

Abstract
Regardless of its cause, liver fibrosis is characterized by the excessive accumulation of extracellular matrix (ECM) in the liver. Hepatic stellate cells (HSCs) are the main producers responsible for the excessive production of ECM and profibrogenic cytokines in fibrotic liver. Therefore, development of HSC-specific delivery systems is essential for the success of antifibrotic agents. The objective of this study is to identify peptide ligands targeting the insulin like growth factor 2 receptor (IGF2R), which is overexpressed on HSCs. We expect to use the peptide ligands for the future development of HSC-targeted drug delivery system.
Protein- and whole cell-based phage display biopannings were conducted to identify phage/peptide candidates. Phage ELISA, cellular uptake and cell viability assay were employed to evaluate the binding affinity and specificity of these peptide ligands to recombinant human IGF2R and HSCs. IGF2R siRNA was used to silence the IGF2R protein expression in human hepatic stellate cells (LX-2) to confirm the specificity of the identified peptide ligands.
Among the identified peptide candidates, the peptide-431 shows the highest binding affinity and specificity to recombinant human IGF2R protein and HSCs. The equilibrium dissociation constant (Kd) of the peptide-431 is 6.19 μM for LX-2 cells and 12.35 μM for rat hepatic stellate cells HSC-T6. Cellular uptake of the peptide-431 in LX-2 cells is significantly reduced after silencing IGF2R with siRNA. The peptide-431 also enhances the uptake of a proapoptotic peptide (KLA peptide) in LX-2 and HSC-T6 cells, indicating that the peptide-431 can be used as a targeting ligand to deliver antifibrotic agents into not only rat but also human HSCs. Dimerization of the peptide-431 further increase its binding affinity to LX-2 cells by approximately nine-fold.
Keywords: liver fibrosis, HSCs, IGF2R, peptide ligand, phage display
Introduction
Liver fibrosis is a global health problem and one of the leading causes of morbidity and mortality in western developed countries. Caused by chronic liver damages, such as hepatitis, alcohol abuse, and nonalcoholic steatohepatitis, liver fibrosis is characterized by abnormal accumulation of extracellular matrix (ECM) in the liver.[1, 2] Various therapeutic agents including small molecular antifibrotic molecules, oligonucleotides and siRNA have been studied for treating liver fibrosis.[3–5] For example, we recently developed an siRNA targeting the poly(rC) binding protein 2 (PCBP2) gene, which is overexpressed and responsible for the stabilization of the collagen α1(I) mRNA in liver fibrogenesis.[5] However, the siRNA has to be specifically delivered to its target cells to exert its therapeutic effect with minimum toxicity in other tissues. Activation of hepatic stellate cells (HSCs) is the crucial step of fibrogensis because HSCs are the main producers responsible for the excessive production of ECM and profibrogenic cytokines in fibrotic liver.[6–9] Therefore, targeted delivery of antifibrotic agents to activated HSCs is essential for the success of liver fibrosis therapy.[4, 10, 11]
The insulin-like growth factor 2 receptor (IGF2R), also known as cation-independent mannose-6-phosphate receptor (M6PR), is a member of the IGF signaling system. IGF2R is a 300 kDa glycoprotein containing three domains, the cytoplasmic domain, transmembrane domain and extracellular domain.[12] The major function of IGF2R is to regulate lysosomal enzymes such as growth factor IGF2 by transporting them into lysosomes, followed by digestion by lysosomal acid hydrolases. IGF2R is expressed in HSCs, and its expression is upregulated during liver fibrogenesis.[13] Moreover, IGF2R can internalize extracellular ligands, and therefore it can be adopted as a target receptor for HSC-specific drug delivery.[11, 14]
Targeted drug delivery, especially active targeting, has attracted a great deal of attention in the past three decades.[15] Various types of targeting moieties, such as antibodies,[16] peptides,[17] aptamers[18] and other small moieties have been employed for active drug targeting.[19] Compared with other targeting moieties, peptides are promising targeting ligands because of their high binding affinity, ease of synthesis, less immunogenicity, and flexibility in chemical conjugation. As a result, peptide ligands have been explored as targeting ligands for a wide variety of drug delivery systems.[17, 20] Peptide ligands can be discovered by phage display technology.[17, 21] The phage biopanning technology is a valuable tool to identify peptide ligands against proteins, cells, or tissues. For example, we recently discovered a prostate cancer cell LNCaP-specific peptide using a whole cell biopanning procedure.[17]
In this study, a combinational biopanning strategy was conducted to identify IGF2R-specific peptides using recombinant human IGF2R protein and rat hepatic stellate cells HSC-T6. The advantage of this combinational biopanning strategy is that it can identify an IGF-2R specific peptide that can also specifically recognize the IGF-2R protein within the content of the complex cell-surface landscape of hepatic stellate cells. The identified peptide shows high affinity and specificity to human and rat hepatic stellate cells. The peptide ligand can also deliver a fused apoptotic peptide to hepatic stellate cells, indicating its promising potential for HSC-specific drug delivery systems.
Materials
The M13 phage display peptide library (Ph.D.™− 12) and bacteria cells ER2738 were purchased from New England Biolabs (Beverly, MA). Recombinant human IGF2R extracellular domain protein and anti-human IGF2R antibody were obtained from R&D SYSEMS® (Minneapolis, MN). Non-enzymatic cell dissociation solution was purchased from MP Biomedicals (Solon, Ohio). All peptides were ordered from United biosystems (Herndon, VA). Cell culture reagents were purchased from Mediatech, Inc. (Manassas, VA) and Thermo Fisher Scientific, Inc. (Pittsburgh, PA).
Cell culture
The rat hepatic stellate cell line (HSC-T6) and spontaneously immortalized human hepatic stellate cell line (LX-2) were kindly provided by Dr. Scott L. Friedman (Mount Sinai School of Medicine, New York University). HSC-T6 and LX-2 cells were cultured in DMEM medium containing 10% Fetal Bovine Serum (FBS), 100 units/mL penicillin and 100 μg/mL streptomycin. All cells were grown at 37°C in a humidified atmosphere containing 5% CO2. The culture medium was changed every other day, and the cells were passaged when they reached 80–90% confluence.
Phage display biopanning
A combinational phage biopanning procedure was conducted using recombinant human IGF2R extracellular domain protein (the 1st, 3rd and 5th rounds) and rat hepatic stellate cells HSC-T6 (the 2nd and 4th round) as shown in Figure 1. Briefly, 1 × 1011 pfu phages from the Ph.D 12 phage library were incubated with immobilized IGF2R protein in a 96-well plateat 4 °C for 2 h under shaking. Unbound phages were removed by washing the immobilized IGF2R protein with PBST (0.1% Tween 20) for three times. Bound phages were recovered by adding 1 mL of elution buffer (0.2 M Glycine-HCL, pH 2.2) and then neutralized with 150 μL of 1 M Tris-HCl (pH 9.1). The recovered phages were then amplified for next round biopanning. The whole-cell biopaning was performed as previously reported.[17] Briefly, HSC-T6 cells were detached with ice cold PBS containing 5 mM EDTA and then suspended in DMEM medium containing 1% BSA at a density of 1 × 107 cells/mL. 2.0 × 1010 phages from the previous round were incubated with the cells at 4 °C for 1 h under shaking. After incubation, 200 μL organic phase composed of dibutyl phthalate and cyclohexanol (9:1, v/v) was added to the cell suspension and centrifuged at 4 °C for 10 min. The bottom of the microcentrifuge tube, which contains the cell/phage complex, was cut off after snap freezing in liquid nitrogen. The bound phages were recovered by infecting ER2738 bacterial cells, followed by amplification for next round biopanning.
Figure 1. Combinational phage biopanning.
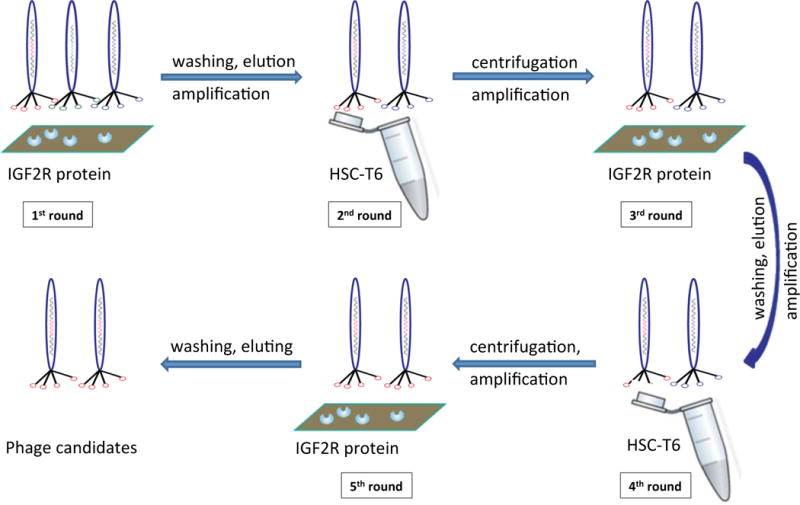
The first, third, and fifth rounds of biopanning were conducted on recombinant human IGF2R protein, while the second and fourth rounds of biopanning were conducted against rat hepatic stellate cells HSC-T6.
Phage DNA sequencing and phage ELISA
Individual phage clones were randomly selected and cultured in 1 mL ER2738 bacteria (growing in early stage) at 37 °C for 4.5 h. Phage DNA was extracted, purified using the DNA clean system (Promega, Madison, WI), and then sequenced with an ABI Genetic Analyzer 3100 (Applied biosystems, USA). The primer used for sequencing is 5′-CCCTCATAGTTAGCGTAACG-3′. The encoded peptide sequence was deduced from the DNA sequence.
Phages expressing inserted peptide were selected for phage ELISA. One microgram of recombinant human IGF2R protein was coated on 96-well ELISA plates at 4°C overnight under gentle shaking. PBS containing 5% BSA was then added for blocking at room temperature for 1 h. 1 × 1010 pfu phages of each phage clone were added and incubated with the IGF2R protein at room temperature for 1 h. After washing to remove the unbound phages, the HRP-conjugated M13 antibody was added and incubated for 1 h. TMB substrate was then added, and the absorbance at 450 nm was measured subsequently with a Beckman DTX 880 multimode Detector (Beckman coulter, Inc., Brea, CA).
Cells detachment study
Human hepatic stellate cells LX-2 were detached using 0.25% trypsin or non-enzymatic cell dissociation solution. The detached cells were incubated with 10 μM 5-FAM labeled peptide-425, peptide-431 and peptide-515 at 37 °C for 1 h under gentle rotation. After washing, the cells were suspended in PBS (500 μL) and subjected to fluorescence analysis on a FACScalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ) using 488-nm laser excitation and a 535-nm emission filter.
Cellular uptake of the selected peptides
LX-2 and HSC-T6 cells were detached using the non-enzymatic cell dissociation solution and suspended in Opti-MEM medium at a density of 1 × 106 cells/mL. Five hundred microliters of the detached cells were incubate with 10 μM 5-FAM labeled peptides at 37 °C under gentle rotation for different time intervals (15 min, 30 min and 60 min). The cells were then subjected to fluorescence analysis using the FACScalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ) as described above.
Binding affinity to hepatic stellate cells
LX-2 and HSC-T6 cells were detached using non-enzymatic cell dissociation solution and suspended in Opti-MEM medium at 1 × 106 cells/mL. 5-FAM labeled peptide-431 at different concentrations were incubated with the cells at 37°C for 1 h under gentle rotation. The Cells were subjected to fluorescence analysis using the FACScalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ) as described above. The Kd values were calculated using GraphPad Prism.
Knockdown of IGF2R using siRNA
Cells were transfected with anti-IGF2R siRNA as previously reported.[22, 23] Briefly, LX-2 cells (2 × 104 cells/well) were seeded in 24-well plates 16 h before the transfection. The anti-IGF2R siRNA was transfected at a final concentration of 10 nM and 50 nM using Lipofectamine® RNAIMAX following the manufactory’s instruction. Twenty-four hours after the transfection, the cells were harvested and total RNA was isolated using GenElute™ Mammalian Total RNA Miniprep Kit (Sigma-Aldrich, Saint Louis MO). The sequences of IGF2R siRNA are 5′- UAUAGAGCAAGCCUGGUCU-3′ (sense strand) and 5′-AGACCAGGCUUGCUCUAUA-3′ (antisense strand) as reported.[24]
Real-time PCR
Real-time PCR was performed using iTaq™ universal SYBR® Green one-step kit (BIO-RAD). The primers used for the study were as follows: hIGF2R 5′-TGT CTC CAT AGA CCT CAC ACC ACT-3′ (forward primer) and 5′-GCT GAA ACC CTG AGC TGC ATT CAT-3′ (reverse primer). 18s ribosomal RNA was used as an internal control and the primers are 5′-GTCTGTGATGCCCTTAGATG-3′ (forward primer) and 5′-AGCTTATGACCCGCACTTAC-3′ (reverse primer).
Western blot
LX-2 cells were transfected with the IGF-2R siRNA for 48 h as described above. The cells were washed with cold PBS and then incubated with ice-cold RIPA buffer containing protease inhibitor cocktail. After 5 minutes incubation on ice, cell lysate was recovered by centrifugation at 4 °C for 20 minutes at 12,000g. The total protein concentration was determined using BCA assay. Equal amounts of total proteins (20 μg) were resolved on a 6% SDS-PAGE gel. The separated proteins were transferred to a PVDF membrane using wet transfer method overnight at 4 °C. The membrane was blocked with 5% non-fat milk at 4°C for 12 h and probed with anti-human IGF-2R monoclonal antibody. The protein was then visualized using horseradish peroxidase conjugated anti-mouse secondary antibody. Images were taken using the FluorChem HD2 Chemiluminescent imaging system (Alpha Innotech, Santa Clara, CA) after incubating with the chemiluminnescent reagent (Millipore Corporation, Billerica, MA) at room temperature for 5 minutes. The same membrane was probed with anti-β-actin antibody as an internal control.
Specificity of the peptide-431 to IGF2R
LX-2 cells were seeded in 4-well champers and transfected with the IGF-2R siRNA for 48 h. The cells were washed with DPBS and incubated with 10 μM peptide-431 at 37 °C for 1h. The cells were then fixed with 10% buffered formalin phosphate solution at room temperature for 10 min, followed by washing with DPBS three times. The cells were mounted in one drop of the VECTASHIELD® Mounting medium with DAPI and imaged with a laser scanning confocal microscope (Leica TCS SP5). LX-2 cells were seeded in 6-well plate and transfected with siRNA for 48 h. The cells were detached by non-enzymetic cells dissociation solution and incubated with 10 μM peptide-431 at 37 °C for 1h. The labeled cells were determined by flow cytometry as described above.
Serum protein binding study
LX-2 cells were detached by non-enzymatic cell dissociation solution and suspended in Opti-MEM medium, FBS-free DMEM medium, 2% FBS DMEM medium and 10% FBS DMEM medium, respectively. The suspended cells were incubated with peptide-431 for 1 h and then subjected to fluorescence analysis using the FACScalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ) as described above.
Cell viability assay
LX-2 and HSC-T6 (1×104 cells per well) cells were cultured in 96-well plates for 12 h in DMEM medium containing 10% FBS. The peptide-431/KLA fusion peptide, the mixture of peptide-431 and KLA peptide, and KLA peptide were incubated with the cells for 48 h. Cell viability was measured using MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay.
Statistics analysis
Data were expressed as the mean ± standard deviation (SD). Difference between any two groups was determined by Tukey’s Multiple Comparison Test following ANOVA. P<0.05 was considered statistically significant.
Results
Phages biopanning
In order to identify peptides that can bind to IGF2R on human and rat hepatic stellate cells, a combinational phage biopanning procedure was conducted. As shown in Figure 1, the first, third, and fifth rounds of biopanning were conducted on recombinant human IGF2R protein, while the second and fourth rounds of biopanning were conducted against rat hepatic stellate cells HSC-T6. In each round, approximately 1011 pfu of phages were incubated with the target, and bound phages were eluted, titered, amplified and then used for the next round of biopanning. Figure 2 shows the number of bound phages after each round of biopanning. After three rounds (1st, 3rd, and 5th rounds) of biopanning on the recombinant human IGF2R protein, the phages from the 5th round pool exhibit much higher affinity to the IGF2R protein compared to the 1st round phages.
Figure 2. The number of recovered phages after each round of biopanning.

Results are represented as the mean ± SD (n=3).
Twenty-eight phage clones were randomly selected after the 4th and 5th rounds of biopanning, followed by amplification and DNA sequencing. As shown in Figure 3A, 16 of the selected phages encode the same peptide sequence, which is named as the peptide-515. One of the phages is peptide insertless. Binding affinity of the selected phage clones encoding inserted peptides was evaluated by ELISA on recombinant human IGF2R protein (Figure 3B). Compared to the control phage, which is peptide insertless, all the selected phages exhibited higher binding affinity.
Figure 3. Sequence (A) and binding efficacy (B) of randomly selected phage clones on human recombinant IGF2R protein.
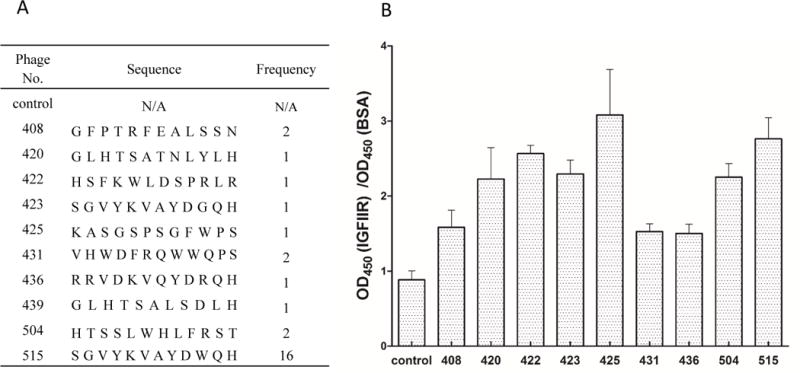
Phage clones were randomly selected after the 4th and 5th round of biopanning. Binding of the phages on IGF2R and BSA was examined with phage ELISA, and the binding ration was calculated. Results are represented as the mean ± SD (n=3).
Cellular uptake of the identified peptides
We next selected the six phages (420, 422, 425, 431, 504, and 515) which show the highest binding affinity to the IGF2R protein and synthesized the encoded peptides for following affinity studies in human and rat hepatic stellate cells. It is known that cell detachment method may change cell condition and thereafter affect cellular uptake of various molecules including peptides. For example, cells treated by non-enzymatic cell dissociation solution show higher peptide uptake compared to trypsinized cells.[25] As a result, we examined cellular uptake of the 5-FAM labeled peptides (425, 431, and 515) in human hepatic stellate cells LX-2 upon the treatment with non-enzymatic cell dissociation solution and trypsin. As Figure 4A&C showed, all the three peptides exhibited higher cellular uptake in non-enzymatic cell dissociation solution, which is consistent with a previous report.[25] We also evaluated the effect of incubation time on cellular uptake of the peptides. 5-FAM labeled peptide-431 was incubated with LX-2 cells for different incubation time points. Cellular uptake of the peptide exhibited a time-dependent increase (Figure 4B&D). Therefore, non-enzymatic cell dissociation solution and one-hour incubation time were selected to evaluate all the peptides (420, 422, 425, 431, 504, and 515) in human hepatic stellate cells LX-2 (Figure 5A&C) and rat hepatic stellate cells HSC-T6 (Figure 5B&D). On both cell lines, the peptide-431 exhibited the highest binding affinity compared to the control peptide and other selected peptides. In addition, the peptide-431 showed much higher cellular uptake in LX-2 (62%) than HSC-6 (12%) cells. This could be due to the fact that the human recombinant IGF2R protein was used in the biopanning procedure, and therefore the peptide ligand has higher binding affinity to IGF2R protein on human HSC than rat HSC.
Figure 4. The effect of cell detachment method (A) and incubation time (B) on cellular uptake of the identified peptides.
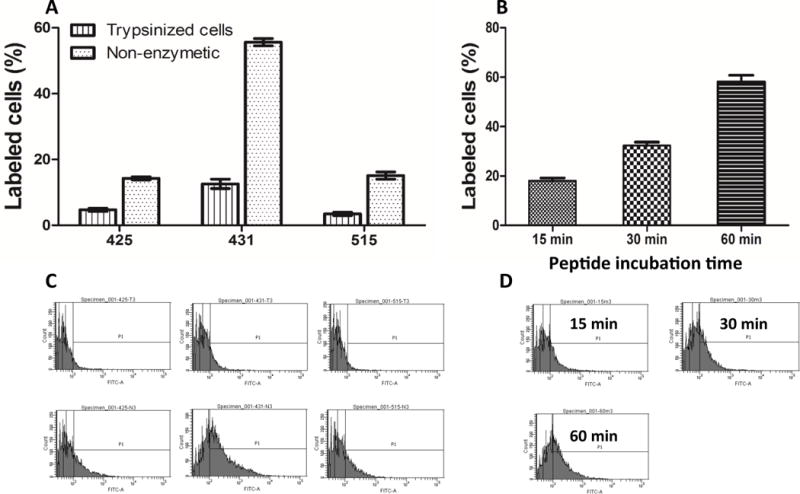
LX-2 cells detached with non-enzymatic cell dissociation solution and trypsin were incubated with 10 μM 5-FAM labeled peptides at 37 °C for 1 hour. The labeled cells were analyzed by flow cytometry after washing with PBS. Results are represented as the mean ± SD (n=3). C: cellular uptake of the identified peptides in LX-2 cells detached with trypsin (up) and non-enzymatic cell dissociation solution (Bottom).
Figure 5. Cellular uptake of the selected peptides in hepatic stellate cells LX-2 (A&C) and HSC-T6 (B&D).
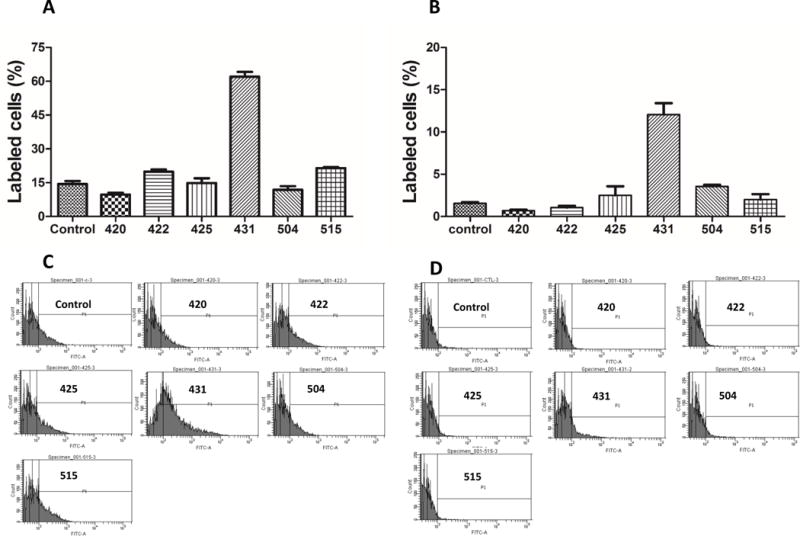
LX-2 and HSC-T6 cells were detached using non-enzymatic cell dissociation solution and then incubate with 10 μM 5-FAM labeled peptides at 37 °C for 1 hour. The labeled cells were analyzed by flow cytometry after washing with PBS. Results are represented as the mean ± SD (n=3).
We next measured the equilibrium dissociation constants (Kd) of the peptides (422, 425, 431, and 515) to LX-2 (Figure 6A) and HSC-T6 (Figure 6B) cells. The cells were incubated with 5-FAM labeled peptides at different concentrations for 1 hour at 37°C. The labeled cells were detected by flow cytometry, and the Kd value was calculated using GraphPad Prism. As Figure 6 demonstrated, the peptide-431 showed the lowest Kd (6.19 μM) in LX-2 cells. It also showed a comparable Kd (12.35 μM) in HSC-T6 cells, suggesting its promising potential as a targeting ligand for both preclinical animal study and future clinical evaluation. The other three peptides (422, 425, and 515) also showed good binding to LX-2 cells. The result is consistent with the cellular uptake result in Figure 5.
Figure 6. Equilibrium dissociation constants (Kd) of the selected peptides in LX-2 (A) and HSC-T6 (B) cells.
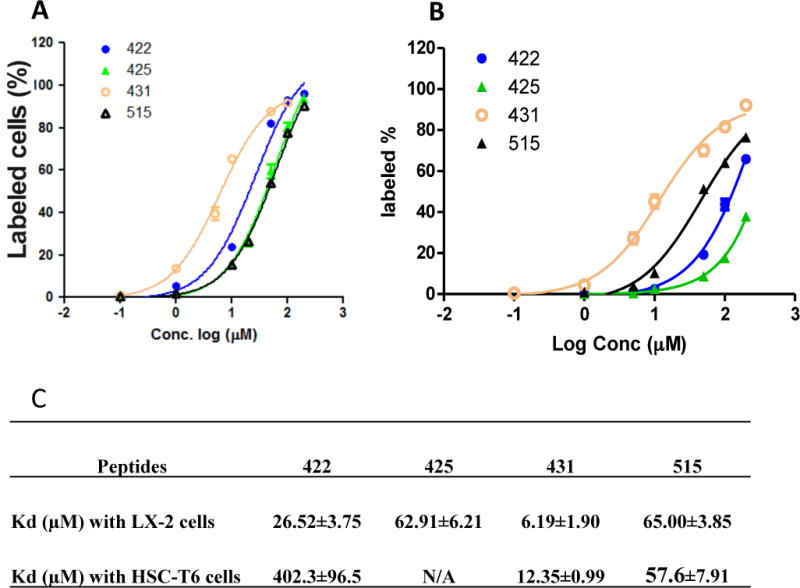
The cells were incubated with 5-FAM labeled peptides at different concentrations at 37°C for 1 hour. The labeled cells were detected by flow cytometry, and the Kd value was calculated using GraphPad Prism. Results are represented as the mean ± SD (n=3).
Specificity of the peptide-431 to IGF2R
In addition to affinity, we also evaluated the specificity of the peptide-431 to IGF2R in LX-2 cells. For this purpose, the expression of IGF2R in LX-2 cells was silenced using siRNA. As shown in Figure 7A&B, the IGF2R siRNA (10 and 50 nM) dramatically knocked down the expression of IGF2R in LX-2 cells at the mRNA and protein levels. After transfection with the siRNA for 24 hours, the mRNA expression of IGF2R was silenced by more than 85%, and the protein expression was silenced to almost negligible levels. Accordingly, the IGF2R siRNA treated cells exhibited lower uptake of the 5-FAM labeled peptide-431 than the cells treated with scrambled siRNA (Figure 8A&B). This result suggests high specificity of the peptide-431 to IGF2R on LX-2 cells.
Figure 7. Knockdown of IGF2R using siRNA.
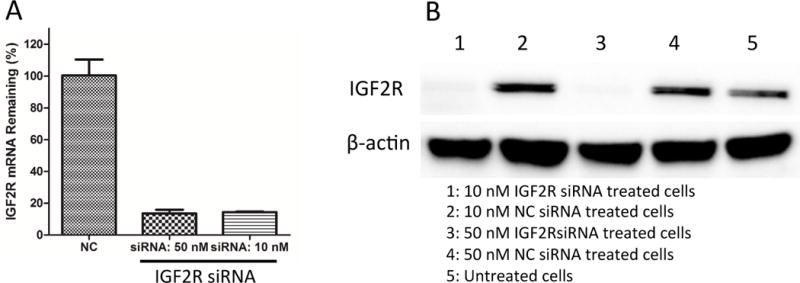
IGF2R siRNA silences IGF2R in LX-2 cells at both the mRNA (A) and protein levels (B). Results are represented as the mean ± SD (n=3).
Figure 8. Specificity of the peptide-431 to IGF2R.
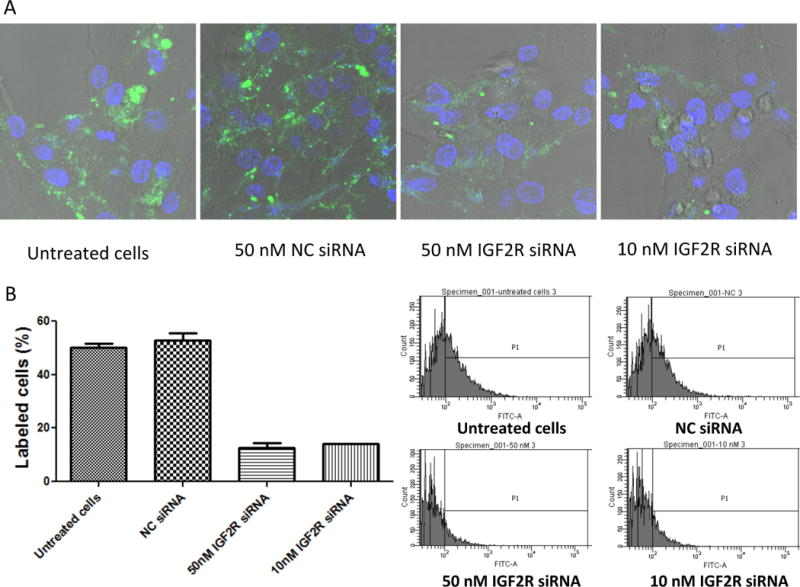
LX-2 cells were treated with the IGF2R siRNA and then incubated with the 5-FAM labeled peptide-431. Cellular uptake of the peptide was evaluated using confocal imaging (A) and flow cytometry (B). Results are represented as the mean ± SD (n=3).
Serum protein binding study
It is important to evaluate the stability of peptides in the presence of serum proteins, which may nonspecifically bind to IGF2R-specific peptides.[26] As a result, we evaluated the cellular uptake of 5-FAM labeled peptide-431 in LX-2 cells in the serum-reduced medium Opti-MEM and DMEM media containing different concentrations of FBS. As illustrated in Figure 9, cellular uptake of the peptide-431 was not significantly affected by the FBS up to 10%, indicating good stability of the peptide in the presence of serum protein.
Figure 9. Serum protein binding of the peptide-431.
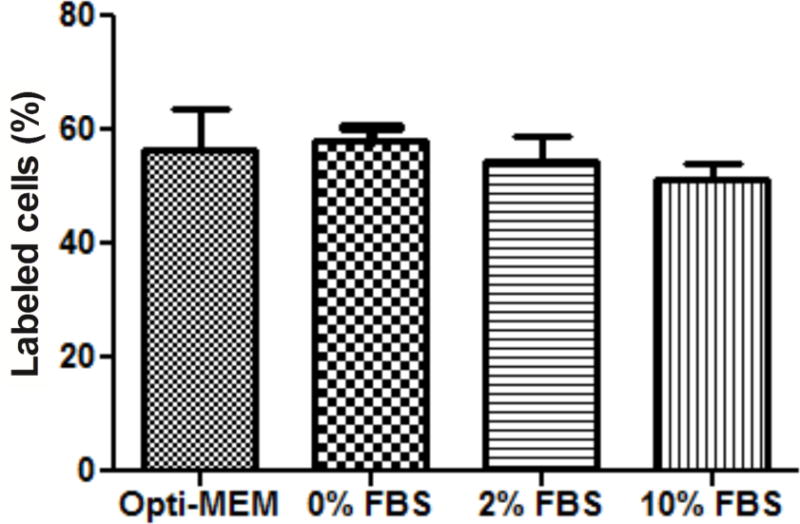
LX-2 cells were detached by non-enzymatic cell dissociation solution and then incubated with the 5-FAM labeled peptide-431 in Opti-MEM medium, and DMEM media containing different concentrations of FBS 37 °C for 1 hour. The labeled cells were analyzed by flow cytometry after washing with PBS. Results are represented as the mean ± SD (n=3).
The peptide-431 enhances the uptake and apoptotic effect of a pro-apoptotic peptide
The objective of this study is to identify an IGF2R-specific peptide ligand that can be used to deliver various anti-fibrotic agents to hepatic stellate cells. Therefore, it is essential to demonstrate that the peptide-431 can efficiently deliver a cargo into hepatic stellate cells. The pro-apoptotic peptide KLAKLAKKLAKLAK (KLA) is able to trigger mitochondrial disruption and induce cell death. However, the KLA peptide itself cannot enter cells to exert its pro-apoptotic activity.[17, 27] We therefore prepared a KLA/peptide-431 fusion peptide and examined its pro-apoptotic activity in LX-2 and HSC-T6 cells (Figure 10). As Figure 10A demonstrated, the KLA peptide alone did not exhibit apoptotic activity due to its inability to enter the cells by itself. Similarly, the mixture of KLA and peptide-431 also did not exhibit apoptotic activity in LX-2 cells. By contrast, the KLA/peptide-431 fusion peptide induced significant cell death in LX-2 cells, indicating that the peptide-431 mediates the cellular uptake of the fusion peptide in LX-2 cells. Similar results were observed in HSC-T6 cells (Figure 10B). These results clearly suggest the promising potential of using the peptide-431 as a targeting ligand to mediate cellular uptake of therapeutic agents.
Figure 10. The peptide-431 enhances the uptake and apoptotic effect of a pro-apoptotic peptide in LX-2 (A) and HSC-T6 (B) cells.
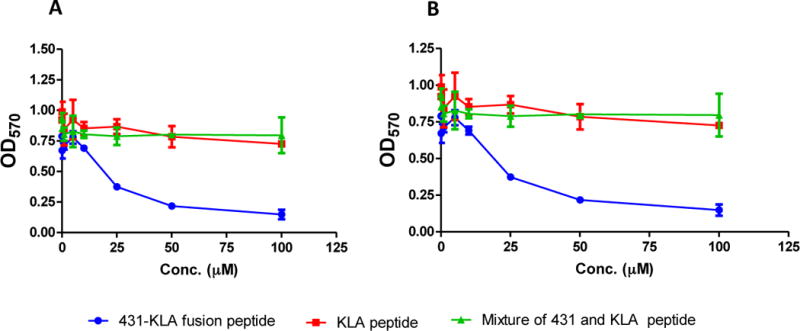
The peptide-431/KLA fusion peptide, the mixture of peptide-431 and KLA peptide, and KLA peptide were incubated with the cells for 48 h. Cell viability was measured using MTT assay. Results are represented as the mean ± SD (n=3).
Dimerization of the peptide-431 improves its binding affinity
In order to improve the binding affinity of the peptide-431, two peptide-431 were linked to a lysine to form the dimeric peptide-431 (Figure 11A). Affinity of the dimeric peptide-431 was evaluated in LX-2 cells as described above. As shown in Figure 11B, Kd value of the dimeric peptide-431 is 700.9 nM, which is approximately nine-fold lower compared to the monomeric peptide-431. This result is in agreement with a previous observation in which a dimeric peptide improves its binding affinity to prostate-specific membrane antigen (PSMA).[28]
Figure 11. Dimerization of the peptide-431 improves its binding affinity to LX-2 cells.
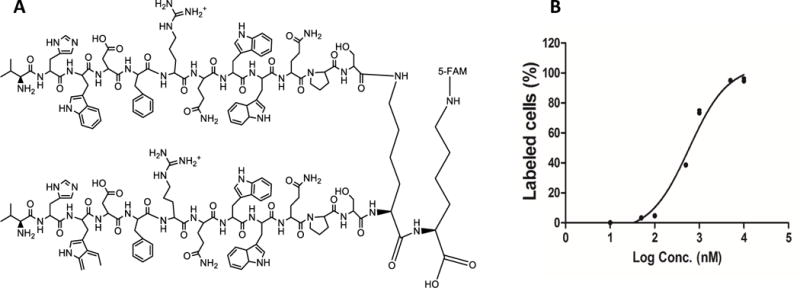
(A) Structure of the dimerized peptide-431. (B) Equilibrium dissociation curve of the dimeric peptide-431 in LX-2 cells. Results are represented as the mean ± SD (n=3).
Discussion
In this study, we identified an IGF2R-specific peptide using a novel combinational biopanning strategy. The selected peptide-431 exhibited a high and specific binding to IGF2R on human and rat HSCs. Moreover, the peptide-431 can deliver a proapoptotic peptide to HSCs and induced cell death, indicating the tremendous potential of using the peptide-431 as a targeting ligand for HSC-specific drug delivery system.
Phage display has been used for peptide ligand identification for various targets. Biopanning of a phage display library can be conducted on recombinant proteins, whole cells or animal tissues. Most of the biopannings are only performed either on a specific protein or on a specific cell line. In this study, we developed a novel biopanning strategy which combines protein-based biopanning with cell-based biopanning to identify IGF2R-specific peptides for HSC targeted drug delivery. Protein-based biopanning is easily controlled and there is no interference from other impurities, which guarantees high specificity of identified ligands. However, a recombinant protein may have a different conformation from its native structure on the cells. Therefore, peptide ligands identified from protein-based biopanning may not bind to the same target on its cells. By contrast, peptide ligands identified from cell-based biopanning can effectively bind to the native target on the cells. The disadvantage of cell-based biopanning is that there are many other proteins, lipids and carbohydrates on cell surface, leading to interfere with the phage biopanning.[17] Therefore, we combined the protein-based biopanning and cell-based biopanning in this study to achieve high specificity as well as binding affinity to IGR2R on hepatic stellate cells. Although the ultimate goal is to identify a peptide ligand that can be used for future clinical study, the peptide ligand should also have a comparable binding affinity to rat IGF2R so that it can be evaluated in preclinical animal studies. For this purpose, we selected human recombinant IGF2R and rat hepatic stellate cell HSC-T6 in the combinational biopanning.
IGF2R is an essential receptor to regulate the mannose-6-phosphate tagged lysosomal enzymes by transferring the enzymes from the secret sites to lysosome/endosome. The 300-kD protein contains 15 extracellular domains, which are the main binding sites for IGF2, M6P and M6P analogues. The domains 3, 5, and 9 form the binding pocket for M6P.[29] To avoid interfering with IGF2R’s native function, the recombinant human IGF2R containing the domains 10–14 was used for the biopanning. We performed a BLAST search (http://blast.ncbi.nlm.nih.gov) to find regions of similarity between human and rat amino acid sequences in the IGF-2R domains 10–14. The BLAST result shows a high similarity between human and rat IGF2R extracellular domain. Approximately 83% amino acids are identical in the hit and the query, and 92% of the amino acids are very similar to each other.
After biopaining, phage ELISA and cellular uptake were conducted to identify several IGF2R-specific peptide ligands. Among them, the peptide-431 shows the highest binding affinity not only in human but also in rat hepatic stellate cells (Figures 5 & 6). The binding affinity of the peptide-431 (Kd 6.19 μM) to LX-2 cells is comparable to the affinity of pentamannose-6-phosphate (PMP) to IGF2R (relative binding affinity IC50 = 5.6 μM). By contrast, the affinity of M6P (relative binding affinity IC50 = 23 μM) to IGF2R is relatively lower.[30, 31] In addition, chemical synthesis of M6P and its analogs is complicated, which limits its application as a targeting ligand for drug delivery systems.[32, 33] Therefore, the peptide-431 is more suitable for HSC targeted drug delivery due to the ease of synthesis and conjugation.
Not only affinity but also specificity is critical for the successful application of a targeting ligand. We therefore evaluated whether binding of the peptide-431 to hepatic stellate cells is mediated by IGF2R (Figure 8). For this purpose, we silenced IGF2R in hepatic stellate cells and then examined cellular uptake of the peptide-431. After knocking down IGF2R, cellular uptake of the peptide-431 was dramatically reduced compared to the control group. This result indicates that the peptide-431 binds to IGF2R on the surface of hepatic stellate cells. We also demonstrated that the peptide-431 is able to deliver the pro-apoptotic KLA peptide into hepatic stellate cells to induce cell death (Figure 10). All these results suggest that the peptide-431 could be used as an IGF2R-specific ligand to deliver various anti-fibrogic agents to hepatic stellate cells.
In general, binding affinity of peptide ligands is lower compared to antibodies and aptamers. However, there are several strategies to improve the binding affinity of peptide ligands. One strategy is to change the stereochemistry of a peptide ligand. For example, a D-amino acid peptide ligand showed a 10-fold improvement in affinity compared to its L-amino acid counterpart.[34] Another strategy is dimerization or trimerization of a peptide ligand. A PSMA-specific peptide was dimerized, and the affinity to PSMA was dramatically enhanced.[28] In our study, binding affinity of the dimeric peptide-431 was improved by 9-fold in comparison to the monomeric peptide-431 (Figure 11). The enhanced binding affinity could be due to the dimmerized extracellular domain of IGF2R.[35] In our future studies, we will evaluate whether different dimerization methods can further enhance its binding affinity.
IGF2R is highly expressed not only in fibrotic HSCs but also in several cancers. For example, IGF2R was found highly expressed in hepatocellular carcinoma (HCC) tissues[36] and melanoma.[37] Therefore, the IGF2Rspecific peptide-431 may be used as a targeting ligand for HCC, melanoma, and brain cells.
In conclusion, we have successfully identified an IGF2R-specific peptide using combinational phage biopanning against human recombinant IGF2R protein and rat hepatic stellate cells HSC-T6. The peptide-431 exhibits high and specific binding to IGF2R on human and rat hepatic stellate cells. In addition, the peptide-431 mediates cellular uptake of an apoptotic peptide in hepatic stellate cells. All the results suggest that the peptide-431 can be a promising ligand for targeted delivery of various anti-fibrotic agents to hepatic stellate cells.
Acknowledgments
This work was supported by an award (1R01AA021510) from the National Institute of Health.
References
- 1.Friedman SL. Liver fibrosis – from bench to bedside. J Hepatol. 2003;38(Suppl 1):S38–53. doi: 10.1016/s0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 2.Cheng K, Mahato RI. Gene modulation for treating liver fibrosis. Crit Rev Ther Drug Carrier Syst. 2007;24(2):93–146. doi: 10.1615/critrevtherdrugcarriersyst.v24.i2.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Negri AL. Prevention of progressive fibrosis in chronic renal diseases: antifibrotic agents. J Nephrol. 2004;17(4):496–503. [PubMed] [Google Scholar]
- 4.Ye Z, et al. Receptor-mediated hepatic uptake of M6P-BSA-conjugated triplex-forming oligonucleotides in rats. Bioconjug Chem. 2006;17(3):823–30. doi: 10.1021/bc060006z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shukla RS, et al. PCBP2 siRNA reverses the alcohol-induced pro-fibrogenic effects in hepatic stellate cells. Pharm Res. 2011;28(12):3058–68. doi: 10.1007/s11095-011-0475-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li D, Friedman SL. Liver fibrogenesis and the role of hepatic stellate cells: new insights and prospects for therapy. J Gastroenterol Hepatol. 1999;14(7):618–33. doi: 10.1046/j.1440-1746.1999.01928.x. [DOI] [PubMed] [Google Scholar]
- 7.Cohen-Naftaly M, Friedman SL. Current status of novel antifibrotic therapies in patients with chronic liver disease. Therap Adv Gastroenterol. 2011;4(6):391–417. doi: 10.1177/1756283X11413002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng K, et al. Biodistribution and hepatic uptake of triplex-forming oligonucleotides against type alpha1(I) collagen gene promoter in normal and fibrotic rats. Mol Pharm. 2005;2(3):206–17. doi: 10.1021/mp050012x. [DOI] [PubMed] [Google Scholar]
- 9.Wallace MC, Friedman SL. Hepatic fibrosis and the microenvironment: fertile soil for hepatocellular carcinoma development. Gene Expr. 2014;16(2):77–84. doi: 10.3727/105221614X13919976902057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng K, et al. Enhanced hepatic uptake and bioactivity of type alpha1(I) collagen gene promoter-specific triplex-forming oligonucleotides after conjugation with cholesterol. J Pharmacol Exp Ther. 2006;317(2):797–805. doi: 10.1124/jpet.105.100347. [DOI] [PubMed] [Google Scholar]
- 11.Ye Z, et al. Targeted delivery of a triplex-forming oligonucleotide to hepatic stellate cells. Biochemistry. 2005;44(11):4466–76. doi: 10.1021/bi047529j. [DOI] [PubMed] [Google Scholar]
- 12.Morgan DO, et al. Insulin-like growth factor II receptor as a multifunctional binding protein. Nature. 1987;329(6137):301–7. doi: 10.1038/329301a0. [DOI] [PubMed] [Google Scholar]
- 13.Beljaars L, et al. Albumin modified with mannose 6-phosphate: A potential carrier for selective delivery of antifibrotic drugs to rat and human hepatic stellate cells. Hepatology. 1999;29(5):1486–93. doi: 10.1002/hep.510290526. [DOI] [PubMed] [Google Scholar]
- 14.Gary-Bobo M, et al. Mannose 6-phosphate receptor targeting and its applications in human diseases. Curr Med Chem. 2007;14(28):2945–53. doi: 10.2174/092986707782794005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shukla RS, Chen Z, Cheng K. Strategies of drug targeting. In: Mitra AK, Lee CH, Cheng K, editors. Advanced Drug Delivery. WILEY; 2013. pp. 105–122. [Google Scholar]
- 16.Liu Y, et al. Gadolinium-loaded polymeric nanoparticles modified with Anti-VEGF as multifunctional MRI contrast agents for the diagnosis of liver cancer. Biomaterials. 2011;32(22):5167–76. doi: 10.1016/j.biomaterials.2011.03.077. [DOI] [PubMed] [Google Scholar]
- 17.Qin B, et al. Identification of a LNCaP-specific binding peptide using phage display. Pharm Res. 2011;28(10):2422–34. doi: 10.1007/s11095-011-0469-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhar S, et al. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci U S A. 2008;105(45):17356–61. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pan X, Lee RJ. Tumour-selective drug delivery via folate receptor-targeted liposomes. Expert Opin Drug Deliv. 2004;1(1):7–17. doi: 10.1517/17425247.1.1.7. [DOI] [PubMed] [Google Scholar]
- 20.Shukla RS, Qin B, Cheng K. Peptides used in the delivery of small noncoding RNA. Mol Pharm. 2014;11(10):3395–408. doi: 10.1021/mp500426r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228(4705):1315–7. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 22.Qin B, et al. Development of cholesteryl peptide micelles for siRNA delivery. J Control Release. 2013;172(1):159–68. doi: 10.1016/j.jconrel.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qin B, Cheng K. Silencing of the IKKepsilon gene by siRNA inhibits invasiveness and growth of breast cancer cells. Breast Cancer Res. 2010;12(5):R74. doi: 10.1186/bcr2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rose PP, et al. Insulin-like growth factor II receptor-mediated intracellular retention of cathepsin B is essential for transformation of endothelial cells by Kaposi’s sarcoma-associated herpesvirus. J Virol. 2007;81(15):8050–62. doi: 10.1128/JVI.00249-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richard JP, et al. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278(1):585–90. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 26.Sanghvi AB, et al. Biomaterials functionalization using a novel peptide that selectively binds to a conducting polymer. Nat Mater. 2005;4(6):496–502. doi: 10.1038/nmat1397. [DOI] [PubMed] [Google Scholar]
- 27.Mai JC, et al. A proapoptotic peptide for the treatment of solid tumors. Cancer Res. 2001;61(21):7709–12. [PubMed] [Google Scholar]
- 28.Aggarwal S, et al. A dimeric peptide that binds selectively to prostate-specific membrane antigen and inhibits its enzymatic activity. Cancer Res. 2006;66(18):9171–7. doi: 10.1158/0008-5472.CAN-06-1520. [DOI] [PubMed] [Google Scholar]
- 29.Bohnsack RN, et al. Cation-independent mannose 6-phosphate receptor: a composite of distinct phosphomannosyl binding sites. J Biol Chem. 2009;284(50):35215–26. doi: 10.1074/jbc.M109.056184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeanjean A, et al. Synthesis and receptor binding affinity of carboxylate analogues of the mannose 6-phosphate recognition marker. Bioorg Med Chem. 2006;14(10):3575–82. doi: 10.1016/j.bmc.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 31.Berkowitz DB, et al. Mono- and bivalent ligands bearing mannose 6-phosphate (M6P) surrogates: targeting the M6P/insulin-like growth factor II receptor. Org Lett. 2004;6(26):4921–4. doi: 10.1021/ol0479444. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Chen G. Chemical synthesis of N-linked glycans carrying both mannose-6-phosphate and GlcNAc-mannose-6-phosphate motifs. J Org Chem. 2011;76(21):8682–9. doi: 10.1021/jo2010999. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y, et al. Synthesis of novel bivalent mimetic ligands for mannose-6-phosphate receptors. Bioorg Med Chem Lett. 2013;23(8):2328–31. doi: 10.1016/j.bmcl.2013.02.068. [DOI] [PubMed] [Google Scholar]
- 34.Zhou N, et al. Exploring the stereochemistry of CXCR4-peptide recognition and inhibiting HIV-1 entry with D-peptides derived from chemokines. J Biol Chem. 2002;277(20):17476–85. doi: 10.1074/jbc.M202063200. [DOI] [PubMed] [Google Scholar]
- 35.Ghosh P, Dahms NM, Kornfeld S. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol. 2003;4(3):202–12. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]
- 36.Zhou Q, et al. Development of IGF signaling antibody arrays for the identification of hepatocellular carcinoma biomarkers. PLoS One. 2012;7(10):e46851. doi: 10.1371/journal.pone.0046851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prakash J, et al. Tumor-targeted intracellular delivery of anticancer drugs through the mannose-6-phosphate/insulin-like growth factor II receptor. Int J Cancer. 2010;126(8):1966–81. doi: 10.1002/ijc.24914. [DOI] [PubMed] [Google Scholar]