

Abstract
Metformin is commonly used to treat type II diabetes, although it may also reduce the risk of cancer and improve the associated prognosis. However, its mode of action in cancer remains unclear. The present study evaluated the effects of metformin on lung adenocarcinoma A549 cells and identified molecular mechanisms of metformin activity. The A549 cells were treated with metformin at different concentrations and cell viability was assayed by using an MTT assay. The cell cycle and the apoptosis rate were assayed by flow cytometry. Nude mice were transplanted with A549 cells and the tumor growth inhibition rate was detected. Once the A549 cells had been treated with 20 mM metformin for 48 h, the cell cycle was arrested in the G0/Gl phase and the apoptosis rate was 20.57±3.16%. The expression of the B-cell lymphoma (Bcl)-2 and Bcl-extra large proteins was downregulated following metformin treatment, while Bax protein expression was significantly increased. Tumor size in the high-dose metformin and cisplatin plus metformin groups was significantly smaller, and the inhibition rates were 41.3 and 72.9%, respectively, compared with the control group. These results indicated that metformin displays anticancer activity against lung adenocarcinoma by causing G1 arrest of the cell cycle and subsequent cell apoptosis through the mitochondria-dependent pathway in A549 cells. Furthermore, it was found that metformin dramatically inhibited lung adenocarcinoma tumor growth in vivo. These data suggest that metformin may become a potential cytotoxic drug in the prevention and treatment of lung adenocarcinoma.
Keywords: metformin, lung adenocarcinoma, apoptosis, mitochondrion-mediated pathway
Introduction
Lung adenocarcinoma is one of the main causes of cancer-related mortality globally, accounting for nearly 30% of cancer-related mortalities worldwide (1). The incidence of lung adenocarcinoma is rising all over the world due to the adoption of lifestyle choices that have an association with cancer, including physical inactivity and smoking. Although advances have been made with regard to early diagnosis and treatment modalities, the prognosis for affected patients remains poor, with a five-year survival rate of only ~15% (2). As a consequence, the prevention of lung cancer is a high priority and urgent efforts are required to identify measures, including drug treatment, which may effectively reduce the risk of lung cancer. Chemotherapy is one of the best approaches for unresectable tumors, but the efficacy of current lung tumor chemotherapy is only modest and the requirement for an optimal lung adenocarcinoma treatment remains. Metformin, a biguanide drug, has been demonstrated to exert anticancer effects (3). The drug reduces the level of glucose by decreasing liver glucose production, thereby increasing fatty acid oxidation and glucose utilization. Notably, previous epidemiological studies suggested that patients with diabetes who were treated with metformin had a lower cancer-related mortality rate and a lower incidence rate of cancer of any type when compared with patients who underwent other treatments (4–7). Additionally, metformin was shown to prevent the induction of carcinogen-induced pancreatic cancer in hamsters that were maintained on high-fat diets (8). The drug was also shown to inhibit the growth of breast and colon carcinoma cells (9,10). Collated evidence from a number of clinical studies has recently been published in a meta-analysis (11). However, the precise mechanisms involved remain incompletely understood. The antitumor activity of metformin may be explained by two mechanisms. Firstly, metformin is able to decrease insulin resistance and lower the levels of circulating insulin by activating AMP-activated protein kinase (AMPK), which causes decreased hepatic gluconeogenesis (10) and increased glucose uptake in the muscle. Secondly, metformin acts as an inhibitor of tumor growth, at least in part by upregulating the activity of AMPK and by downstream suppression of signaling through the mammalian target of rapamycin (mTOR) (12). Several other potential mechanisms for the anticancer action of metformin have also been demonstrated, including the suppression of HER2 oncoprotein expression, the downregulation of cyclin D1 expression and p53 activation (9,13–15). However, there have been few studies evaluating the potential utility of metformin in in vivo models of cancer, and the method by which metformin induces apoptosis remains unknown. The present study describes experiments that were performed to investigate the hypothesis that metformin exhibits direct anti-proliferative actions on lung adenocarcinoma cells in vitro and in vivo.
Materials and methods
Chemicals and reagents
Metformin was obtained from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in phosphate-buffered saline (PBS). Cell culture chemicals and materials were obtained from Invitrogen Life Technologies (Burlington, ON, Canada). Anti-β-actin, anti-B-cell lymphoma (Bcl)-2, anti-Bax and anti-caspase-3 were purchased from Cell Signaling Technology (Beverly, MA, USA). Horseradish peroxidase-conjugated anti-rabbit immnoglobulin (Ig)G, anti-mouse IgG and enhanced chemiluminescence (ECL) reagents were obtained from Amersham Pharmacia Biotech (Piscataway, NJ, USA).
Cell lines and culture conditions
The human lung adenocarcinoma A549 cells were obtained from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). The A549 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mmol/l L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C, in an atmosphere of 5% CO2. The cells were passaged by 0.25% Trypsin-EDTA when they reached 80% confluence.
Cell proliferation assay
An MTT assay was used to evaluate the effect of metformin on the lung adenocarcinoma cells. Briefly, ~10,000 cells were seeded into 96-well tissue culture plates and then treated with different doses (0, 5, 10, 20 and 50 mmol/l) of metformin for 24, 48 and 72 h, respectively. MTT reagent was then added to each well, and the cells were further incubated for 6 h. Absorbance was measured in an automated microplate reader (ELX 800; BioTek Instruments, Inc., Winooski, VT, USA) at 450 nm.
Cell morphological analysis
The A549 cells were treated with 10.0 mM metformin or 0.1% dimethyl sulfoxide (control) for 48 h. The cells were then incubated with 10 µg/ml Hoechst 33342 and observed by fluorescence microscope (DMIRB; Leica, Wetzler, Germany).
Flow cytometry
The A549 cells were starved of serum for 24 h and then treated with different doses (0, 10 and 20 mmol/l) of metformin for 48 h. The cells were then washed with PBS (pH 7.4) and fixed with 70% ice-cold ethanol at 4°C overnight. After fixation, the cells were stained with propidium iodide (PI) at 1 mg/ml for 30 min at room temperature. The cell cycle was analyzed by flow cytometry (FACScan; BD Biosciences, Franklin Lakes, NJ, USA).
For cell apoptosis detection, the apoptotic rate of A549 cells was analyzed using an Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China). A total of 1×105 cells/well were seeded into six-well plates and cultured in DMEM at 37°C overnight. Subsequent to starvation for 12 h, the cells were treated with different doses of metformin (0, 10 and 20 mmol/l) in complete medium for 48 h, digested with 2.5 mg/ml trypsin, washed twice with PBS and suspended with 300 µl binding buffer (Nanjing KeyGen Biotech Co., Ltd.). The cells were then incubated with 2 µl Annexin V and 5 µl PI for 15 min at room temperature, and the distribution of viable, early apoptotic, late apoptotic and necrotic cells was detected using a FACSCaliber flow cytometer (BD Biosciences). Cells that were negative for the Annexin V-FITC and PI were considered to be viable cells, cells that were positive for Annexin V-FITC, but negative for PI were considered to be early apoptotic cells, cells that were positive for Annexin V-FITC and PI were considered to be late apoptotic cells, while cells that were negative for both Annexin V-FITC and PI were considered to be necrotic. The sum of the early and late apoptotic cells constituted the total number of apoptotic cells, which was presented as the percentage of the total cells.
Mitochondrial and cytosolic fractionation
The Cell Mitochondria Isolation kit (Beyotime Institute of Biotechnology, Haimen, China) was used to perform the isolation of the mitochondria and cytosol, according to the manufacturer's instructions. Samples of cytosol and mitochondria were dissolved in lysis buffer, and proteins were subjected to western blotting, respectively.
Western blot analysis
The A549 cells were lysed in a radioimmunoprecipitation assay buffer (9.1 mM dibasic sodium phosphate, 1.7 mM monobasic sodium phosphate, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 0.2 mM sodium vanadate, 0.2 mM phenylmethylsulfonyl fluoride and 0.2 U/ml aprotinin). Clarified protein lysates (50 g) were resolved electrophoretically on denaturing SDS-polyacrylamide gels (10%), and transferred to nitrocellulose membranes. The membranes were then blocked with 1% bovine serum albumin at room temperature for 1 h and then incubated with the indicated specific primary antibodies for 3 h. Proteins were visualized with Horseradish peroxidase (HRP)-conjugated secondary antibodies. To corroborate equal loading, membranes were stripped and reprobed using an antibody specific for β-actin. Finally, antigen-antibody complexes were detected using the ECL system.
A549 tumor xenograft
A total of 6×106 A549 cells were injected into the right flank of 30 BALB/c nude mice (supplied by the Experimental Animal Department of Binzhou medical College, Shandong, China). Seven days later, 25 mice with tumors ~100 mm3 in size were randomly distributed into the following five groups: Control group (PBS), low-dose metformin (40 mg/kg/day) group, high-dose metformin (200 mg/kg/day) group, cisplatin (5 mg/kg/day) group and metformin (40 mg/kg/day) plus cisplatin (5 mg/kg/day) group. Tumor volume (mean values and 95% confidence intervals) was measured every three days after the initial injection. After 18 days, the mice were sacrificed by cervical dislocation and the tumor weights were measured. The study was approved by the ethics committee of Binzhou Medical College (Binzhou, China).
Statistical analysis
All experiments, except that of the nude mice study, were repeated at least three times and the values are expressed as the mean ± standard deviation. Statistical significance was determined by Student's t-test. P<0.05 was considered to indicate a statistically significant difference.
Results
Metformin inhibits human lung carcinoma A549 survival in a dose- and time-dependent manner
The viabilities of A549 cells treated with different concentrations of metformin (0, 5, 10, 20 and 50 mmol/l) for different time periods (0, 24, 48 and 72 h) were determined using MTT assay. As shown in Fig. 1A, the cell viabilities were decreased in a dose- and time-dependent manner. Cisplatin is the most active treatment for lung carcinoma, however, the sensitivity of tumor cells to cisplatin varies. The present results indicated that metformin could increase the cytotoxicity of cisplatin in A549 cells (Fig. 1B).
Figure 1.
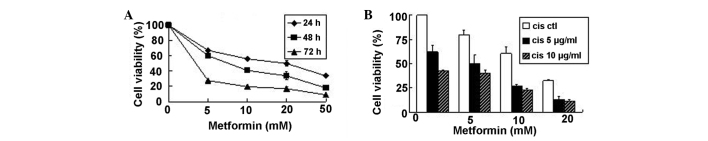
Effects of metformin on the proliferation of A549 cells. (A) Metformin inhibited the proliferation of the cells in a dose- and time-dependent manner. The A549 cells were exposed to increasing doses of metformin for 24, 48 and 72 h respectively, and cell proliferation was measured using the MTT assay. (B) The addition of metformin potentiated the cytotoxicity induced by cisplatin. A549 cells were cultured for 48 h in the presence of increasing amounts of cisplatin with and without metformin, and cell proliferation was measured using the MTT assay. The results are shown as the mean ± standard deviation of triplicate experiments. Ctl, control; cis, cisplatin.
Metformin induces cell cycle arrest in the G0/G1 phase in human lung adenocarcinoma cells
To identify the effect of metformin on the cell cycle of human lung carcinoma cells, the cell cycle distribution of A549 cells treated with different concentrations of metformin (0, 10 and 20 mmol/l) for 48 h was determined using a PI staining assay. As shown in Fig. 2, metformin induced cell cycle arrest in the G0/G1 phase in the A549 cells. Treatment with different doses of metformin (10 or 20 mM) for 48 h resulted in an increase in A549 cells in the G0/G1 population, to 65.9 and 82.6%, respectively, compared with 54.6% in the untreated control group (Fig. 2).
Figure 2.

Metformin blocks the cell cycle in the G0/G1 phase. Flow cytometry analysis of proliferating A549 cells 48 h after the treatment with metformin (10 and 20 mM). Fractions of cells in the G0/G1, S and G2/M phases of the cell cycle are indicted. Untreated cells were used as controls. *P<0.05 vs. control. CON, control; Met, metformin.
Metformin induces apoptosis in human lung adenocarcinoma cells
Hoechst-33342 and Annexin-V/PI staining assays were combined to investigate whether metformin can induce A549 cell apoptosis. First, the apoptotic morphology of the A549 cells treated with 10.0 mM metformin for 24 h was observed, and the metformin-treated cells were shown to manifest brighter, granular, blue fluorescence and more apoptotic bodies compared with control group (Fig. 3A). The percentage of apoptotic cells was determined by cell flow cytometric analysis following PI staining. Compared with the untreated cells, the cells treated with metformin for 48 h underwent apoptosis in a dose-dependent manner (Fig. 3B).
Figure 3.

Metformin induces apoptosis of A549 cells. (A) A549 cells were treated with 10 mM metformin for 48 h. Images of the cells were captured under a light microscope, or the cells were stained with Hoechst 33342 and visualized under a fluorescence microscope. (B) A549 cells were treated with metformin at different doses for 48 h and stained with Annexin V-propidium iodide. The percentage of apoptotic cells was measured by FACScan analysis (*P<0.05 vs. control; **P<0.01 vs. control). The results are shown as the mean ± standard deviation of triplicate experiments. CON, control; PI, propidium iodide; Met, metformin.
Metformin induces apoptosis mainly through the mitochondria-mediated pathway
The treatment with metformin decreased the expression of Bcl-2 and Bcl-extra large (Bcl-xl), and increased the expression of Bax in a dose-dependent manner in the A549 cells, as determined by western blot analysis (Fig. 4A). Downstream of the apoptosis signaling pathways, there is significant cleavage activation of caspase-3 and poly(ADP-ribose) polymerase (PARP) (Fig. 4B). A significant release of cytochrome c from the mitochondria to the cytosol was observed after the cells were treated with metformin (Fig. 4C). These results indicate that metformin induces apoptosis mainly through the mitochondria-mediated internal pathway in lung carcinoma cells.
Figure 4.
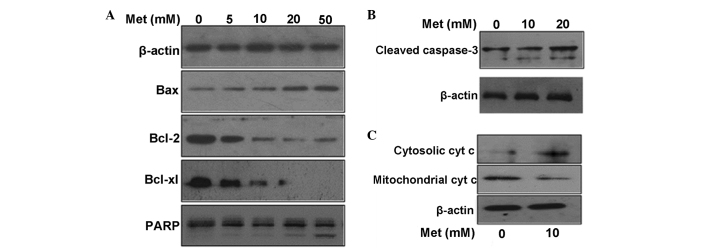
Metformin induces apoptosis of A549 cells mainly through the mitochondia-mediated pathway. (A) Effect of metformin on the expression of apoptosis-regulating proteins, as assessed by western blotting. Protein fractions of total cells were assessed for expression of Bcl-2, Bcl-xl, Bax and PARP. (B) Effect of metformin on the activation of caspase-3 in A549 cells. The antibody for caspase-3 recognized the cleaved form (17 and 19 kDa). (C) Effect of metformin on cytochrome c distribution in the mitchondria and cytosol. The mitchondria and cytosol were fractionated with a Cell Mitchondria Isolation kit. Data represent one of three independent experiments. β-actin was used as a loading control. Met, metformin; Bcl, B-cell lymphoma; Bcl-xl, Bcl-extra large; PARP, poly(ADP-ribose) polymerase.
Metformin suppresses tumor proliferation in vivo
In order to determine whether metformin could affect tumor growth, the effect of metformin on tumor growth inhibition was studied in vivo via intraperitoneal injection. It was found that tumor growth was markedly inhibited in the high-dose metformin and metformin plus cisplatin groups (Fig. 5A). Compared with the control group, the tumor xenograft treated with metformin (200 mg/kg/day) was signifiantly decreased in size (Fig. 5B). In addition, there was a signifiant decrease in tumor size in the metformin (40 mg/kg/day) plus cisplatin group, when compared with the other groups (Fig. 5C). The tumor volume of the high-dose metformin and metformin plus cisplatin groups was 611.4±51.5 and 281.6±21.1 mm3, respectively, compared with the control group (1,042.4±39.7 mm3). The tumor growth inhibition rate was 41.3% in the high-dose metformin group (P<0.01) and 72.9% in the metformin plus cisplatin group (P<0.01) compared with the control (Fig. 5D). Body weight reduction is an indicator of drug toxicity. In the high-dose metformin and metformin plus cisplatin groups, body weight was 22.31±1.69 and 23.83±2.39 g, respectively (Fig. 5E), which was comparable to the control group.
Figure 5.

Metformin decreases A549 xenograft tumor proliferation. (A) The final tumor weight at necropsy at 18 days post-administration. (B) Effect of metformin on the body weight of mice during 18 days of treatment. (C) Examples of tumor regression in metformin-treated and control (CON) animals: 1, animals of control group; 2, animals of metformin (200 mg/kg/day) group. (D) Tumor from A549 xenograft administered different treatments for 18 days: 1, control group; 2, metformin (40 mg/kg/day) group; 3, metformin (200 mg/kg/day) group; 4, cisplatin group; 5, metformin (40 mg/kg/day) plus cisplatin group. (E) Tumor growth kinetics during administration of metformin for 18 days. Data are presented as the mean ± standard deviation (n=5). **P<0.01 vs. control. DDP, cisplatin; Met, metformin.
Discussion
Metformin has a long history of human use, with less toxicity and a relatively low cost when compared with other antidiabetic drugs. The drug is now widely used as a first-line treatment for type II diabetes (16). Notably, numerous studies have indicated that metformin could protect patients with type II diabetes from cancer and inhibit cancer cell proliferation in vitro (17–22). According to the present study, metformin is a potent inhibitor of cell proliferation in the A549 cell line. For example, inhibition of cell proliferation was observed in the A549 cells treated with 20 mM metformin for 48 h, with a 63% decrease in cell viability. The study then analyzed whether metformin affects the cell cycle. To determine this, proliferating A549 cells were treated with metformin for 48 h at different concentrations. As shown in Fig. 2, an increasing number of cells accumulated in the G0/G1 phase. In parallel, a reduced percentage of cells was observed in the S and G2/M phases. In order to determine whether metformin induces apoptosis in A549 cells, an Annexin V-fluorescein isothiocyanate labeling assay and found that metformin at concentrations of <5 mM did not affect the amount of Annexin V-positive cells (data not shown). However, metformin at a concentration of >5 mM increased the percentage of cells positive for Annexin V. These results indicate that metformin can induce apoptosis, but only at high concentrations.
There are two classic apoptotic pathways in mammalian cells, namely the mitochondria-mediated apoptotic pathway and the death receptor-mediated apoptotic pathway, and there is cross-talk between the two. To further clarify the apoptotic molecular mechanisms of human lung carcinoma cells induced by metformin, the total proteins of the A549 cells treated with different concentrations of metformin for 24 h were analyzed using a western blot assay. Using the present data, it may be concluded that metformin induces the apoptosis of A549 cells in vitro mainly via the mitochondria-mediated internal pathway, which is initiated by a range of apoptosis-inducing signals that cause an imbalance in the major apoptosis regulators, the proteins of the Bcl-2 family, such as Bcl-2, Bcl-xl and Bax. Bax, a pro-apoptotic protein, accumulates on the mitochondria subsequent to being activated, and triggers the permeability of the outer mitochondrial membrane to increase. As a result, the mitochondria release cytochrome c. This, in turn, activates downstream death programs, such as caspase 3 and PARP. These observations suggested that modulation of the mitochondria-mediated pathway may be an important mechanism underlying the biological effects of metformin. The present study results are similar to those in the study by Gotlieb et al (23), which reported that metformin significantly inhibits the ovarian cancer cell line growth and potentiates the effects of cisplatin. Additionally, the inhibition of growth by metformin was partially abolished by the AMPK inhibitor, compound C. The study also discovered that metformin-induced anti-proliferation is associated with the phosphorylation of AMPK and the decrease of p-p70S6K (23). Another study found that metformin was able to induce apoptosis in vitro in colon cancer cells, but only in those cells that were p53-deficient (9). A subsequent study found that metformin in combination with paclitaxel resulted in mainly G2 arrest and decreased human telomerase reverse transcriptase mRNA expression. Metformin was demonstrated to potentiate the effects of paclitaxel in endometrial cancer cells by inhibiting cell proliferation and modulating the mTOR pathway (24). In another study, metformin was found to decrease high-fat-induced cardiac cell death by inhibiting ceramide synthesis. However, proton and lactate accumulation are induced by metformin, which leads to caspase-3-independent cell damage at high concentrations (25). These studies indicate that metformin mediates its effects through a range of mechanisms.
In vivo, the present data showed that the high-dose metformin and metformin plus cisplatin groups could significantly decrease A549 tumor growth without any significant side-effects, including weight loss, hair loss, dysphoria or lethargy. The present results are of particular significance since this is the first time that metformin has been shown to inhibit lung adenocarcinoma growth in a xenograft model. Similar to the present study, a study by Huang et al reported that metformin delayed the onset of tumors, but in mice deficient in the phosphatase and tensin homolog tumor suppressor (26). Ben Sahra et al found that metformin inhibited prostatic tumor growth in vivo (27). Another study also found that metformin could prevent pancreatic cancer in hamsters fed a high-fat diet and exposed to pancreatic carcinoma (8).
Thus, in summary, the present study is a preliminary study suggesting that metformin selectively induces the apoptosis of lung carcinoma A549 cells via the mitochondria-mediated internal pathway. Although metformin has been demonstrated to inhibit proliferation in vitro in prostate, breast, colon and ovarian cancer cell lines, the present study observed its effect on lung adenocarcinoma for the first time. The results support the development of pre-clinical experiments to further evaluate the potential role of combining metformin with chemotherapy as a new treatment for lung adenocarcinoma. Further long-term studies of metformin are required in patient populations of similar and larger sizes in order to confirm these observations.
Acknowledgements
This study was supported by a grant from the National Science and Technology Special Foundation for Major Infectious Diseases Prevention and Control (No. 2008Ex 10002019).
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Mulshine JL, Sullivan DC. Clinical practice. Lung cancer screening. N Engl J Med. 2005;352:2714–2720. doi: 10.1056/NEJMcp042630. [DOI] [PubMed] [Google Scholar]
- 3.Dowling RJ, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment. BMC Med. 2011;9:33. doi: 10.1186/1741-7015-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evans JM, Donnelly LA, Emslie-Smith AM, et al. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29:254–258. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 6.Lee MS, Hsu CC, Wahlqvist ML, et al. Type 2 diabetes increases and metformin reduces total, colorectal, liver and pancreatic cancer incidences in Taiwanese: a representative population prospective cohort study of 800,000 individuals. BMC Cancer. 2011;11:20. doi: 10.1186/1471-2407-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu Z, Jiang W, Thompson MD, et al. Metformin as an energy restriction mimetic agent for breast cancer prevention. J Carcinog. 2011;10:17. doi: 10.4103/1477-3163.83043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schneider MB, Matsuzaki H, Haorah J, et al. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology. 2001;120:1263–1270. doi: 10.1053/gast.2001.23258. [DOI] [PubMed] [Google Scholar]
- 9.Buzzai M, Jones RG, Amaravadi RK, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 10.Dowling RJ, Zakikhani M, Fantus IG, et al. Metformin inhibits mammalian target of rapamycin dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 11.Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS One. 2012;7:e33411. doi: 10.1371/journal.pone.0033411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ben Sahra I, Laurent K, Loubat A, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–3586. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 14.Zhuang Y, Miskimins WK. Cell cycle arrest in Metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1 and requires p27Kip1 or p21Cip1. J Mol Signal. 2008;3:18. doi: 10.1186/1750-2187-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin suppresses HER2 (erbB-2)oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle. 2009;8:88–96. doi: 10.4161/cc.8.1.7499. [DOI] [PubMed] [Google Scholar]
- 16.Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334:574–579. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- 17.Wang LW, Li ZS, Zou DW, et al. Metformin induces apoptosis of pancreatic cancer cells. World J Gastroenterol. 2008;14:7192–7198. doi: 10.3748/wjg.14.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–7511. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cantrell LA, Zhou C, Mendivil A, et al. Metformin is a potent inhibitor of endometrial cancer cell proliferation - implications for a novel treatment strategy. Gynecol Oncol. 2010;116:92–98. doi: 10.1016/j.ygyno.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodwin PJ, Stambolic V, Lemieux J, et al. Evaluation of metformin in early breast cancer: a modification of the traditional paradigm for clinical testing of anti-cancer agents. Breast Cancer Res Treat. 2011;126:215–220. doi: 10.1007/s10549-010-1224-1. [DOI] [PubMed] [Google Scholar]
- 21.Xiong Y, Lu QJ, Zhao J, Wu GY. Metformin inhibits growth of hepatocellular carcinoma cells by inducing apoptosis via mitochondrion-mediated pathway. Asian Pacific J Cancer Prev. 2012;13:3275–3279. doi: 10.7314/APJCP.2012.13.7.3275. [DOI] [PubMed] [Google Scholar]
- 22.Soranna D, Scotti L, Zambon A. Cancer risk associated with use of metformin and sulfonylurea in type 2 diabetes: a meta-analysis. Oncologist. 2012;17:813–822. doi: 10.1634/theoncologist.2011-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gotlieb WH, Saumet J, Beauchamp MC, et al. In vitro metformin anti-neoplastic activity in epithelial ovarian cancer. Gynecol Oncol. 2008;110:246–250. doi: 10.1016/j.ygyno.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 24.Hanna RK, Zhou C, Malloy KM, et al. Metformin potentiates the effects of paclitaxel in endometrial cancer cells through inhibition of cell proliferation and modulation of the mTOR pathway. Gynecol Oncol. 2012;125:458–469. doi: 10.1016/j.ygyno.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.An D, Kewalramani G, Chan JK, et al. Metformin influences cardiomyocyte cell death by pathways that are dependent and independent of caspase-3. Diabetologia. 2006;49:2174–2184. doi: 10.1007/s00125-006-0338-9. [DOI] [PubMed] [Google Scholar]
- 26.Huang X, Wullschleger S, Shpiro N, et al. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008;412:211–221. doi: 10.1042/BJ20080557. [DOI] [PubMed] [Google Scholar]
- 27.Ben Sahra I, Laurent K, Loubat A, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–3586. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]