DNA into HEK293 cells for lentiviral package. Lipofectamine 2000 (Thermo Fisher Scientific, Inc.) was used to transfect DNA into HEK293 for gene overexpression. Protein expression was determined by western blot analysis.
In vitro kinase assay
Glutathione S-transferase-Rheb was purified from BL21(DE3) competent Escherichia coli (New England Biolabs, Ipswich, MA, USA) and subjected to a kinase assay in kinase buffer (25 mM Tris, pH 7.5; 10 mM MgCl2; 2 mM DTT; 5 mM β-glycerophosphate; and 0.1 mM Na3VO4) at 30°C for 30 min. Tris, MgCl2, dithiothreitol, β-glycerophosphate and Na3VO4 were all from Sigma-Aldrich (Shanghai, China).
Xenograft assay
PRAK+/+ and PRAK−/− mouse embryonic fibroblasts (MEFs), HRas and adenovirus early region 1A (E1A)-expressing lentiviral plasmids were kindly provided by Professor Jiahuai Han (Xiamen University, Xiamen, China). A tumor formation assay was performed as previously described (8). Briefly, MEFs were infected with HRas- and adenovirus E1A-expressing lentiviruses. Lentiviruses were packaged in HEK293 cells, and at 48 h post-transfection, the cell media were collected and filtered through a 0.45 µm membrane (Millipore, Billerica, MA, USA), and then mixed with fresh medium (1:1) and 4 ng/ml polybrene (Sigma-Aldrich). Transformed MEFs (2×106) were injected subcutaneously into the flanks of female immunodeficient athymic nude mice (age, 6–8 weeks; Biomodel Organisms, Shanghai, China) in 100 µl phosphate-buffered saline (PBS; Thermo Fisher Scientific, Inc.). Mice were housed under specific pathogen-free condition, caged individually and given ad libitum access to food and water. All animal experiments were conducted with the approval of the Institutional Animal Care and Use Committee of Fujian Medical University (Fujian, China). Tumors were measured at least every 2 days without anaesthetizing. Mice were sacrificed using carbon dioxide for euthanasia when the tumor diameter was >1 cm.
Patient samples
A total of 70 HCC patients were enrolled in the present study at the Liver Center of Fujian Province (Mengchao Hepatobiliary Hospital of Fujian Medical University; Fujian, China), between January 2011 and July 2012. HCC and corresponding adjacent normal liver tissue samples were obtained from patients who underwent hepatectomy surgery. Patients were not included in the present study if they had previously received preoperative chemotherapy or radiation therapy. Peritumoral liver tissues were obtained from regions >3 cm from the tumor site. Immediately following surgery, tissue samples were fixed in neutral-buffered formalin and embedded in paraffin (Sinopharm Chemical Reagent, Shanghai, China) for immunohistochemical studies. HCC diagnoses were confirmed through pathological studies; in addition, the peritumoral liver tissue samples were all confirmed to be normal. Clinical information was collected from patient records. Tumor stage was determined using the Barcelona Clinic Liver Cancer (BCLC) staging system (16) and tumor differentiation was graded according to the Edmondson grading system (17). The present study was approved by the Institute Research Ethics Committee of Fujian Medical University. Written informed consent was obtained from each patient according to the committee regulations.
Tissue microarrays (TMAs) and immunohistochemistry
According to the protocol described by Kononen et al (18) for TMA, a modified method was developed for the preparation of paraffin TMAs, as previously described (19). HCC and corresponding peritumoral tissue from 70 patients was formalin-fixed and paraffin-embedded. Next, 3 tissue cores (diameter, 0.75 mm) from each sample block were exhumed by a holing needle and then arrayed on the recipient block. Immunohistochemistry was performed using the Elivision™ Plus two-step system (Maixin Biotech Co., Ltd., Fuzhou, China) according to the manufacturer's instructions, as previously described (19). TMA slides were blocked with 10% normal goat serum (Maixin Biotech Co., Ltd.) at 37°C for 30 min and incubated with a 1:500 dilution of rabbit polyclonal anti-human phospho-Rheb antibody for 1 h at 37°C followed by three washes with PBS containing 0.05% Triton X-100 (Beyotime Institute of Biotechnology, Shanghai, China). The slides were incubated with polymerized horseradish peroxidase-conjugated anti-mouse/rabbit immunoglobulin G (Maixin Biotech Co., Ltd.), followed by 3,3′-diaminobenzidine; slides were then counterstained with hematoxylin (Harbin Green Specimen Technology Development, Harbin, China). Negative control sections were incubated with pre-immune serum. The percentage of hepatocytes or tumor cells showing cytoplasmic staining was scored for each case as previously described (20,21). Cells were scored broadly, according to the staining intensity and the distribution of stained cells. Staining intensities were scored as follows: No staining, 0; weak staining, 1; moderate staining, 2; and strong staining, 3. The distribution of stained cells was scored as follows: 0%, 0; <25%, 1; 25–50%, 2; and >50%, 3. The final staining score was obtained by adding the scores of staining intensity and distribution score of stained cells. A score of 0–2 points was considered negative and scores of 3–6 points were considered positive. The staining results were evaluated by two independent pathologists (Professor Shengbing Zang and Miss Xueting Fang) who were blinded to the clinicopathological features of the samples; all differences in interpretation were re-evaluated to reach a consensus. Tissue scoring was performed in triplicate for each tumor and high levels of homogeneity for staining intensity and percentage of positive cells were obtained.
Statistical analysis
Statistically significant differences between and among groups were assessed using the χ2 test with SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference between values.
Results
Rheb phosphorylation was induced through Ras overexpression in a PRAK-dependent manner
In order to explore the role of Rheb phosphorylation in the antitumor barrier, it was investigated whether Rheb phosphorylation was induced by certain oncogenes. Ras overexpression is known to activate PRAK (8) and PRAK was reported to directly phosphorylate Rheb at serine 130 (15); therefore, it was hypothesized that Rheb phosphorylation may be induced by Ras overexpression. The results of the present study confirmed that the co-expression of Ras with PRAK and Rheb in HEK293 cells markedly increased the signal of Rheb phosphorylation at serine 130 (Fig. 1A). In addition, the results revealed that Ras-induced Rheb phosphorylation was dose-dependent (Fig. 1B). By contrast, little Rheb phosphorylation was detected in cells co-expressing Ras and Rheb without PRAK. Subsequently, it was investigated whether PRAK activity was required for Ras-induced Rheb phosphorylation. As shown in Fig. 2A, kinase-dead PRAK mutant (PRAK-KM) and dominant-negative PRAK mutant (PRAK-DN) were able to abolish Ras-induced Rheb phosphorylation. In addition, the kinase assay demonstrated that PRAK-KM abolished PRAK activity on Rheb in vitro (Fig. 2B). These data indicated that Rheb phosphorylation may be induced through Ras overexpression, which is dependent on PRAK and requires its kinase activity.
Figure 1.
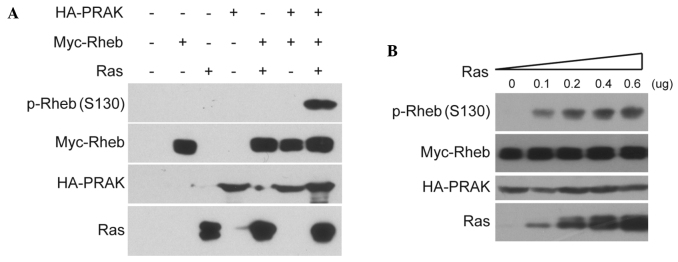
Rheb phosphorylation is induced by Ras overexpression. (A) HEK293 cells were transfected with 0.2 µg HA-PRAK, 0.2 µg Myc-Rheb or 0.6 µg HRas alone or in combination. Phosphorylation of Rheb was determined using anti-phospho-Rheb (S130) antibodies. (B) HEK293 cells were transfected with HA-PRAK, Myc-Rheb and increasing concentrations of HRas (0, 0.1, 0.2, 0.4 and 0.6 µg). Phosphorylated Rheb levels were then determined. p-Rheb, phosphorylated Ras homolog enriched in brain; PRAK, p38-regulated/activated protein kinase; HA, hemagglutinin.
Figure 2.
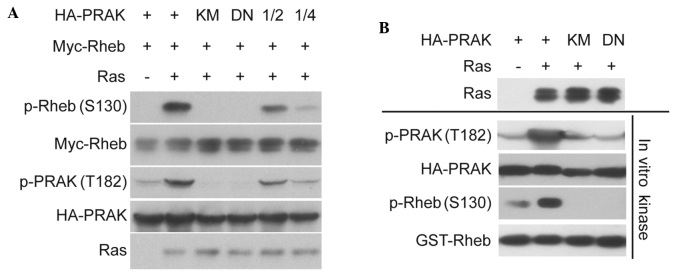
Rheb phosphorylation induced by Ras overexpression requires PRAK kinase activity. (A) HA-PRAK, HA-PRAK-KM or PRAK-DN were co-transfected with Myc-Rheb and HRas into HEK293 cells. Phosphorylation of Rheb was determined. (B) HA-PRAK, HA-PRAK-KM or HA-PRAK-DN were co-transfected with HRas into HEK293 cells. HA-PRAK was then pulled down and incubated with glutathione S-transferase-Rheb for in vitro kinase assay. Phosphorylation of Rheb was determined. Rheb, Ras homolog enriched in brain; PRAK, p38-regulated/activated protein kinase; KM, kinase-dead mutant HA-PRAK; DN, dominant-negative mutant HA-PRAK; p-, phosphorylated; HA, hemagglutinin; 1/2, 0.1 µg HA-PRAK for DNA transfection; 1/4, 0.05 µg HA-PRAK.
Rheb phosphorylation is involved in PRAK-mediated tumor suppression
Ras overexpression is a cellular model for tumor development and PRAK is known to act as tumor suppressor in this process (8). In addition, Rheb phosphorylation was reported to attenuate its activity on mTORC1 activation (15), which is pivotal for tumor growth. Therefore, it was proposed that PRAK-mediated Rheb phosphorylation may also be associated with PRAK-mediated tumor suppression. In the present study, PRAK+/+ and PRAK−/− MEFs were infected with HRas and E1A. Cells were then injected into nude mice and tumor formation was monitored periodically (at least every 2 days). The results revealed that tumor formation of PRAK+/+ MEFs was slower compared with that of PRAK−/− MEFs (Fig. 3A and B). In addition, the phosphorylation of Rheb, ribosomal protein S6 and extracellular signal-regulated kinase (ERK) were determined in tumor tissues. As shown in Fig. 3C, phosphorylation of Rheb was detected in tumors derived from PRAK+/+ MEFs, but not in those from PRAK−/− cells. By contrast, phosphorylation of S6 in PRAK+/+ tumors was decreased compared with that in PRAK−/− tumors, which supported the hypothesis that Rheb phosphorylation may inhibit mTORC1 activity. Phosphorylation of ERK1/2 was unchanged. In conclusion, these data indicated that Rheb phosphorylation may be involved in PRAK-mediated tumor suppression.
Figure 3.

Rheb phosphorylation is involved in PRAK-mediated tumor suppression. (A) Primary MEF cells from E13.5 WT (PRAK+/+) and KO (PRAK−/−)embryos were transduced with HRas and E1A, then inoculated subcutaneously into six nude mice (left flank, PRAK+/+; right flank, PRAK−/−). Image of only 5 mice was captured at 24 days post-inoculation, since 1 mouse died before. (B) Tumor volumes were monitored daily. (C) Following ~25 days, mice were sacrificed and tumors were harvested. Phosphorylation of Rheb, S6 and Erk were determined. Rheb, Ras homolog enriched in brain; PRAK, p38-regulated/activated protein kinase; MEF, mouse embryonic fibroblasts; WT, wild type; KO, knock-out; p-, phosphorylated.
Rheb phosphorylation is compromised in human HCC tissues
In order to investigate Rheb phosphorylation in cancer development, immunohistochemistry was performed on TMAs containing 70 HCC tissue samples and their peritumoral liver tissue samples in order to assess Rheb phosphorylation levels. Cytoplasmic staining of Rheb phosphorylation was moderate or strong in peritumoral liver tissue samples (Fig. 4A), while the tumor cells of certain HCC tissue samples demonstrated loss of expression of Rheb phosphorylation (Fig. 4B). The presence of Rheb phosphorylation in HCC tissue samples (62.86%) was significantly reduced compared with that in peritumoral liver tissue samples (98.57%) (c2=28.679, P<0.001). In addition, the expression of Rheb in HCC tissue samples was examined. The results demonstrated that peritumoral liver tissue samples (Fig. 4C) and HCC tissue samples (Fig. 4D) exhibited moderate or strong cytoplasmic staining of Rheb. The presence of Rheb in HCC tissue samples (92.86%) was not significantly different from that in peritumoral liver tissue samples (98.57%) (c2=2.786, P=0.10). Furthermore, the association between Rheb phosphorylation and selected clinicopathological parameters of HCC patients was evaluated. As shown in Table I, Rheb phosphorylation did not vary significantly with gender, age, tumor number, serum α-fetoprotein levels, hepatitis B virus infection, histological differentiation or the presence of cirrhosis or portal vein thrombus. However, it was demonstrated that patients with low Rheb phosphorylation were prone to larger tumors (>5 cm) and advanced BCLC stage. In conclusion, these data indicated that low Rheb phosphorylation was associated with poor proliferation and progression of HCC.
Figure 4.

Downregulation of Rheb phosphorylation in human HCC tissues. Immunohistochemical staining was performed using the Elivision™ Plus two-step immunohistochemical technique with3,3′-diaminobenzidine staining; original magnifications, x400. (A) Strong immunoreactivity of Rheb phosphorylation was detected in peritumoral liver tissue. (B) Rheb phosphorylation was lost in HCC tissue. Strong immunoreactivity of Rheb was detected in (C) normal peritumoral liver and (D) HCC tissues. (p-)Rheb, (phosphorylated-)Ras homolog enriched in brain; HCC, hepatocellular carcinoma.
Table I.
Associations between Rheb phosphorylation and clinicopathologic parameters.
| p-Rheb expression | |||||
|---|---|---|---|---|---|
| Variable | n | Positive, n (%) | Negative, n (%) | χ2 | P-value |
| Gender | |||||
| Male | 61 | 40 (65.6) | 21 (34.4) | 1.500 | 0.221 |
| Female | 9 | 4 (44.4) | 5 (55.6) | ||
| Age (years) | |||||
| ≤50 | 33 | 19 (57.6) | 14 (42.4) | 0.746 | 0.388 |
| >50 | 37 | 25 (67.6) | 12 (32.4) | ||
| Tumor size (cm) | |||||
| ≤5 | 17 | 15 (88.2) | 2 (11.8) | 6.194 | 0.013 |
| >5 | 53 | 29 (54.7) | 24 (45.3) | ||
| Tumor number | |||||
| Single | 54 | 34 (63.0) | 20 (37.0) | 0.001 | 0.973 |
| Multiple | 16 | 10 (62.5) | 6 (37.5) | ||
| α-fetoprotein (ng/ml) | |||||
| ≤400 | 30 | 21 (70.0) | 9 (30.0) | 1.147 | 0.284 |
| >400 | 40 | 23 (57.5) | 17 (42.5) | ||
| Cirrhosis | |||||
| Yes | 48 | 31 (64.6) | 17 (35.4) | 0.195 | 0.659 |
| No | 22 | 13 (59.1) | 9 (40.9) | ||
| HBsAg | |||||
| Positive | 60 | 36 (60.0) | 24 (40.0) | 1.469 | 0.226 |
| Negative | 10 | 8 (80.0) | 2 (20.0) | ||
| Differentiation | |||||
| Well/moderate | 37 | 25 (67.6) | 12 (32.4) | 0.746 | 0.388 |
| Poor | 33 | 19 (57.6) | 14 (42.4) | ||
| BCLC stage | |||||
| 0/A | 12 | 12 (100.0) | 0 (0.0) | 8.558 | 0.003 |
| B/C | 58 | 32 (55.2) | 26 (44.8) | ||
| Portal vein thrombus | |||||
| Yes | 39 | 25 (64.1) | 14 (35.9) | 0.059 | 0.809 |
| No | 31 | 19 (61.3) | 12 (38.7) | ||
(p-)Rheb, (phosphorylated-)Ras homolog enriched in brain; HBsAg, hepatitis B surface antigen; BCLC, Barcelona Clinic Liver Cancer.
Discussion
Tumor formation is a consequence of oncogene activation and tumor suppressor inactivation; when activated, oncogenes drive cell growth and proliferation. In order to maintain cellular homeostasis, cells develop barriers to tumor development, most of which are mediated by tumor suppressors (22). Inactivation of tumor suppressors deregulates intracellular barriers and leads to epigenetic and metabolic reprogramming in favor of cancer development (1,23). Unveiling the mechanisms underlying barrier establishment and deregulation may therefore further current understanding of the process of tumor development and may aid the development of novel anticancer therapies.
Ectopic expression of activated Ras was reported to evoke a fabricated network of intracellular signaling pathways. Ras promotes cell proliferation through the Ras-phosphoinostide 3-kinase-mTORC1 (24) and Ras-mitogen-activated protein kinase (25) pathways. In addition, Ras has been reported to elicit cellular antitumorigenic defenses through the Ras-PRAK-p53 signaling pathway (8). Inactivation of PRAK was demonstrated to attenuate its antitumor barrier, promoting tumor formation (15). In addition to p53, Rheb was also identified as a substrate of PRAK (15). However, the role of PRAK-mediated Rheb phosphorylation in cancer development remained to be elucidated. The present study demonstrated that PRAK-mediated Rheb phosphorylation was induced by Ras overexpression and that Rheb phosphorylation may be involved in PRAK-mediated tumor suppression. Therefore, it was proposed that Rheb phosphorylation may be an alternative mechanism underlying PRAK-mediated tumor suppression in addition to p53 activation.
In conclusion, although Rheb overexpression was observed in skin tumors (13), prostate cancer (21,26) and lymphomas (27), the present study did not identify any significant differences in Rheb expression between cancer and peritumoral tissue in HCC. However, it was revealed that Rheb phosphorylation levels in tumor tissue samples were markedly reduced compared with those in peritumoral tissue samples. In addition, the clinicopathological data indicated that Rheb phosphorylation was associated with the poor proliferation and progression of HCC, supporting the hypothesis that Rheb phosphorylation may be an intracellular barrier to cancer development.
Acknowledgements
The present study was supported by grants from the National Natural Science Foundation of China (no. 31100995), the Natural Science Foundation of Fujian Province (no. 2011J01187) and the Scientific Research Project of Education Department of Fujian Province (no. 2013B009).
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Bae HJ, Chang YG, Noh JH, Kim JK, Eun JW, Jung KH, Kim MG, Shen Q, Ahn YM, Kwon SH, et al. DBC1 does not function as a negative regulator of SIRT1 in liver cancer. Oncol Lett. 2012;4:873–877. doi: 10.3892/ol.2012.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dimauro T, David G. Ras-induced senescence and its physiological relevance in cancer. Curr Cancer Drug Targets. 2010;10:869–876. doi: 10.2174/156800910793357998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 5.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dörken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 6.Ferbeyre G. Barriers to Ras transformation. Nat Cell Biol. 2007;9:483–485. doi: 10.1038/ncb0507-483. [DOI] [PubMed] [Google Scholar]
- 7.New L, Jiang Y, Han J. Regulation of PRAK subcellular location by p38 MAP kinases. Mol Biol Cell. 2003;14:2603–2616. doi: 10.1091/mbc.E02-08-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun P, Yoshizuka N, New L, Moser BA, Li Y, Liao R, Xie C, Chen J, Deng Q, Yamout M, et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell. 2007;128:295–308. doi: 10.1016/j.cell.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 9.Yamagata K, Sanders LK, Kaufmann WE, Yee W, Barnes CA, Nathans D, Worley PF. Rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J Biol Chem. 1994;269:16333–16339. [PubMed] [Google Scholar]
- 10.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Babcock JT, Quilliam LA. Rheb/mTOR activation and regulation in cancer: Novel treatment strategies beyond rapamycin. Curr Drug Targets. 2011;12:1223–1231. doi: 10.2174/138945011795906589. [DOI] [PubMed] [Google Scholar]
- 12.Wazir U, Newbold RF, Jiang WG, Sharma AK, Mokbel K. Prognostic and therapeutic implications of mTORC1 and Rictor expression in human breast cancer. Oncol Rep. 2013;29:1969–1974. doi: 10.3892/or.2013.2346. [DOI] [PubMed] [Google Scholar]
- 13.Lu ZH, Shvartsman MB, Lee AY, Shao JM, Murray MM, Kladney RD, Fan D, Krajewski S, Chiang GG, Mills GB, et al. Mammalian target of rapamycin activator RHEB is frequently overexpressed in human carcinomas and is critical and sufficient for skin epithelial carcinogenesis. Cancer Res. 2010;70:3287–3298. doi: 10.1158/0008-5472.CAN-09-3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng H, Liu A, Liu B, Li M, Yu H, Luo X. Ras homologue enriched in brain is a critical target of farnesyltransferase inhibitors in non-small cell lung cancer cells. Cancer Lett. 2010;297:117–125. doi: 10.1016/j.canlet.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Zheng M, Wang YH, Wu XN, Wu SQ, Lu BJ, Dong MQ, Zhang H, Sun P, Lin SC, Guan KL, et al. Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nat Cell Biol. 2011;13:263–272. doi: 10.1038/ncb2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Llovet JM, Di Bisceglie AM, Bruix J, Kramer BS, Lencioni R, Zhu AX, Sherman M, Schwartz M, Lotze M, Talwalkar J, Gores GJ. Panel of Experts in HCC-Design Clinical Trials: Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst. 2008;100:698–711. doi: 10.1093/jnci/djn134. [DOI] [PubMed] [Google Scholar]
- 17.Edmondson HA, Steiner PE. Primary carcinoma of the liver: A study of 100 cases among 48,900 necropsies. Cancer. 1954;7:462–503. doi: 10.1002/1097-0142(195405)7:3<462::AID-CNCR2820070308>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 18.Kononen J, Bubendorf L, Kallioniemi A, Bärlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G, Kallioniemi OP. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med. 1998;4:844–847. doi: 10.1038/nm0798-844. [DOI] [PubMed] [Google Scholar]
- 19.Shengbing Z, Feng LJ, Bin W, Lingyun G, Aimin H. Expression of KiSS-1 gene and its role in invasion and metastasis of human hepatocellular carcinoma. Anat Rec (Hoboken) 2009;292:1128–1134. doi: 10.1002/ar.20950. [DOI] [PubMed] [Google Scholar]
- 20.Eom M, Han A, Lee MJ, Park KH. Expressional difference of RHEB, HDAC1, and WEE1 proteins in the stromal tumors of the breast and their significance in tumorigenesis. Korean J Pathol. 2012;46:324–330. doi: 10.4132/KoreanJPathol.2012.46.4.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kobayashi T, Shimizu Y, Terada N, Yamasaki T, Nakamura E, Toda Y, Nishiyama H, Kamoto T, Ogawa O, Inoue T. Regulation of androgen receptor transactivity and mTOR-S6 kinase pathway by Rheb in prostate cancer cell proliferation. Prostate. 2010;70:866–874. doi: 10.1002/pros.21120. [DOI] [PubMed] [Google Scholar]
- 22.Prieur A, Peeper DS. Cellular senescence in vivo: A barrier to tumorigenesis. Curr Opin Cell Biol. 2008;20:150–155. doi: 10.1016/j.ceb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 23.Menendez JA, Alarcón T. Metabostemness: a new cancer hallmark. Front Oncol. 2014;4:262. doi: 10.3389/fonc.2014.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 25.Aksamitiene E, Kiyatkin A, Kholodenko BN. Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: A fine balance. Biochem Soc Trans. 2012;40:139–146. doi: 10.1042/BST20110609. [DOI] [PubMed] [Google Scholar]
- 26.Nardella C, Chen Z, Salmena L, Carracedo A, Alimonti A, Egia A, Carver B, Gerald W, Cordon-Cardo C, Pandolfi PP. Aberrant Rheb-mediated mTORC1 activation and Pten haploinsufficiency are cooperative oncogenic events. Genes Dev. 2008;22:2172–2177. doi: 10.1101/gad.1699608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mavrakis KJ, Zhu H, Silva RL, Mills JR, Teruya-Feldstein J, Lowe SW, Tam W, Pelletier J, Wendel HG. Tumorigenic activity and therapeutic inhibition of Rheb GTPase. Genes Dev. 2008;22:2178–2188. doi: 10.1101/gad.1690808. [DOI] [PMC free article] [PubMed] [Google Scholar]