

Abstract
The transcription factor PU.1 is a member of the ETS family, which is expressed in a wide variety of hematopoietic lineages. Accumulating evidence has indicated that PU.1 plays a key role in hematopoiesis, and reduced expression of PU.1 leads to the pathogenesis of human myeloid leukemia. As a multi-functional factor, PU.1 is also required for mixed lineage leukemia (MLL) stem cell potential and the development of MLL. However, the function of PU.1 in human non-MLL leukemia and its molecular mechanism remains poorly understood. In the present study, PU.1 siRNA was demonstrated to efficiently inhibit the transcription level of oncogene MEIS1 in the human acute myeloid non-MLL leukemia U937 cell line. In addition, PU.1, as a positive regulator of MEIS1, performed a crucial role in maintaining cell proliferation. Using electrophoretic mobility shift assay, chromatin immunoprecipitation analysis and luciferase reporter assay, previously unexplored evidence that PU.1 activated the MEIS1 promoter through a conserved binding motif in vitro and in vivo was further defined. Overall, the present study provides insight into the molecular mechanism of the contribution of PU.1 to the pathogenesis of non-MLL U937 cells, which is mediated by direct regulation of MEIS1 transcription. The present data reveal the possibility of developing an alternative therapy for non-MLL leukemia by targeting PU.1-mediated MEIS1 gene activation.
Keywords: PU.1, MEIS1, leukemia, transcription
Introduction
Hematopoiesis is a complex process that generates multiple lineages of various blood cell types with distinct functions (1). The hematopoietic stem cells (HSCs) constantly renew themselves to prevent exhaustion of the stem cell pool (2). Transcription factors play a pivotal role in this orchestrated process by manipulating the expression of lineage-specific genes. The ETS family member PU.1, encoded by SPI1, is one of the most important regulators involved in normal hematopoiesis, particularly in myeloid differentiation, as PU.1 regulates the expression of almost all myeloid genes, including granulocyte macrophage colony-stimulating factor (CSF) receptor α (GM-CSFRα), macrophage CSF receptor (M-CSFR) and granulocyte CSF receptor (G-CSFR) (3–5). PU.1 is required for the commitment and maturation of myeloid lineages, and the expression of PU.1 increases during granulocytic and monocytic differentiation (6,7). Previous studies have revealed that the introduction of PU.1 at high levels induced macrophage differentiation in primary fetal liver progenitors. Inversely, deficient PU.1 expression severely impaired hematopoietic development or led to leukemia (8). PU.1 knockout mice present an early block in myeloid differentiation and lack of mature myeloid cells (6), while graded reduction in PU.1 expression to 20% of wild-type expression has been demonstrated to induce acute myeloid leukemia (AML) in all mice (9). Accordingly, altered PU.1 function is possibly involved in leukemogenesis, as the PU.1 gene mutation has been described in certain patients with AML (10). Furthermore, certain oncogenic fusion proteins, such as AML1-eight twenty-one and promyelocytic leukemia-retinoic acid receptor α, are also associated with PU.1 inhibition (11–14). Thus, as a tumor suppressor, PU.1 expression facilitates commitment to myeloid differentiation and its downregulation may be crucial in the pathogenesis of AML.
However, PU.1 exerts various functions at distinct hematopoietic stages. Although PU.1 is expressed at low levels in the early stage of hematopoiesis, it has an indispensable function in the maintenance of the HSC pool (6,15). Loss of PU.1 expression in mice leads to a weakening in the self-renewal capacity of long-term HSCs, which are then outcompeted by normal HSCs in bone marrow (8). This role of PU.1 provides insights into possible processes occurring in leukemia stem cells. Previous studies have reported that PU.1 is also required for the initiation and maintenance of AML stem cells induced by monocytic leukemia zinc-finger protein fusion proteins (4). Similarly, PU.1 is the immediate cause for maintaining the leukemic phenotype in MEL cells by promoting repopulation of transformed erythroblastic cells and blocking the terminal differentiation program towards erythrocytes, which are also reversed by downregulation of the expression of PU.1 (6). Collectively, PU.1 is crucial for not only lineage differentiation, but also the leukemic process.
Previous studies have revealed that PU.1 is essential for leukemia harboring mixed lineage leukemia (MLL) gene rearrangements (16), which is characterized by high expression of the homeobox oncogene MEIS1 (17). In addition, there is a positive association between the expression of PU.1 and MEIS1 in MLL patients, and the regulation of MEIS1 by PU.1 is central to the pathogenesis of leukemia harboring MLL rearrangements (16). However, the function of PU.1 and its mechanism in non-MLL remains unclear. In the present study, in order to investigate the role of PU.1 in acute myeloid not harboring MLL rearrangements, the human acute myeloid leukemia U937 cell line was selected, as this cell line exhibits a relative higher expression level of endogenous MEIS1, compared to the other two non-MLL cell lines. Our current work reveals the regulatory function and molecular mechanism of PU.1, which facilitates the development of targeted therapies with potential to correct the inappropriate MEIS1 expression for non-MLL leukemia.
Materials and methods
Cell cultures and transfection
U937 cells (American Type Culture Collection, Manassas, VA, USA) were maintained in RPMI-1640 with 10% fetal bovine serum (FBS). 293T cells (American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS. PU.1 siRNA (accession no., NM_003120; catalog nos., SASI_Hs02_00335096, SASI_Hs02_00335097 and SASI_Hs02_00335098) and MEIS1 siRNA (accession no., NM_002398; catalog no., SASI_Hs01_00088989) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The cells were transfected with siRNA using X-treme GENE siRNA (Roche, Indianapolis, IN, USA), according to the manufacturer's instructions.
Viable cell count
A total of 45 µl single cell suspension was mixed with 5µl Trypan Blue (0.4%; Invitrogen Life Technologies, Carlsbad, CA, USA) and incubated for 5 min at room temperature. Next, the unstained (viable) cells were counted using a hemocytometer (Hausser Scientific, Horsham, PA, USA) under a light microscope (Professional Infinity Planachromatic Binocular Upright Microscope; VWR, Philadelphia, PA, USA).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total cellular RNA was extracted using TRIzol reagent (Invitrogen Life Technologies), according to the manufacturer's instructions. Reverse transcription was performed according to the manufacturer's instructions (Promega, San Luis Obispo, CA, USA). qPCR was performed using SYBR Green qPCR Master mix (Fermentas, Pittsburgh PA, USA) on a MyiQ thermocycler (Bio-Rad, Hercules, CA, USA). PGK, a housekeeping gene with constitutive expression, was used as an internal control to normalize the RNA level. The primer sequences used are listed in Table I.
Table I.
Primer sequences.
| Primer | Direction | Sequence | Usage |
|---|---|---|---|
| MEIS1 promoter upstream | F | 5′-TAAGACGCGACCTGTTATGGC-3′ | ChIP-qPCR |
| R | 5′-CCAGAATGCTAGAACCCGGA-3′ | ChIP-qPCR | |
| MEIS1 promoter | F | 5′-GCATTGTGTAAGACGCGACCTG-3′ | ChIP-qPCR |
| R | 5′-CGACCAGAATGCTAGAACCCGGAAG-3′ | ChIP-qPCR | |
| MEIS1 intron 1 | F | 5′-TGCTGACATACAGCGATCCC-3′ | ChIP-qPCR |
| R | 5′-CACTCACACTGGCAGGCTTG-3′ | ChIP-qPCR | |
| MEIS1 intron 2 | F | 5′-TCAGGATGCAATGGTGAGCA-3′ | ChIP-qPCR |
| R | 5′-TAAGGCCCTCATCACTCCCA-3′ | ChIP-qPCR | |
| PGK1 | F | 5′-AGAGCCCAGAGCGACCCTT-3′ | RT-PCR |
| R | 5′-AAAAGCCATTCCACCACCAAT-3′ | RT-PCR | |
| MEIS1 | F | 5′-ATGTGACAATTTCTGCCACCG-3′ | RT-PCR |
| R | 5′-CCTGAACGAGTAGATGCCGTG-3′ | RT-PCR | |
| PU.1 | F | 5′-GAGCCCCCCACTGGAGGT-3′ | RT-PCR |
| R | 5′-TGGTACAGGCGGATCTTCTTCT-3′ | RT-PCR | |
| GFI1 | F | 5′-GAGCCTGGAGCAGCACAAAG-3′ | RT-PCR |
| R | 5′-TCCCACAGATCTTACAGTCAAAGC-3′ | RT-PCR | |
| WT Probe | F | 5′-CCACTACTTCCGGGTTCTAGC-3′ | EMSA |
| R | 5′-GCTAGAACCCGGAAGTAGTGG-3′ | EMSA | |
| MT Probe | F | 5′-CCACTACGCGAGGGTTCTAGC-3′ | EMSA |
| R | 5′-GCTAGAACCCTCGCGTAGTGG-3′ | EMSA |
F, forward; R, reverse; WT, wild type; MT, point mutated; ChIP-qPCR, chromatin immunoprecipitation-quantitative polymerase chain reaction; RT-PCR, reverse transcription-PCR; EMSA, electrophoretic mobility shift assay.
Chromatin immunoprecipitation (ChIP)
The cells were cross-linked with 1% formaldehyde for 10 min at room temperature, and the reaction was subsequently stopped with 0.125 M glycine. The cells were washed with phosphate-buffered saline and then lysed in cell lysis buffer. The nuclei were recovered by centrifugation and then lysed in nuclear lysis buffer. Chromatin was sonicated and precleared overnight with 50 µl of rabbit immunoglobulin (Ig)G (catalog no., sc-3888; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and 40 µl of protein A/G-agarose (Invitrogen Life Technologies). Precleared lysate was incubated with 5 µg of purified rabbit anti-PU.1 antibody (catalog no., sc-352X; Santa Cruz Biotechnology, Inc.). An aliquot of precleared lysate (10%) was reserved as input. Immunoprecipitates were washed and eluted with 100 mM NaHCO3 and 1% SDS. Cross-links were reversed at 65°C for 12 h. RNA and protein were digested with RNase A and proteinase K. Isolated DNA was purified by MinElute Reaction Cleanup kit (Qiagen, Valencia, CA, USA). The amount of purified DNA was subjected to qPCR using SYBR Green Master Mix (Applied Biosystems, Grand Island, NY, USA). The data are shown as fold enrichment over input DNA. The primer sequences were listed in Table I.
Electrophoretic mobility shift assay (EMSA)
In total, 107 cells were harvested and resuspended with 400 µl cold buffer A, which consisted of 10 mM Hepes (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 0.5 mM phenylmethylsulfonyl fluoride (PMSF) and 0.5 mM DTT. Subsequent to being maintained on ice for 10 min, the cell suspension was centrifuged with 4,000 × g for 10 sec and the supernatant fraction was discarded. The pellet cells were resuspended in 80 µl cold buffer B, which consisted of 20 mM Hepes (pH 7.9), 25% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM PMSF and 0.5 mM DTT, in a 1.5 ml eppendorf tube and incubated on ice for 20 min for high-salt extraction. Cellular debris was removed by centrifugation at top speed (12,500 × g) for 30 min at 4°C, and the supernatant was reserved as nuclear extract.
Double-stranded probes were generated by annealing the following oligomers to their respective complementary sequences: Wild-type, 5′-CCACTACTTCCGGGTTCTAGC-3′; and point mutated, 5′-CCACTACGCGAGGGTTCTAGC-3′. Electrophoretic mobility shift assay (EMSA) was performed using the Lightshift Chemiluminescent EMSA kit (Thermo Fisher Scientific, Inc., Rockford, IL, USA), according to the manufacturer's instructions. For supershift bands, the same rabbit IgG or rabbit anti-PU.1 antibody were added to the EMSA reaction.
Luciferase reporter assay
The U937 cells were cultured in 12 well plates and transfected with 0.2 µg of luciferase reporter plasmids (pGL3 or pGL3-wild type MEIS1 promoter or pGL3-mutated MEIS1 promoter) using Fugene HD (Roche), following the manufacturer's instructions. Plasmid pCMV-LacZ was co-transfected as an internal control. The activity of β-galactosidase and luciferase was measured 48 h subsequent to transfection using Galacto-Light Plus (Applied Biosystems) and luciferase assay system (Promega), respectively. The luciferase activity of each sample was normalized to the β-galactosidase. The transfection was performed in triplicate wells and replicated with similar results in three independent experiments.
Results
PU.1 and MEIS1 each play a crucial role in the proliferation of human AML U937 cells
High expression of MEIS1 is one of the characterizations of leukemia harboring MLL gene rearrangements, whereas a limited expression level is generally demonstrated in leukemia without MLL gene fusion (17). In the present study, a panel of leukemic cell lines was initially compared using RT-qPCR, and the human AML U937 cell line was selected to explore the function of PU.1 and MEIS1 in leukemia without MLL gene rearrangements, as this cell line demonstrated a relatively increased expression of MEIS1 compared with the two other non-MLL cell lines (Fig. 1A). In the present study, which aimed to investigate the biological effects, three PU.1 short interfering sequences and one MEIS1 short interfering sequence were applied to knock down the expression of PU.1 and MEIS1. Subsequently, trypan blue staining and cell counting were used to assess the number of viable cells at 1–5 days after transfection. As shown in Fig. 1B, MEIS1 knock down markedly inhibited the rate of cell growth after 3 days, compared with the cells transduced with scrambled control siRNA. Notably, PU.1 exerted the similar function as suppressed cell proliferation with a one-day delay. These suggest that PU.1 and MEIS1 are each required for cell maintenance and MEIS1 may be a downstream gene of PU.1 in non-MLL leukemia.
Figure 1.
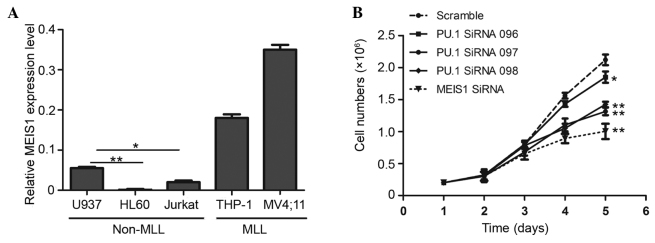
PU.1 and MEIS1 are essential for the proliferation of U937 cells. (A) Non-MLL leukemia cells in the study exhibit limited expression levels of MEIS1, as determined by reverse transcription-quantitative polymerase chain reaction. The U937 cell line demonstrated a relatively increased expression of MEIS1 compared with other non-MLL cell lines. The data were normalized, with PGK acting as an internal control. *P<0.05; **P<0.01. (B) Cell proliferation curve of PU.1 and MEIS1 knockdown cells. The U937 cells were transfected with PU.1 or MEIS1-specific siRNA or scramble siRNA. The cell numbers were counted at the indicated time points. The data are expressed as the mean ± standard deviation of three experiments. *P<0.05, **P<0.01 vs. scramble. MLL, mixed lineage leukemia; siRNA, small interfering RNA.
Deregulation of MEIS1 upon loss of PU.1 expression in the human U937 cell line
In order to confirm the regulatory function of PU.1 on MEIS1, the U937 cells were transfected with PU.1 siRNA. The RT-PCR results revealed that the knockdown was efficient, with a 50–80% reduction of PU.1 RNA expression in the cells transfected with PU.1 siRNA compared with the cells transfected with scrambled control siRNA. As expected, the downregulated PU.1 significantly inhibited leukemia oncogene MEIS1 expression in U937 cells (Fig. 2A). However, the expression of Gfi1, a well-known agonist gene of PU.1 (18), was markedly increased (Fig. 2B), and was considered to be the PU.1-knockdown experiment monitor control. The present data identified that PU.1 was positively involved in MEIS1 transcription in U937 cells.
Figure 2.
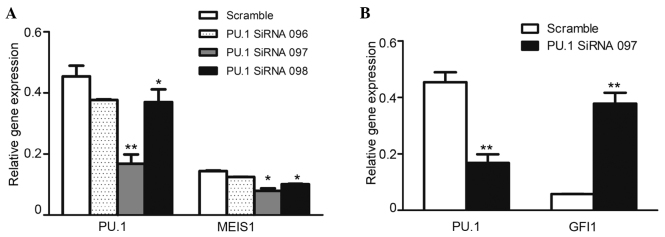
Knockdown of PU.1 downregulates the expression of MEIS1 and increases the expression of GFI1. PU.1 siRNA was transfected into the U937 cell line. Reverse transcription-quantitative polymerase chain reaction was used to analyze the gene expression level and knockdown efficiency of PU.1. The data were normalized to the level of PGK mRNA expression. The data are expressed as the mean ± standard error of three experiments, compared with the scramble siRNA control (*P<0.05; **P<0.01). siRNA, small interfering RNA.
PU.1 protein is enriched by the MEIS1 promoter locus in vivo
To understand the mechanism of transcriptional regulation of MEIS1 by PU.1, evolutionary conserved genomic sequences of the MEIS1 promoter (hg19 version) were identified using the UCSC Genome Browser (Genome Bioinformatics Group, University of California Santa Cruz, Santa Cruz, CA, USA). To test the possible recruitment of PU.1 to this conserved promoter region in vivo, ChIP-qPCR primers were designed to amplify various locations. As Fig. 3 revealed, marked enrichment of PU.1 at the promoter region of MEIS1 was detected, but no visible binding in upstream of the promoter and two intron regions were found. PU.1 significantly bound to the MEIS1 promoter region with ~10 fold enrichment over input DNA, indicating that MEIS1 may be directly regulated by PU.1 in the U937 cell line.
Figure 3.
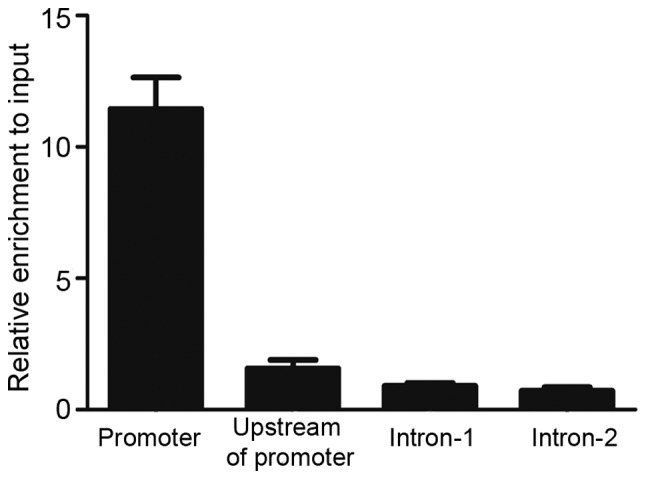
PU.1 is enriched in MEIS1 promoter regions in the U937 cell line. Crosslinked chromatin was immunoprecipitated with antibody against PU.1 and the precipitated DNA was amplified using primer pairs spanning various regions, as indicated. The data are expressed as enriched folds in different regions vs. the input DNA.
Predicted PU.1 binding site is essential for MEIS1 promoter activity
To identify whether the PU.1 binding region is of functional importance, the MEIS1 promoter (898 bp upstream to 2 bp downstream of transcription start site) was cloned into the pGL3-basic vector. Additional point mutation was performed by PCR mutagenesis (CTTCCG to CGCGAG) (Fig. 4A), based on the locus of the PU.1 enrichment peak determined by the present ChIP-qPCR data. The luciferase reporter assay was then performed in U937 cells transfected with pGL3-basic vectors inserted with the wild-type or mutated MEIS1 promoter region. The results revealed that the wild-type promoter was able to evidently increase the downstream luciferase activity by 30-fold (Fig. 4B), compared with the empty pGl3-basic vector. By contrast, mutating the PU.1 binding site entirely reduced this increase of the promoter activity. These data reveal that this binding site contributes strongly to the activity of the MEIS1 promoter and indicate that a key transcription factor exists at this promoter locus.
Figure 4.
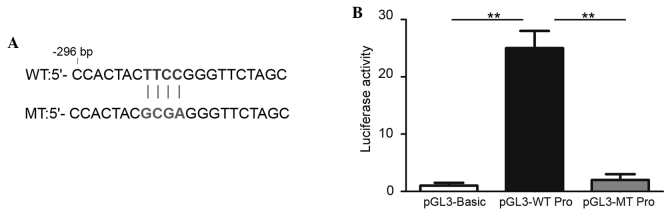
Predicted PU.1 binding site is required for the activity of the MEIS1 promoter. (A) The WT and MT sequences are reported. (B) The U937 cells were transfected with luciferase reporter gene vector pGL3-Basic, pGL3-WT MEIS1 promoter or pGL3-MT MEIS1 promoter. The luciferase assay was used to evaluate promoter activities. The data are expressed as the mean ± standard error of three experiments (**P<0.01). WT, wild type; MT, point mutated.
Functional MEIS1 promoter binding site is occupied by PU.1 protein
In vitro, it was determined that the transcription factor PU.1 bound to this bio-functional site in the MEIS1 promoter by EMSA with nuclear exacts from non-MLL leukemia cells. Using biotin-labeled oligonucleotide probes corresponding to the putative binding region between nucleotides −296 and −276 of the promoter (Fig. 4A), which is upstream of the MEIS1 transcription start site, specific bands were detected that were readily competed off with wild type cold probes, but not with mutated cold probes (Fig. 5A). In addition, the obtained bands were supershifted by PU.1-specific antibody, but not isotype IgG in 293T cells overexpressing PU.1 (Fig. 5B). Overall, the present data indicated that the regulatory function of PU.1 on MEIS1 is mediated by direct protein-DNA binding in the promoter region of the MEIS1 gene.
Figure 5.

PU.1 binds to the predicted binding sequence in the MEIS1 promoter region. (A) Nuclear extracts were prepared and incubated with biotin-labeled or cold probes containing the region −296 to −276 bp upstream of the MEIS1 transcription start site prior to gel electrophoresis. The specific binding bands were competed by cold WT probes, but not cold MT probes. (B) The PU.1 binding bands were super shifted by the antibody to PU.1, but not isotype IgG. WT, wild type; MT, point mutated; Ig, immunoglobulin.
Discussion
Numerous factors that activate the expression of MEIS1 genes in leukemia have been identified, such as the MLL fusion protein (19), Hoxa9 (20) and E74-like factor 1 (21). The majority of previous studies have focused on leukemia with MLL rearrangements (17,19,22,23), as this leukemia is characterized by high expression of the homeobox gene MEIS1. Downregulation of MEIS1 in MLL rearranged acute leukemia results in the reduced expression of genes associated with cell cycle entry and inhibition of cell proliferation (17), and also impairs engraftment (22), indicating that MEIS1 gene activation is a key event in leukemia with MLL rearrangements. Compared with the leukemia harboring MLL rearrangement, MEIS1 demonstrates decreased or limited expression in non-MLL rearranged leukemia, which confers certain challenges to the associated studies. At present, the regulation and bio-function of MEIS1 in leukemia without MLL rearrangement remains unknown. The present preliminary data reveals that the limited expression of MEIS1 also functions as an essential oncogene in the human acute leukemia U937 cell line, a non-MLL leukemia cell line. In addition, the present results provide evidence that the activity of MEIS1 is regulated tightly by the transcription factor PU.1.
The transcription factor PU.1 is a hematopoietic-specific ETS family member involved in the development of all hematopoietic lineages (6,8) and acts as an activator and repressor to regulate the transcription of various genes (24,25). Traditionally, PU.1 functions as a tumor suppressor in the majority of leukemia types. Dysregulation of PU.1 leads to loss of lineage development and leukemia in vitro and in vivo (9,26–28). Previous studies, however, have also demonstrated that PU.1 is required for the repopulation or self-renewal capacity of normal hematopoietic stem cells, and sustained PU.1 levels also balance cell cycle-associated regulators to prevent the exhaustion of adult HSC (8,15). These studies indicated that the presence of PU.1 activity may be required to favor the growth of myeloid leukemia stem cells. Previous studies have reported that PU.1 demonstrated an essential expression and activated a well-known oncogene MEIS1 pathway in MLL, accompanied by MEIS1 overexpression (16). In addition, the expression of the PU.1 gene and the survival rate appeared to be inversely associated in human AML samples with MLL rearrangement (16). Despite the requirement of PU.1 in the development of MLL, as a pro-tumor gene, the key function of PU.1 may not be limited to MLL in AML.
In the current study, it was identified that knockdown of PU.1 caused inhibition of cell proliferation in the human non-MLL U937 cell line and the function of PU.1 was mediated by MEIS1 transcriptional regulation. This result is consistent with the observation in MLL, indicating that PU.1 may function as a pro-tumor gene ubiquitously, or not specifically in MLL leukemia. In the present study, which aimed to investigate the regulatory mechanism, the activity of putative PU.1 binding site in MEIS1 promoter region as a positive regulatory motif in U937 cells was confirmed by the Luciferase report system. Importantly, the present data initially revealed that PU.1 exhibited strong enrichment in the MEIS1 promoter region using in vivo and in vitro assays. Notably, this PU.1 binding genomic locus in U937 cells varies from the regulation via intron regions in MLL (16), indicating that MEIS1 regulatory sites by PU.1 may be multiple in various type of leukemia.
Overall, the present study identifies that transcription factor PU.1 is required for cell proliferation in U937 cells and its biological function, at least in part, is mediated by regulating the expression of target oncogene MEIS1 directly. This indicates that the potential tumor activator effect of PU.1 may be a universal phenomenon and interference of PU.1 expression may be an alternative target for non-MLL acute myeloid leukemia treatment. The present finding may potentially lead to a novel direction for non-MLL studies. However, the roles of PU.1 in other non-MLL cell lines as well as other subtypes of leukemia remain to be addressed in the future.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (grant no., 81100381).
References
- 1.Orkin SH, Zon LI. Hematopoiesis: An evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kehrl JH. Hematopoietic lineage commitment: role of transcription factors. Stem Cells. 1995;13:223–241. doi: 10.1002/stem.5530130304. [DOI] [PubMed] [Google Scholar]
- 3.Gangenahalli GU, Gupta P, Saluja D, Verma YK, Kishore V, Chandra R, Sharma RK, Ravindranath T. Stem cell fate specification: Role of master regulatory switch transcription factor PU.1 in differential hematopoiesis. Stem Cells Dev. 2005;14:140–152. doi: 10.1089/scd.2005.14.140. [DOI] [PubMed] [Google Scholar]
- 4.Aikawa Y, Katsumoto T, Zhang P, Shima H, Shino M, Terui K, Ito E, Ohno H, Stanley ER, Singh H, et al. PU.1-mediated upregulation of CSF1R is crucial for leukemia stem cell potential induced by MOZ-TIF2. Nat Med. 2010;16:580–585. doi: 10.1038/nm.2122. 1p following 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Houston IB, Huang KJ, Jennings SR, DeKoter RP. PU.1 immortalizes hematopoietic progenitors in a GM-CSF-dependent manner. Exp Hematol. 2007;35:374–384. doi: 10.1016/j.exphem.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Mak KS, Funnell AP, Pearson RC, Crossley M. PU.1 and Haematopoietic Cell Fate: Dosage Matters. Int J Cell Biol. 2011;2011:808524. doi: 10.1155/2011/808524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedman AD. Transcriptional control of granulocyte and monocyte development. Oncogene. 2007;26:6816–6828. doi: 10.1038/sj.onc.1210764. [DOI] [PubMed] [Google Scholar]
- 8.Iwasaki H, Somoza C, Shigematsu H, Duprez EA, Iwasaki-Arai J, Mizuno S, Arinobu Y, Geary K, Zhang P, Dayaram T, et al. Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood. 2005;106:1590–1600. doi: 10.1182/blood-2005-03-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nutt SL, Metcalf D, D'Amico A, Polli M, Wu L. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. J Exp Med. 2005;201:221–231. doi: 10.1084/jem.20041535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renneville A, Roumier C, Biggio V, et al. Cooperating gene mutations in acute myeloid leukemia: A review of the literature. Leukemia. 2008;22:915–931. doi: 10.1038/leu.2008.19. [DOI] [PubMed] [Google Scholar]
- 11.Seshire A, Rößiger T, Frech M, et al. Direct interaction of PU.1 with oncogenic transcription factors reduces its serine phosphorylation and promoter binding. Leukemia. 2012;26:1338–1347. doi: 10.1038/leu.2011.331. [DOI] [PubMed] [Google Scholar]
- 12.Mueller BU, Pabst T, Fos J, et al. ATRA resolves the differentiation block in t (15;17) acute myeloid leukemia by restoring PU.1 expression. Blood. 2006;107:3330–3338. doi: 10.1182/blood-2005-07-3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vangala RK, Heiss-Neumann MS, Rangatia JS, et al. The myeloid master regulator transcription factor PU.1 is inactivated by AML1-ETO in t (8;21) myeloid leukemia. Blood. 2003;101:270–277. doi: 10.1182/blood-2002-04-1288. [DOI] [PubMed] [Google Scholar]
- 14.Wang K, Wang P, Shi J, et al. PML/RARalpha targets promoter regions containing PU.1 consensus and RARE half sites in acute promyelocytic leukemia. Cancer Cell. 2010;17:186–197. doi: 10.1016/j.ccr.2009.12.045. [DOI] [PubMed] [Google Scholar]
- 15.Staber PB, Zhang P, Ye M, et al. Sustained PU.1 levels balance cell-cycle regulators to prevent exhaustion of adult hematopoietic stem cells. Mol Cell. 2013;49:934–946. doi: 10.1016/j.molcel.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou J, Wu J, Li B, et al. PU.1 is essential for MLL leukemia partially via crosstalk with the MEIS/HOX pathway. Leukemia. 2014;28:1436–1448. doi: 10.1038/leu.2013.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar AR, Li Q, Hudson WA, Chen W, Sam T, Yao Q, Lund EA, Wu B, Kowal BJ, Kersey JH. A role for MEIS1 in MLL-fusion gene leukemia. Blood. 2009;113:1756–1758. doi: 10.1182/blood-2008-06-163287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahl R, Iyer SR, Owens KS, Cuylear DD, Simon MC. The transcriptional repressor GFI-1 antagonizes PU.1 activity through protein-protein interaction. J Biol Chem. 2007;282:6473–6483. doi: 10.1074/jbc.M607613200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeisig BB, Milne T, García-Cuéllar MP, Schreiner S, Martin ME, Fuchs U, Borkhardt A, Chanda SK, Walker J, Soden R, et al. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol Cell Biol. 2004;24:617–628. doi: 10.1128/MCB.24.2.617-628.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu YL, Fong S, Ferrell C, Largman C, Shen WF. HOXA9 modulates its oncogenic partner Meis1 to influence normal hematopoiesis. Mol Cell Biol. 2009;29:5181–5192. doi: 10.1128/MCB.00545-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiang P, Lo C, Argiropoulos B, et al. Identification of E74-like factor 1 (ELF1) as a transcriptional regulator of the Hox cofactor MEIS1. Exp Hematol. 2010;38:798–798. doi: 10.1016/j.exphem.2010.06.006. 808 e1-e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orlovsky K, Kalinkovich A, Rozovskaia T, et al. Down-regulation of homeobox genes MEIS1 and HOXA in MLL-rearranged acute leukemia impairs engraftment and reduces proliferation. Proc Natl Acad Sci USA. 2011;108:7956–7961. doi: 10.1073/pnas.1103154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong P, Iwasaki M, Somervaille TC, So CW, Cleary ML. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev. 2007;21:2762–2774. doi: 10.1101/gad.1602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta P, Gurudutta GU, Saluja D, Tripathi RP. PU.1 and partners: Regulation of haematopoietic stem cell fate in normal and malignant haematopoiesis. J Cell Mol Med. 2009;13:4349–4363. doi: 10.1111/j.1582-4934.2009.00757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dakic A, Metcalf D, Di Rago L, Mifsud S, Wu L, Nutt SL. PU.1 regulates the commitment of adult hematopoietic progenitors and restricts granulopoiesis. J Exp Med. 2005;201:1487–1502. doi: 10.1084/jem.20050075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenbauer F, Wagner K, Kutok JL, et al. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat Genet. 2004;36:624–630. doi: 10.1038/ng1361. [DOI] [PubMed] [Google Scholar]
- 27.Suraweera N, Meijne E, Moody J, Carvajal-Carmona LG, Yoshida K, Pollard P, Fitzgibbon J, Riches A, van Laar T, Huiskamp R, et al. Mutations of the PU.1 Ets domain are specifically associated with murine radiation-induced, but not human therapy-related, acute myeloid leukaemia. Oncogene. 2005;24:3678–3683. doi: 10.1038/sj.onc.1208422. [DOI] [PubMed] [Google Scholar]
- 28.Walter MJ, Park JS, Ries RE, Lau SK, McLellan M, Jaeger S, Wilson RK, Mardis ER, Ley TJ. Reduced PU.1 expression causes myeloid progenitor expansion and increased leukemia penetrance in mice expressing PML-RARalpha. Proc Natl Acad Sci USA. 2005;102:12513–12518. doi: 10.1073/pnas.0504247102. [DOI] [PMC free article] [PubMed] [Google Scholar]