

Abstract
Adoptive immunotherapy, or the infusion of lymphocytes, is a promising approach for the treatment of cancer and certain chronic viral infections. The application of the principles of synthetic biology to enhance T cell function has resulted in substantial increases in clinical efficacy. The primary challenge to the field is to identify tumor-specific targets to avoid off-tissue, on-target toxicity. Given recent advances in efficacy in numerous pilot trials, the next steps in clinical development will require multicenter trials in order to establish adoptive immunotherapy as a mainstream technology.
Keywords: adaptive immunity, adoptive immunotherapy, gene transfer, chimeric antigen receptor
Introduction
Adoptive immunotherapy, or cell therapies, is undergoing a period of growth and enthusiasm following encouraging data of clinical efficacy. Virus- directed cell therapies are under investigation for the treatment of chronic viral infections like HIV and for viruses that cause morbidity and mortality in the immunosuppressed setting of bone marrow transplantation. In addition, cell therapies are poised to take a prominent role in both hematologic malignancies and solid tumors. Here we will review the history and rationale of immunotherapy and advances in understanding the principles of T cell transfer that are thought to impact clinical results. We will also discuss strategies and methods that are important in developing appropriate, effective, reliable, and scalable culture systems. Our current understanding of methodologies of engineering cells to re-direct them to specific targets, endowing immune cells with additional functions and safety features, and combining cells with other immune and targeted therapies is discussed in this review (Figure 1). Finally, we will illustrate how immune monitoring and biomarkers can determine the effects and fate of cell therapies in the clinical setting, and conclude with a brief discussion on what elements will be required to establish a new pillar of medical treatments built around personalized cell therapies.
Figure 1. Adoptive transfer of autologous, genetically engineered, in vitro–expanded T cells: The “seed,” the “soil” and the “fertilizer”.
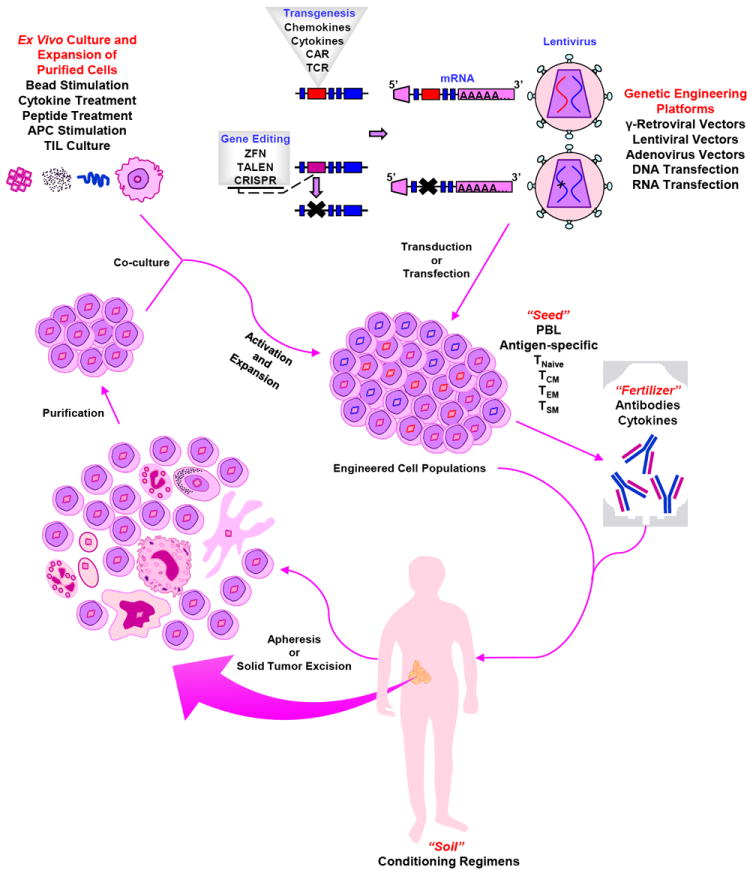
Autologous cells are harvested from the patient by apheresis. Following purification, cells undergo polyclonal in vitro activation and expansion, as well as genetic modification to form the “seed.” Engineered cell populations (“seeds”) are re-infused (“planted”) into the pre-conditioned patient (“soil”), along with antibodies and/or cytokines (“fertilizer”).
History and rationale for adoptive immunotherapy
Given the abilities of T cells to recognize and kill target cells, it is not surprising that most investigations of adoptive T cell therapy have targeted chronic viruses and cancer.
Viruses
Cell and gene therapy strategies have been proposed from the earliest days of the HIV epidemic (1) (2). The first clinical use of chimeric-antigen receptor (CAR) modified T cells was in HIV infection. In this setting, the CAR was composed of the receptor for the HIV envelope protein, namely the extracellular and transmembrane portions of the CD4 protein, fused to the TCR zeta signaling molecule (CD4z CAR). The proposed mechanism of action was for transduced T cells to lyse HIV-envelope expressing T cells. Between 1998 and 2005, three clinical studies evaluated the CD4z CAR expressed in autologous CD4+ and CD8+ T cells via a retroviral vector in subjects with active viremia (3) or in T cell–reconstituted patients with chronic HIV-1 infection (4). These studies showed that infusion of re-directed T cells was feasible and safe; in addition, T cells trafficked to reservoirs of infection (mucosa) and had modest effects on viremia. A decade later, analysis of the data collected from these protocols in a long-term follow-up study demonstrated the safety of retroviral modification of human T cells and the long-term persistence of CAR-modified T cells, with an estimated half-life of at least 17 years (5). This study added to the literature indicating that T cells were not as susceptible to retrovirus-mediated insertional mutagenesis as hematopoietic stem cells. In 2009, the remarkable story of the “Berlin patient” was published (6); this was the first report of a patient being functionally cured of HIV infection following an allogeneic hematopoietic stem cell transplant for acute myelogenous leukemia. The donor was homozygous for the CCR5 Δ32 mutation, which confers genetic resistance to HIV infection. This has challenged the field to develop cell-therapy based approaches that do not require myeloablative chemotherapy or allogeneic donors. One approach has been to develop gene therapy strategies to reduce CCR5 expression, either through shRNA encoded by lentiviral vectors (7) or through gene-editing strategies using zinc-finger nucleases (ZFN) to disrupt the CCR5 gene in T cells (8). In these cases, autologous gene-modified T cells are reinfused with the goal of reconstituting the T cell repertoire in HIV-infected patients. Interpretation of T cell effects on viremia and control of HIV may be affected by ongoing treatment with highly active anti-retroviral therapy (HAART), and carefully designed trials with scheduled, carefully monitored, treatment interruptions are underway.
Patients with hematologic malignancy undergoing allogeneic bone marrow transplantation are also at high risk for viral illness, particularly from reactivation of chronic viruses such as CMV, EBV, and HHV6; primary adenovirus infection can also cause acute and severe illness in this immunocompromised population. Although pharmacologic treatments for these viruses are available, they often have limited efficacy, must be administered recursively, and have significant side effects. For these reasons, several transplant centers have focused on developing donor-derived virus-specific T cells that can be administered as a donor lymphocyte infusion, either prophylactically or as treatment (9) (10). Because of the limitations in approaching healthy donors and single-patient manufacturing lots of virus-specific T cells, some centers have developed ‘third-party’ T cell banks derived from a panel of donors selected to span the most common HLA alleles (11) (12) (13). The Baylor group has pioneered the use of T cell lines that are specific for three to five viruses simultaneously, and have administered these to patients either as donor-derived or as third-party derived lymphocyte infusions (11, 14-16). Importantly, the incidence and severity of graft vs host disease has been limited or tolerable in all of these studies. These forms of adoptive immunotherapy are the most clinically advanced, with publication of Phase II, multicenter trials 11.
Cancer
Immunotherapy for cancer has a long and somewhat checkered history; the first observations of immune system engagement having anti-tumor effects are often attributed to William Coley, who observed regression of sarcoma following severe bacterial infections in the 1890s (17). However, it was the seminal finding that allogeneic immune reconstitution after bone marrow transplant had anti-leukemic effects (18) that definitively identified the anti-cancer effects of immune cells. Allogeneic bone marrow transplant remains the most potent, widely available form of cellular immune therapy and offers curative potential for hematologic malignancies. Researchers soon noted that the major mediators of the graft-vs-leukemia effect were T cells (19), while a contribution by NK cells was noted later (20) but can also be quite potent (21). There is a strong rationale to combine T cell therapy with NK cell therapy because NK cells do not cause graft vs host disease while they may limit resistance to T cell therapy through the emergence of MHC class I deficient tumor cells.
In the case of relapse after allogeneic transplantation, withdrawal of immunosuppressive therapy and/or donor lymphocyte infusions (DLI) are considered standard therapies but do have the potential to cause or worsen graft vs host disease. Ex vivo activation and culture of donor lymphocytes has also been clinically evaluated and appears to have modest benefit over standard DLI (22), particularly in hematologic malignancies aside from chronic myeloid leukemia (CML). Although CML was formerly one of the most common indications for transplant, it tends to be the most response to immune manipulations such as DLI; in the modern era, it is most often treated with tyrosine kinase inhibitors such as imatinib, dasatinib, and nilotinib. The limitation of these inhibitors is that they are expensive and while they result in long term remissions in most cases, they are not curative. The major opportunity for research in CML is to combine targeted agents such as kinase inhibitors with adoptive transfer therapy, with the goal of developing a curative regimen.
In solid tumors, investigators have hypothesized that tumor-infiltrating lymphocytes are the result of a naturally-occurring, yet ineffective, T cell response to the tumor. The observation of tumor-infiltrating lymphocytes (TILs) has spawned three forms of immune-based clinical interventions designed to convert TILs into effective cells: First, systemic administration of cytokines and immunologically active proteins such as IL-2 and interferon, which are currently approved for melanoma; second, systemic administration of antibody therapies aimed to modify T cell activation and relief of checkpoint blockade, such as ipilimumab (anti-CTLA-4), anti-PD-1 and anti-PD-L1, anti-4-1BB and anti-CD40, to name a few. Checkpoint blockade therapy has had remarkable results not only in melanoma (23), but also in tumors such as lung cancer that have been previously thought to be ‘immunologically silent’ (24, 25). Even more encouraging, simultaneous blockade of two checkpoints (CTLA-4 and PD-1) in melanoma significantly improved the response rate and time to response over either therapy alone (26). The third TIL strategy is direct isolation and ex vivo activation of the tumor-infiltrating lymphocytes and has been tested in multiple early-phase studies and results in durable responses in melanoma (27). In the majority of presenting cases, however, this approach cannot be undertaken, either because surgical material is not available or contains insufficient numbers of TILs, or the patient cannot tolerate the conditioning regimen or the time required for manufacturing of their TIL product, preventing the ability to conduct randomized controlled studies.
As discussed below, recent advances in the use of genetically engineered T cells, and an understanding of the principles underlying effective T cell therapy, have produced encouraging results in the use of T cell therapies for viruses, hematologic malignancies, and solid tumors. T cell therapy is now poised to advance from Phase I trials to more Phase II and Phase III trials, and for the first time is being actively clinically developed by multiple biotech and pharmaceutical companies, with the goal of offering a standardized, quality-controlled, regulatory-body approved treatment for the integration of cell therapies to the treatment of patients worldwide.
Principles of T cell transfer: the soil, the fertilizer, the seed
T cell transfer and engraftment into the host is a complex biologic process, and can be optimized by a thorough understanding of the role of the host immune system (the soil), growth factors and the balance against inhibitory cells (the fertilizer), and the transferred T cell product (the seed) (Figure 1).
Preparation of the soil (host conditioning)
Evidence from bone marrow transplantation and adoptive therapy trials of TILs demonstrated that ‘conditioning,’ or lymphodepleting the host enhanced engraftment of the transferred T cells. There are multiple hypotheses as to why conditioning, or preparing the ‘soil’ for the incoming T cells, is attractive, particularly in the setting of malignancy. These include reduction of tumor burden (thus improving the effector:target ratio in vivo), reducing the population of inhibiting regulatory T cells (28), and inducing production of homeostatic cytokines to facilitate proliferation of the transferred T cells (29). Typical regimens for host conditioning include cyclophosphamide with or without fludarabine; some centers also use total body irradiation. All of these techniques, particularly the most intense ones that combine chemotherapies and irradiation (30), appear to improve persistence of the transferred T cells and the clinical responses in the setting of cancer. Notably, however, host conditioning has not been required in the setting of HIV infection to enable long term persistence of transferred T cells (5); similarly low numbers of virus-specific T cells can persist and expand in the post-transplant setting (31). Furthermore, recent reports with CAR-modified T cells administered in the absence of host conditioning have had clinical effects in both hematologic and solid tumors (32, 33).
Fertilizer: cytokines and T-cell modulating antibodies
Cytokines provide important growth and homeostatic signals to T cells, with IL-2, IL-7, and IL-15 being particularly well-studied. Recombinant human IL-2 as a single agent is FDA-approved for metastatic melanoma, though because of its toxicity, it is only administered in select centers. Furthermore, the biologic role of IL-2 is physiologically complicated; low-dose IL-2 is thought to maintain regulatory T cells and has been used to control graft vs host disease (34). Thus, it is not clear that administration of IL-2 will help the transferred cytotoxic T cells over the native regulatory T cells. Strong pre-clinical data support the use of IL-7 and IL-15, both of which are also being explored in clinical trials. IL-15 in particular is thought to relieve the inhibition of regulatory T cells while providing support for adoptively transferred T cells (35).
The combination of adoptive cell therapies with newer agents, including checkpoint blockade and/or small molecule targeted therapies is still in its nascent stages but is bound to generate excitement. Many combinations can be envisioned: co-administration of agonistic CD40 antibodies (36) or 4-1BB antibodies (37) to mediate co-stimulation, or with checkpoint blockade such as anti-PD-1 or anti-CTLA-4, is likely to improve both the effects of the transferred T cells and to stimulate the native T cell responses to tumors. Typically, small-molecule drugs aimed at aberrant signaling in the tumor effects rapid but short-lived tumor responses, while immunotherapy approaches take longer to eliminate tumor but are potentially long-lived. Combinations of treatment with T cell transfer coupled with small molecule drugs targeting tumor mutations (such as BRAF inhibitors in melanoma (38), or Bruton’s tyrosine kinase or Bcl-2 inhibitors in lymphoma) have the exciting potential to make cancer treatment chemotherapy-free. (Tables 1-3 and Figure 2)).
Table 1.
Summary of current clinical trials of CD19 CAR T cells
| Target Antigens | Cancers | CAR Signaling Domain | Combinatorial/Engineering Strategies (Biologicals, Drugs) | Phase/ID | Sponsor |
|---|---|---|---|---|---|
| CD19 | B cell malignancies relapsed post-allo-HSCT, T cells from donor | CAR: CD28-CD3ζ | None | I; NCT01087294 | National Cancer Institute |
|
| |||||
| CD19 | NHL; CLL | CAR: CD28-CD137-CD3ζ or CD28-CD3ζ | None | I; NCT01853631 | Baylor College of Medicine |
|
| |||||
| CD19 | Relapse/refractory CLL, NHL, or ALL | CAR: CD3ζ | None | I/II; NCT01865617 | Fred Hutchinson Cancer Research Center |
|
| |||||
| CD19 | ALL; DLBCL; MCL; NHL; CLL relapsed post allo-HSCT | CAR: CD28-CD3ζ | CMV- or EBV-specific T cells derived from donor CD62L+ TCM | I/II; NCT01475058 | Fred Hutchinson Cancer Research Center |
|
| |||||
| CD19 | ALL; CLL; NHL | CAR: CD137-CD3ζ | None | NP; NCT01864889 | Chinese PLA General Hospital |
|
| |||||
| CD19 | ALL; CLL; NHL | CAR: CD28-CD3ζ | Ipilimumab | I; NCT00586391 | Baylor College of Medicine |
|
| |||||
| CD19 | CLL; Small lymphocytic lymphoma; MCL; | CAR: CD28-CD3ζ | |||
| Follicular lymphoma; Large cell lymphoma | IL-2 | I/II; NCT00924326 | National Cancer Institute | ||
|
| |||||
| CD19 | Auto-HSCT for NHL followed by T cell infusion | CAR: CD28-CD3ζ | none | I; NCT01840566 | Memorial Sloan-Kettering Cancer Center |
|
| |||||
| CD19 | B cell leukemia; B cell lymphoma | CAR: 4-1BB-CD3ζ | None | I; NCT01626495 | CHOP/University of Pennsylvania |
|
| |||||
| CD19 | NHL; CLL | CAR: CD28-CD3ζ | CD19 CAR transduced PBLs and EBV-specific CTLs | I; NCT00709033 | Baylor College of Medicine |
|
| |||||
| CD19 | Pediatric relapsed B cell ALL | CAR: CD28-CD3ζ | None | I; NCT01860937 | Memorial Sloan-Kettering Cancer Center |
|
| |||||
| CD19 | CD19+ malignancies | CAR: CD28-4-1BBζ | None | NP; NCT01029366 | University of Pennsylvania |
|
| |||||
| CD19 | ALL; CLL; NHL | CAR: CD28-CD3ζ | CD19 CAR transduced tri-virus-specific CTLs (CMV, EBV, and adenovirus) | I/II; NCT00840853 | Baylor College of Medicine |
|
| |||||
| CD19 | Pediatric leukemia and lymphoma | CAR: CD28-CD3ζ | None | I; NCT01593696 | National Cancer Institute |
| CD19 | Relapsed ALL post-allo-HSCT | CAR: CD28-CD3ζ | CD19 CAR transduced EBV-specific CTLs | I; NCT01430390 | Memorial Sloan-Kettering Cancer Center |
|
| |||||
| CD19 | Auto-HSCT for NHL followed by T cell infusion | CAR: CD28-CD3ζ | TCM-enriched CD8+ T cells | I/II; NCT01318317 | City of Hope/National Cancer Institute |
|
| |||||
| CD19 | ALL | CAR: CD3ζ | CD19 CAR transduced EBV-specific CTLs | I/II; NCT01195480 | University College, London |
|
| |||||
| CD19 | Leukemia | CAR: CD28-CD3ζ; 4-1BB-CD3ζ | None | I/II; NCT00466531 | Memorial Sloan-Kettering Cancer Center/University of Pennsylvania |
|
| |||||
| CD19/EGFRt | Auto-HSCT for NHL followed by T cell infusion | CAR: CD28-CD3ζ | TCM-enriched T cells; (cetuximab as possible suicide system) | I; NCT01815749 | City of Hope |
|
| |||||
| CD19/EGFTt | Pediatric ALL | CAR: CD28-CD3ζ | None | I; NCT01683279 | Seattle Children’s Hospital |
allo-HSCT, allogeneic hematopoietic stem cell transplant; NHL, non-Hodgkin lymphomas; CLL, Chronic lymphocytic leukemia; ALL, Acute lymphoblastic leukemia; DLBCL, Diffuse large B-cell lymphoma; MCL, Mantle cell lymphoma; MALT, Mucosa-associated lymphoid tissue; CMV, Cytomegalovirus; EBV, Epstein–Barr virus; TCM, Central memory T cells; PBTLs, Peripheral blood T lymphocytes; CTLs, Cytotoxic T lymphocytes; NP, Information not provided.
Table 3.
Summary of recent clinical trials involving genetically-redirected T cells
| Target Antigens | Cancers | Receptor | Combinatorial/Engineering Strategies (Biologicals, Drugs) | Phase/ID | Sponsor |
|---|---|---|---|---|---|
| WT1 | AML; CLL | TCR | None | I/II; NCT01621724 | University College, London |
| WT1 | AML, MDS, or CML | TCR | Aldesleukin; Virus-specific CD8+ T Cells | I/II; NCT01640301 | Fred Hutchinson Cancer Research Center/University of Washington Cancer Consortium |
| NY-ESO-1 | Melanoma | TCR | None | I/II; NCT01350401 | Adaptimmune |
| NY-ESO-1/LAGE-1 | Multiple myeloma | TCR | None | I/II; NCT01892293 | Adaptimmune |
| NY-ESO-1/MAGE-A3/6 | Multiple myeloma | TCR | None | I/II; NCT01352286 | Adaptimmune |
| CEA | Metastatic cancers | IgCD28TCR | None | II; NCT01723306 | Roger Williams Medical Center |
| MART-1 | Metastatic melanoma | TCR | Administration of MART-126.35-Pulsed Dendritic Cells and IL-2 | II; NCT00910650 | Jonsson Comprehensive Cancer Center |
AML, Acute myelogenous leukemia; CLL, Chronic lymphocytic leukemia; MDS, myelodysplastic syndromes; CML, Chronic myelogenous leukemia; TCR, T cell receptor.
Figure 2. Durable tumor regression may be achieved by combining immunotherapy with targeted therapeutic strategies.
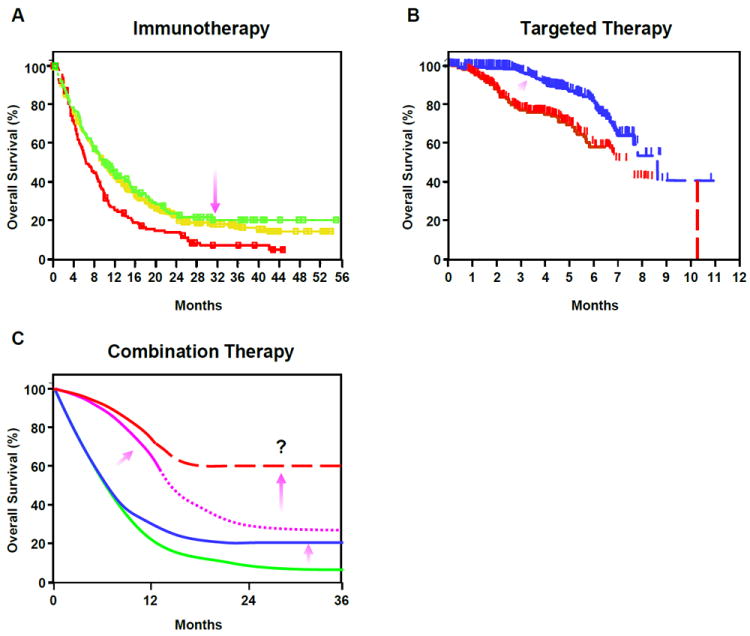
(A) Patients with metastatic melanoma exhibit improved survival with Ipilimumab treatment(23) (Kaplan-Meir survival plot). Treatments are as follows: ipilimumab-alone (green line); gp100-alone (red line); ipilimumab + gp100 (yellow line). Purple arrow indicates how immunotherapy raises the tail of curve, indicating prolonged effects. (B) Kaplan-Meir survival plot showing that improved survival can be achieved in melanoma with Vemurafenib therapy (204). Patients were treated with either Dacarbazine (red line) or Vemurafenib (blue line). Purple arrow indicates direction of change effected by targeted therapy; overall survival is improved early but effects are transient. (C) Hypothetically, the combination of immunotherapy with targeted treatment may increase survival in patients with metastatic cancers. The green line depicts a typical survival curve for standard cancer therapy; the blue line represents survival with immunotherapy alone; the purple line represents the effect of a targeted therapy alone survival plot; the dashed red line depicts the potential enhanced survival that can be achieved using immunotherapy combined with targeted therapy. Arrows highlight the impact of the above therapeutic regimens on the tail of the curve or the spread between curves for different treatments. Thus, immunotherapy requires more time, but increases survival; Targeted therapy works rapidly, but is not durable. Combining these strategies may ultimately improve the fraction of patients with long-term survival.
Seed: the cell product
Because of the complexity of T cell activation, differentiation, and homeostasis, several groups have investigated the optimal cell population to serve as the ‘seed’ for adoptive cell therapy.
It is now clear that T cells that have been cultured extensively, whether stimulated with autologous dendritic cells, artificial antigen presenting cells (APCs), or cloned and passaged on allogeneic feeder cells, have a terminally differentiated phenotype with a loss of in vivo engraftment and proliferative capacity, and limited in vivo function. Reprogramming of the T cell (39, 40) may overcome these effects, but is associated with its own complex culture system that will be difficult to adopt widely. Currently, culture systems that rely on repetitive antigen stimulation to generate a T cell product are not easily scalable, efficient or reliable enough to generate functional T cells for immunotherapy, except perhaps as third party donor banks, which are expensive to generate due to the heterogeneity of HLA types in the population.
In an effort to maintain the persistence and function of adoptively transferred T cells, some investigators have used T cells specific to a chronic virus such as EBV or CMV and re-directed them to tumor-associated antigens (41, 42). Others have explored using phenotypically defined populations that are expected to proliferate and survive for longer, such as central memory T cells (43) or naïve T cells (44), to improve engraftment in pre-clinical studies. Recent data has identified and characterized an early-differentiated, stem-cell memory T (TSCM) (45). These cells constitute the most undifferentiated human T-cell compartment exhibiting bona fide memory functions, and can survive for extended periods even after the loss of cognate antigens. This type of T cell is thought to persist and support memory T cell functions, which would make it an ideal candidate for long-term control of cancer in addition to engaging it for viral vaccine purposes (46). However, it is not clear that the frequencies of the TSCM peripheral blood samples are consistent in large numbers of diverse cancer patients, and it will require validated and clinically approved systems to isolate these cells to form the basis of a cell therapy product. (Figure 3)
Figure 3. Phenotypic and functional changes in T cells during progressive differentiation driven by chronic antigen stimulation.
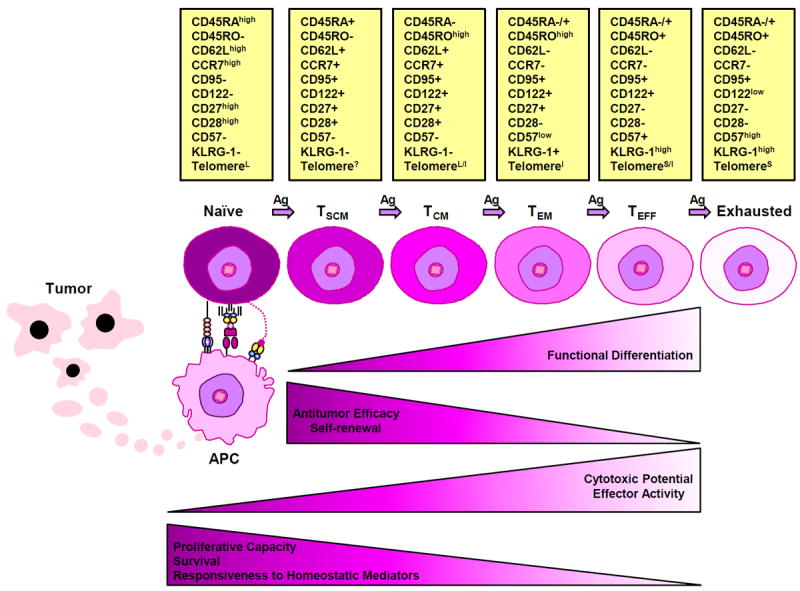
A state of persistent and frequent antigenic stimulation, such as that induced by tumor burden, facilitates a progressive differentiation pathway whereby naïve T cells become terminally differentiated effectors. Changes in the phenotypic markers that characterize progressive T cell differentiation are depicted as: expressed (+), not expressed (-), expressed at high levels (high), expressed at low levels (low); long telomere length (L), unknown telomere length (?); long/intermediate telomere length (L/I), intermediate telomere length (I), short/intermediate telomere length (S/I) and short telomere length (S). Together with the gradual shortening of telomere length, T cells lose their proliferative and self-renewal capacities, as well as responsiveness to homeostatic mediators and ultimately become exhausted. Although cytotoxic potential/effector functions increase with persistent antigen stimulation and T cells must be fully differentiated to possess anti-tumor activity, experimental evidence suggests that in the context of adoptive cell therapy, increasing differentiation state is inversely correlated with anti-tumor efficacy (205).
Finally, although the efficacy of adoptive cell therapy is most often attributed to CD8+ T cells, there are reports of pure CD4+ T cell populations mediating tumor regression (47). Furthermore, immune effector cell types other than T cells can and have been used in cell transfer protocols. For example, NK-based trials have been published in the autologous (48) and allogeneic settings (20), and re-direction or engagement of NK cells is an area of active research.
Strategies of ex vivo T cell culture
An inherent barrier to widespread clinical application remains the manufacturing difficulties and the access to robust and efficient methods for the expansion of input T lymphocytes. Our laboratory has developed methods for the efficient activation, expansion, and gene transduction of T lymphocytes. (Figure 4) Additionally, desired properties of adoptive immunotherapies include 1) demonstrated potency against tumor or infectious organism, 2) efficient engraftment enabling a high effector to target ratio, 3) long term persistence and memory.
Figure 4. Clinical application of gene-modified cell therapies.
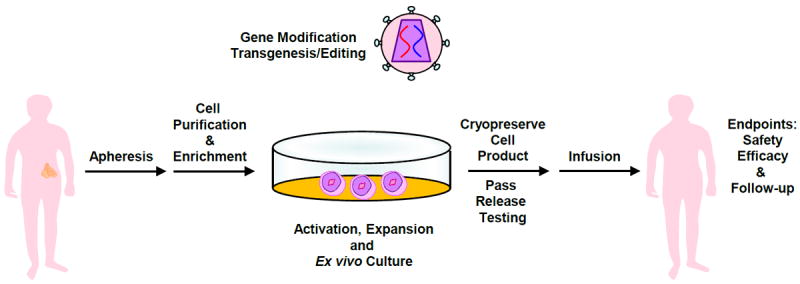
Cells of interest are isolated from the whole blood of a patient, followed by enrichment, activation and expansion. At the time of activation, the lentiviral vector is added. On the final day of culture, cells are harvested and cryopreserved in an infusible media. The patient is infused with gene-modified cells and endpoint assays are conducted at designated time intervals. At the conclusion of active monitoring, in the US, the patient is transferred to a destination protocol for long-term follow-up as per FDA guidelines.
T Cell Therapy and Ex Vivo Culture Methods
The clinical application of T-cell based therapeutics has gained extensive momentum within the past 30 years due to a number of critical discoveries that included the identification of T cell antigens that have also been tested as cancer vaccines (49). There have been a large number of studies that suggest that DCs, when appropriately activated and induced to present tumor-associated antigens can elicit tumor-specific T cell immunity. This dendritic cell therapeutic approach is currently being pursued by several biotechnology companies (50-53), but has limitations in that the ability to generate dendritic cells varies from patient to patient and this variability may result in short-term or insufficient T cell activation to generate an effective immune response.
Magnetic Bead-Based Artificial Antigen Presenting Cells
With recognition that both a primary specificity signal via the T Cell Receptor (TCR) (Signal 1) and a costimulatory/regulatory signal via the CD28 receptor (Signal 2) are simultaneously required for the generation of full T-cell effector function and a long-lasting immune response (54), we developed efficient and reproducible methods of mimicking the signal provided to T cells by dendritic cells, but without delivering a negative costimulatory signal. With artificial Antigen Presenting Cells (aAPC), T cells to be grown rapidly ex vivo to clinical scale for therapeutic applications. The technology enables direct T cell activation, instead of indirect activation via vaccines, which can be modulated by the nature of cell dose as necessary to achieve a clinical response (55, 56).
The first generation of off-the-shelf aAPC covalently linked clinical grade anti-human CD3 and anti-CD28 monoclonal antibodies to magnetic Dynal beads (Life Technologies) which serve to crosslink the endogenous CD3 and CD28 receptors on the T cell. This bead-based aAPC enables the most efficient reported growth of human polyclonal naïve and memory CD4+ T cells (56). In terms of cell function, the expanded cells retain a highly diverse TCR repertoire and, by varying the culture conditions, can be induced to secrete cytokines characteristic of T helper 1 (Th1) or T helper 2 (Th2) cells (57). One important advantage of this bead-based system is that it does not cross-react with CTLA-4 and therefore provides unopposed CD28 stimulation for more efficient expansion of T cells. Another, unanticipated discovery was that crosslinking of CD3 and CD28 with bead-immobilized antibody renders CD4+ T lymphocytes highly resistant to HIV infection. This is due to the down-regulation of CCR5, a necessary co-receptor for the internalization of HIV, as well as the induction of high levels of β-chemokines, the natural ligands for CCR5 (58-60), and allows for the efficient culture of CD4+ T cells from HIV-infected study subjects. Ex vivo expansion may also indirectly enhance T cell activity by removing T cells from a tumor-induced immunosuppressive milieu (61-64). Other key features are that exogenous growth factors or feeder cells are not needed to enable the T cell stimulation and expansion, as with previous methods.
Cell-based Artificial Antigen Presenting Cells
Cell-based artificial Antigen Presenting Cell (aAPC’s) lines have been derived from the chronic myelogenous leukemia line K562 (65-67). K562 cells do not express Major Histocompatibility Complex (MHC) or T costimulatory ligands, and these cells may represent a DC precursor that retains many other attributes that make DCs such effective APCs, such as cytokine production, adhesion molecule expression and macropinocytosis. These cells have been transduced with a library of lentiviral vectors that allows for the customized expression of stimulatory and costimulatory molecules that can used activate and expand different subset of T cells, and be further modified to amplify antigen specific T cells in culture. These aAPCs offer the advantage of expression of molecules additionally to CD3 and CD28 on their surface. The K562 aAPCs have been transduced with vector to express the antibody Fc-binding receptor and the costimulatory molecule 4-1BB. The expression of CD64, the high affinity Fc receptor, on K562 aAPC’s allows the flexibility of loading antibodies directed against T cell surface receptors. CD3 and CD28 antibodies are added to the cells and are bound by the Fc receptor to yield a cell that expresses CD3, CD28 and 4-1BB. These cell-based aAPC’s have proved to be more efficient at activating and expanding T cells, especially CD8+ and antigen-specific T cells, than the magnetic bead-based aAPC (66-68). In addition, the cells are capable of stimulating CD4 cells efficiently.
Thus, K562 cells may represent ideal cell scaffolds to which the desired MHC molecules, costimulatory ligands, and cytokines can be introduced in order to establish a DC-like aAPC. Advantages of this artificial dendritic cell platform include high levels of MHC expression, a wide array of costimulatory ligands and the ability to engage in cytokine crosstalk with the T cell. This mimics the advantages of natural dendritic cells, without recognized disadvantages including the need to derive natural DCs from either G-CSF mobilized CD34+ cells or monocytes, patient specific differentiation, limited life span, and limited replicative capacity. Moreover, these cells have been injected into humans as part of a tumor vaccine (69), signifying that these cells can be used in a GMP manner. Additionally, our laboratory and our collaborators have now developed either bead or cell-based aAPCs optimized for Th2 cells (57) (70), and for T regulatory cells (71).
Manufacturing Process
Independent of which of the above aAPC’s is used, the manufacturing procedure remains similar, starting with an apheresis product. Alternatively, T cells can be derived from a blood draw, bone marrow, ascites, or tumor infiltrating lymphocytes. The pheresis product may be washed out of collection buffer in a Haemonetics CellSaver5 or other automated cell washing device, or directly loaded in the Terumo Elutra™ Cell Separation System for depletion of monocytes and isolation of lymphocytes. The depletion of CD4+, CD8+, or CD25+ T cells can be accomplished using a Miltenyi CliniMACS®. This instrument is an electromechanical device intended to isolate certain cell subsets via large scale magnetic cell selection in a closed and sterile system. Before selection, the washed cells from a pheresis product are magnetically labeled by using particles conjugated with anti-CD4, anti-CD8, or anti-CD25 mAb. A single-use tubing set, including separation columns, is then attached to the CliniMACS® Instrument and the cell preparation bag, containing the labeled cells. After starting the selection program, the system automatically applies the cell sample to the separation column, performs a series of washing steps depending on the program chosen and finally elutes the purified target cells.
The lymphocyte fraction from the Elutra™ Cell Separation System, or enriched T cells are cultured in a nutrient media and stimulated to divide and grow via the addition of the antibody coated magnetic beads or of irradiated and antibody pre-loaded K562 aAPC’s, each of which is described above. Gene transduction of anti-CD3/anti-CD28 aAPC stimulated T cells with retroviral, lentiviral, or adenoviral vectors is very efficient (72-74). The whole mixture of cells, growth media, vector and aAPC is added to a gas permeable plastic bag or alternative culture vessel. Tubing leads on the bags and a variety of connecting devices allow the cells to be grown in a closed system with minimal risk of contamination. After gene vector washout, if needed, on the Baxter CytoMate and during log phase cell growth, cultures are transferred to the WAVE Bioreactor, where cell concentrations may reach 1 × 107 cells/ml or higher. The advantage of the WAVE is that T cells can be grown at higher densities, which saves labor on processing and during the cell harvest. The cultures are maintained for 9-11 days prior to harvesting and preparation for reinfusion or cryopreserved for later infusion. At harvest, magnetic bead-based aAPC are removed, the cells are washed, resuspended and cryopreserved in an infusible solution. If the cells are to be infused fresh, in process samples are taken for microbiological testing, viability, and cell phenotype by flow cytometry for the release. Testing is repeated on the final product, although results for some tests are not available until after the cells are infused.
Genetic engineering platforms
T lymphocytes can be modified by gene transfer methods to permanently or transiently express therapeutic genes to enhance and expand the therapies. Importantly, genetic engineering can also be used to endow lymphocytes with several other features, including an increased proliferative potential (75), a prolonged in vivo persistence (76), an improved capacity to migrate to tumor tissues (77), or to recognize an entirely new antigen. Re-direction of antigen-specificity is usually based on either a TCR of known specificity (78) or a synthetic receptor such as a chimeric antigen receptor (CAR), which recognizes antigen through antibody-derived complementarity-determining regions, but signal through TCR-associated molecules (79).
Current clinical trials of permanently modified T cells employ viral and non-viral based approaches. Retroviral (gamma-retroviral and lentiviral) vectors can be used to transduce cells without producing any immunogenic viral proteins, with the transgene becoming a permanent part of the host cell genome. Retrovirus-based gene delivery has proven to be an extremely useful tool in gene therapy research and is commonly used in trials of T cell therapies. Non-viral DNA transfection or transposons are also used for permanent gene expression in gene-modified T cell based therapies. Gene delivery by using adenoviral vector or RNA transfection enables the transgene expression for up to 1 week; these approaches have promise for when transient transgene expression is desirable (Figure 5).
Figure 5. Vector systems for T adoptive therapy.
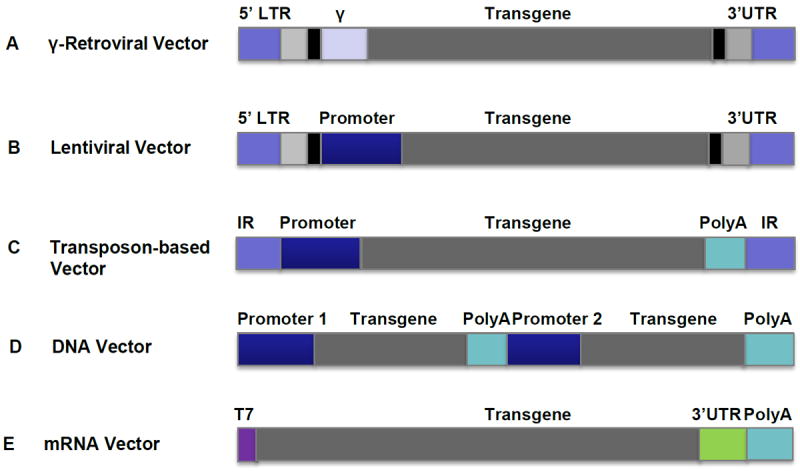
(A) γ-retroviral vectors are one of the most commonly used vector systems for permanent gene expression in T cells for adoptive immunotherapy in the clinic. Clinical grade vector produced under Good Manufacturing Practices (GMP) viral vectors can be produced by stable packaging cell lines. vectors. GMP Viral vectors can be produced by stable packaging lines. (B) Lentiviral vectors are becoming more popular vector systems for permanent gene expression in T cells for adoptive immunotherapy clinical trials, due to their advantages of transducing non-dividing cells, high transduction efficiency and potentially safer integrating profile over γ-Retroviral vectors. (C) Non-viral transposase-mediated gene transfer, in which gene integration is achieved by the provision of the transposase enzyme, can mediate permanent gene expression in T cells. (D) DNA can be directly transfected into T cells by electroporation and integrated; genetically modified T cells can be cloned and expanded. (E) T cells can be directly transfected by electroporation with in vitro transcribed mRNA without integrating. This is a transient transgene system and the transgene can be expressed in T cells for up to 1 week. Repeated infusions of the mRNA-transfected T cells are required. All vectors are depicted as linear DNA. Internal repeat (IR); Long terminal repeat (LTR); Packaging Signal (ψ); Untranslated region (UTR); Poly(A) tail (PolyA).
Gamma-Retroviral vectors
Currently, most retroviral vectors are derived from murine or avian retroviruses. The Moloney murine leukemia retrovirus (gamma-retrovirus) has been extensively studied as a vector, and can package up to 8 kb of genetic material. Vectors that are derived from gamma - retrovirus have been most useful for long-term gene expression because of their ability to integrate into the host genome, which results in permanent expression of the transgene with low intrinsic immunogenicity (80). The first human trial of immunotherapy with gene-modified T cells was reported in 1990 in patients with advanced melanoma using tumor-infiltrating lymphocytes modified by retroviral gene transduction (81); in this case, the retrovirus was used to encode neomycin resistance and served only to track the fate of the infused T cells. Also, in 1990, two girls suffering from adenosine deaminase severe combined immunodeficiency (ADA SCID) were treated by T lymphocytes transduced with a gammaretrovirus expressing the ADA gene, which led to the reconstitution of the patient’s immune system with those gene-corrected T cells (82). Since then, retroviral vectors have been the major tools for permanent transgene expression, and have been widely used as vehicles to deliver genes into different type of cells for gene therapy, including T lymphocytes (83, 84). Hematopoietic stem cells (HSCs), which have the potential to self-renew and differentiate into all blood lineages, were initially thought to be the most desirable targets for retroviral gene modification for the treatment of genetic disorders and other diseases (85-91). Although initial results were encouraging, adverse events were observed in trials for SCID-X1 and X-chromosome linked chronic granulomatous disease (X-CGD) due to vector integrations in the vicinity of well-characterized proto-oncogenes (87, 92, 93). It is believed that the target cells most vulnerable to insertional-mediated transformation are primitive progenitor cells, and that more mature cells are less prone to this event (94-97). T lymphocytes remain major targets for retroviral-based gene modification, not only to deliver therapeutic genes but also to be redirected for a specific tumor-associated antigens (3, 78, 98-100). Unlike HSCs, T lymphocytes are less susceptible to transformation (101). Although insertional mutagenesis can contribute to immortalization of retrovirally transduced mature T cells in vitro, it is a rare event, and occurs in the setting of a synergistic effect between activation of a proto-oncogene, such as LMO2, and robust signaling through T cell homeostatic cytokines, such as IL-2 or IL-15 (102, 103). Whether a T cell stimulating signal generated by a transduced TCR or CAR can synergize with activation of a proto-oncogene caused by retroviral insertional mutagenesis to mediate transformation of the transduced T cells remains to be elucidated. Malignant transformation has not been observed thus far in clinical trials of retroviral-based gene transfer into mature T cells (3, 5, 72, 104).
Lentiviral vectors
Lentiviral gene transfer is relatively new and shares many features of the retroviral system. Lentiviruses are distinct members of the retroviruses family. Lentiviral vectors have been constructed from several types of lentiviruses, but the most commonly used is the human immunodeficiency virus (HIV), because its molecular biology has been extensively studied (83, 105).
Lentiviral vectors resemble gamma-retroviral vectors in their ability to stably integrate into the target cell genome, resulting in persistent expression of the gene of interest; they can accommodate up to 10 kb transgene material, and the immunogenicity of the vector is low. However, in contrast to gamma-retroviral vectors, lentiviral vectors have the advantage of being able to transduce non-dividing cells (106), they have broader tissue tropisms, and have a potentially safer integration site profile (107, 108). Furthermore, lentiviral vectors are less susceptible to gene silencing by host restriction factors (109). These distinctive features broaden the possible applications of lentiviral vectors, especially in settings where gamma-retroviral vectors are not suitable. Lentiviral vectors have been safely used in human clinical trials to engineer HSCs and T lymphocytes and there have been no oncogenic events observed (110) (73, 89, 90, 111). However, clonal expansion and dominance of hematopoietic progenitors has been reported in a clinical trial in which HSCs were genetically modified with a lentiviral vector that expressed the beta-globin gene for the treatment of thalassemia (90). Therefore, genotoxicity of insertional mutagenesis is still a potential safety concern for lentiviral vectors.
From a manufacturing perspective, it is notable that stable packaging cell lines are easily established for gamma-retroviral vectors, whereas this is still challenging for lentiviral vectors due to the toxicity of envelope proteins; this limitation obligates researchers to generate vectors from inefficient, transient multi-plasmid transfections. In addition to the risk of insertional mutagenesis, another potential safety issue applicable to both gamma-retroviral and lentiviral vectors is the possibility of generating replication competent retroviruses (RCR). Although new generations of vectors have been designed to reduce the production of RCR, these additional steps result in decreased efficiency of vector production.
Adenovirus vectors
Adenovirus vectors are capable of transducing both dividing and quiescent cells; they can accommodate relatively large transgenes, production of high-titer vector stocks is relatively easy, and the vectors are non-oncogenic due to their lack of integration into the host genome. Adenovirus vectors are widely used in clinical trials, especially for cancer targeted gene therapies. Application of adenovirus vectors in T cell-based therapy is limited by the transient transgene expression and the immunogenicity of the vector. Chimeric adenoviral vectors Ad5–F35 were reported to mediate gene transfer up to 10% of resting T cells and 30–45% of T cells after activation with phytohaemagglutinin (112). Ad5F35 vectors could result in gene transfer in more than 90% of T cells after activation by CD3 and CD28 specific antibodies (8, 74). Adenovirus vectors have promise as gene delivery vehicles to T cells in clinical situations where duration of transgene expression of less than a week is required, and there is no foreseeable need for repeated cell infusions. Examples of such situations can be envisioned for gene editing strategies such as zinc-finger nucleases (ZFN) or transcription activator-like effector nucleases (Talens), or Clustered Regularly Insterspaced Short Palindromic Repeats(CRISPR)-mediated specific gene silencing (8).
DNA transfection and Transposon-based gene delivery
Non-viral based DNA transfection methods remain popular as vectors for gene therapy due to their low immunogenicity and a low risk of insertional mutagenesis. The first clinical trial testing the adoptive transfer of T cells engineered using electroporation was recently reported (113), and although this approach was safe, the cells were short-lived after transfer, probably owing to the long-term culture of the cells that was required to select sufficient numbers of permanently integrated T cell clones for the treatment.
Transposon-based systems can integrate transgenes much more efficiently than plasmids that do not contain an integrating element (114, 115). Sleeping Beauty (SB) was shown to provide efficient stable gene transfer and sustained transgene expression in primary cell types, including human hematopoietic progenitors, mesenchymal stem cells, muscle stem/progenitor cells (myoblasts), pluripotent stem cells and T cells (116). Various transposase-based systems are now entering clinical trials to test the safety and feasibility of this approach to engineer T cells (117). Non-viral vectors have several advantages over viral vectors as a modality to engineer cells, including lower costs and perceived safety benefits. However, the safety profile of these approaches is still uncertain, since the relative genotoxicity of transposons is unknown. Approaches to achieve site-specific integration and DNA editing are under development, and if these prove to be efficient, they should allay concerns regarding lymphocyte engineering using non-site-specific integration approaches.
RNA transfection
Thus far we have discussed viral transduction or plasmid DNA transfection of T cells, which can result in stable genomic integration, allowing for constitutive, permanent expression of the transgenes. Safety concerns, such as genotoxicity, potential generation of RCR, and the difficulty in predicting off-tumor toxicities are potential limiting factors for the widespread use of gene-modified T cells, particularly as a first approach in the clinical setting. Moreover, there are clinical situations in which multiple infusions of the engineered T cells would be required (i.e. to overcome the lack of persistence or the immunosuppressive influence of the tumor microenvironment), making manufacturing a clinical dose T cell product difficult to achieve and expensive. When transient expression of the transgene is desired, such as to identify potential off-tumor toxicities, or recursive infusions are planned, RNA transfection of T cells is an attractive approach because it is relatively inexpensive, fast, and the transfection efficiencies can easily approach 100%. RNA-based electroporation of human T lymphocytes using in vitro-transcribed mRNA mediates transient expression of proteins for approximately 1 week. The self-limiting transgene expression can provide a safety check for off-tumor, on-target or off-target toxicities, or other unwanted side effects, as the engineered T cells are essentially a ‘biodegradable’ product. Furthermore, there is no genotoxicity concern as the introduced mRNA does not integrate into the host genome. RNA electroporation has been used to deliver message for TCR or CAR, chemokine receptors or cytokines (118-121). In one study, T cells modified by CAR RNA were evaluated in a side-by-side comparison with retrovirus modified T cells; RNA engineering was at least as efficient as retroviral gene transfer (122). Alternatively, for transposon-based gene delivery systems, transposase enzymes can be delivered as mRNA thereby avoiding the possibility of genomic integration (123); gene editing strategies based on ZFN, Talens or CRISPR can be also delivered by RNA transfections. In pre-clinical animal studies, multiple injections of CAR RNA modified T cells can mediate regression of disseminated tumors (124-126). Clinical trials of treating solid tumors with RNA-electroporated CAR T cells have been initiated by several groups (33) and the safety and efficacy results will provide valuable information for further cancer treatments using genetically modified T cells.
T cells re-directed with specific T cell receptors (TCRs)
Transduction of T cells with a specific TCR has the advantage of redirecting the T cell to an intracellular antigen. Given that most oncogenic proteins are intracellular, development of a panel of TCRs specific to oncogenic driver proteins has great appeal. However, a library of MHC-restricted antigen-specific TCR reagents would need to be characterized and available to treat patients, who have diverse HLA alleles. Furthermore, the low-affinity of most tumor-directed TCRs is thought to significantly impact their efficacy. This is one reason that most peptide-cancer vaccines alone or in combination with adjuvants or professional antigen-presenting cells have produced underwhelming clinical responses despite in vitro evidence of tumor-directed T cell responses (127-129).
Several groups have explored retroviral transduction of native T cell receptors with the goal of re-directing a bulk of T cells to an intracellular antigen. Potentially significant obstacles were hypothesized for the reason that when a T cell transcribes the chains for two different TCRs, there are four potential combinations of T cell receptors that can be expressed at the cell surface (native-alpha/beta, transduced alpha/beta, and native/transduced ‘mispaired’ heterodimers). This is problematic for two crucial reasons: (1) the native/transduced heterodimers have unknown specificity and potential autoimmune consequences, which has been demonstrated to occur in some mouse models (130), and (2), there is dilution of the signal transduction apparatus, since the availability of CD3 complex molecules is limiting. Early studies of HIV-directed TCRs encountered low levels of expression of the transduced TCR, along with mispairing; this combination resulted in decreased efficacy in vitro and heterogeneous populations of T cells (131). Several groups have described methods to favor pairing of the transduced TCRs by engineering the transduced TCR chains, including (1) partial murinization of the constant regions(132-134), (2) the addition of disulfide bonds (130, 135-138), (3) altering the knob-in-hole directional interaction of the endogenous TCR constant regions (139), and (4) adding signaling domains to the intracellular portions of the transduced TCR (140). Another interesting approach is to knockdown the endogenous TCR with gene editing or shRNA (141); a third party bank of T cells that also have endogenous HLA knocked down can also be envisioned (142). Most of these modified TCR designs are still in pre-clinical development. (Figure 6)
Figure 6. Strategies to improve the function of transgenic TCRs.
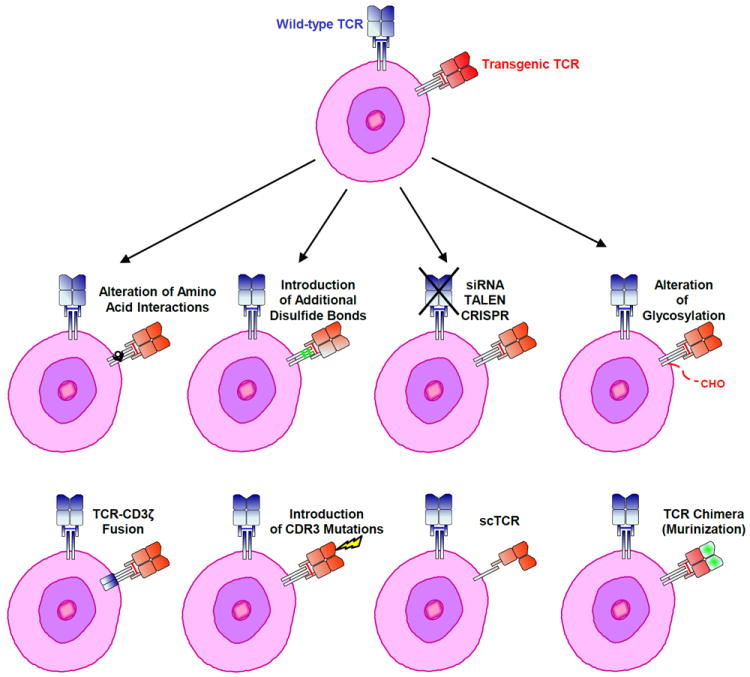
Expression of a transgenic TCR may be improved by preventing the formation of mixed dimers between endogenous and ectopic engineered TCR chains. These strategies include the alteration of amino acid interactions, the creation of TCR-CD3 ζ fusions, single chain (sc) receptors, TCR chimeras (murinization) and the introduction of additional disulfide bonds or glycosylation. Mutating amino acids in the complementarity determining region 3 (CDR3) and knocking down/out endogenous TCR expression may also increase transgenic TCR activity.
Nevertheless, trials of native TCR-transduced T cells have been reported, and though some have resulted in significant anti-tumor responses (99), others have noted significant on-target toxicity (78, 143) and off-target toxicity (144), particularly when the TCR has relatively high affinity for its cognate antigen. In one clinical study of a high affinity transduced TCR to the MART-1 melanocyte antigen, off-tumor toxicity was observed in the form of significant uveitis and otitis, based on destruction of pigmented cells in the eye and inner ear, respectively (78). In a second example of off-tumor on-target toxicity, a TCR specific for the MAGE-A3 cancer testis antigen was known to cross-react with an epitope derived from the related antigen MAGE-A12; in clinical trials, neurologic toxicity was observed and found to be a result of previously unrecognized expression of MAGE-A12 in the brain (143). The dose-limiting toxicity of TCRs directed to carcinoembryonic antigen (CEA) was colitis (145). Although testing for on-target toxicity can be relatively straightforward, for example by RT-PCR from archived normal tissues, testing for potential off-target toxicities of TCRs is significantly more challenging. Typically, the starting point for identifying off-target toxicity requires testing of the new TCR against a panel of live cultured cells that are representative of human tissues to serve as targets. In one case, despite extensive pre-clinical testing on panels of cell lines, cardiac toxicity of a MAGE-A3 directed TCR could not be replicated in vitro until beating cardiac myocytes derived from induced pluripotent cells were used as targets (144, 146); in this case, the cause of ‘off-target’ toxicity turned out to be the result of TCR cross-reactivity to an unrelated peptide derived from titin(144, 146). Interestingly, the effects of TCR-transduced T cells that have been encountered clinically have not been a result of the predicted effects of mispairing and poor signaling; rather, the toxicities have been related to TCR affinity and specificity, and demonstrate the high potency of TCR-transduced T cell products. Clinical trials with native-TCR and engineered-TCR transduced T cells directed to a number of HLA-restricted antigens are underway in hematologic malignancies and solid tumors (Table 3), with results showing early promise (147).
T cells re-directed with chimeric antigen receptors
The first generation of chimeric antigen receptors (CARs) were engineered receptors comprising an scFv (where the variable portions of the light and heavy chains of a high-affinity antibody are connected by a linker sequence), a transmembrane domain and the signaling domain of CD3zeta (79, 148). Since then, second generation CARs have included the costimulatory domains derived from CD28, 4-1BB, or OX40 to optimize T cell activation, and these have improved function in vivo particularly against more aggressive tumors that do not express costimulatory molecules (149); third ‘generation ’ CARs also include the signaling domains of a third molecule such as TNF receptor family members such as 4-1BB or Ox40; these have less potent cytotoxic activity but persist longer in vivo (150-152). Because CARs are antibody-based, high-affinity single-chain variable fragments (scFv) derived from antibody sequences typically have been directed to native surface antigens, which restricts suitable targets to proteins or epitopes displayed on the surface of the target cell. (Figure 7)
Figure 7. T cells can be redirected to possess specificity for tumors.
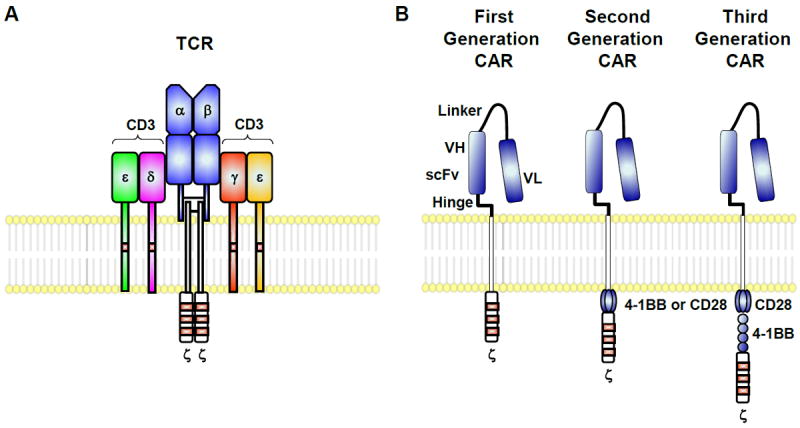
(A) Endogenous T cells express a single TCR. (B) Alternatively, these genes can be engineered to express chimeric tumor antigen–specific receptors or “CARs” that target surface antigens in an MHC-independent manner. T bodies express an extracellular portion that is usually derived from an antibody and intracellular signaling modules derived from T cell signaling proteins. First generation CARs contain CD3ζ, while second generation CARs possess a co-stimulatory endodomain (e.g., CD28 or 4-1BB) fused to CD3ζ. Third generation CARs consist of two co-stimulation endodomains linked to CD3ζ.
The exact mechanisms of how CARs function are still unknown (153), but it does appear that CARs homodimerize independently of the TCR, and only become part of the CD3 complex if the transmembrane domain selected is that of CD3zeta (154). This is not entirely surprising given that the transmembrane or intracellular signaling domains typically dimerize (CD8alpha, CD28), although some of them (4-1BB) are thought to form trimers in their native conformation. There is also some evidence to suggest that the spacing and conformation of binding to the epitope between the target and the CAR-T cell is important in optimizing CAR-induced signaling (155), whereas affinity appears to have less of a role in CAR-T cells (particularly compared to TCR-redirected T cells).
Similarly to TCRs, CAR-directed T cells do seem to maintain an exquisite sensitivity to low levels of the cognate antigen. This is perhaps surprising because the signaling domains of CARs are not expected to amplify signal to the same degree as TCR triggering of the entire CD3 receptor complex. Indeed, in vitro data suggests that CAR-T cells have a threshold for signaling at about 50 molecules/target, whereas native T cells require engagement of 1-10 molecules for TCR triggering (156, 157). Recent results suggest that the TCR has multiple modes of downstream signaling that regulate discrete functional events, and that the number of ITAMS recruited to the synapse regulate these distinct signaling pathways (158). Clinical investigation of CAR-T cells have confirmed this low threshold for activation: in trials of CAR-T cells directed to the tumor antigen Her2/neu or carbonic anhydrase IX, subjects have experienced severe toxicity based on low-level expression of the target antigen in the lung or the biliary tract, respectively (159, 160).
Currently, most pre-clinical investigation of new forms of CAR-T cells involve xenogeneic immunodeficient models, where human tumor is implanted either subcutaneously or orthotopically, and human T cells are injected either into the tumor or intravenously, either simultaneously or after tumor engraftment. These models are limited in that the tumor microenvironment is not replicated in the animal, the remaining arms of the immune system are absent or debilitated, and there is generally no possibility of evaluating off-tumor expression of target. In the case of CD19, for example, pre-clinical models did not predict that CAR-T cells would cause the degree of cytokine release or macrophage activation that has been observed clinically (32). Although syngeneic models (161, 162) may overcome some of the limitations of the xenogeneic models, the active cell under evaluation is the engineered T cell, and mouse and human T cells do exhibit significant mechanistic differences that affect the evaluation of the engineered CAR molecule; for example, mouse T cells are much more dependent on CD28 signaling, and 4-1BB signaling has modest effects at best (163, 164).
There are number of clinical trials of CAR-T cells directed to a variety of antigens underway (Tables 1-2) (165). Several centers have directed effort to CD19-directed CAR-T cells (Table 1), and to other B cell markers such as immunoglobulin light chains and CD20 (Table 2), in part because hematopoietic cells have been extensively characterized, and the expression of their surface molecules is often lineage-dependent. Multiple reviews of the CD19 CAR-T cell trials have been written (166-169), and are only discussed in aggregate here. One issue that has complicated the interpretation of the CD19 CAR-T cell trials is that each center has developed their own CAR, with different single-chain variable fragments to effect antigen binding, different signaling domains, different modes of introduction of the CAR gene into T cells, different conditioning regimens, and different post-CAR-T infusion interventions. However, in sum, it has become apparent that some B cell malignancies are more consistently clinically responsive to CD19-CAR T than others; for example, trials in chronic lymphocytic leukemia (CLL) have yielded very mixed clinical results (111, 170-173), whereas trials in acute lymphocytic leukemia (ALL) have yielded impressive responses in multiple centers (32, 100, 174). In one case of pediatric ALL, a patient relapsed with CD19-negative disease, indicating that the CD19-CAR T effected very strong selective pressure on cells expressing the CD19 target (32). In contrast, tumor cells from all the non-responding patients with CLL appear to retain CD19 expression. The fate and length of persistence of the CAR-T cells seems to have a significant impact on the clinical responses, but the determinants of these variables are still unclear. It is likely that factors such as the input cell population and the tumor microenvironment play a prominent role in determining CAR-T persistence and therefore on clinical efficacy, even when other variables (type of CAR-T and manufacturing process) are controlled.
Table 2.
Summary of current clinical trials of CAR T cells for cancer
| Target Antigens | Cancers | CAR Signaling Domain | Combinatorial/Engineering Strategies (Biologicals, Drugs) | Phase/ID | Sponsor |
|---|---|---|---|---|---|
| CD20 | ALL; CLL; NHL | CAR: 4-1BB-CD3ζ | None | NP; NCT01735604 | Chinese PLA General Hospital |
| CD30 | NHL; HL | CAR: CD28-CD3ζ | None | I; NCT01316146 | Baylor College of Medicine |
| CD30 | NHL; HL | CAR: CD28-CD3ζ | CD30 CAR transduced EBV-specific CTLs | I; NCT01192464 | Baylor College of Medicine |
| CD33 | Relapsed adult myeloid leukemia; chemotherapy refractory adult myeloid leukemia | CAR: CD137- CD3ζ | None | I/II; NCT01864902 | Chinese PLA General Hospital |
| CD138 | Relapsed and/or chemotherapy resistant multiple myeloma | CAR: CD137-CD3ζ | None | I/II; NCT01886976 | Chinese PLA General Hospital |
| cMet | Metastatic breast cancer; Triple negative breast cancer | CAR: 4-1BB-CD3ζ | None | I; NCT01837602 | University of Pennsylvania |
| EGFRvIII | Malignant glioma; Glioblastoma; Brain cancer | CAR: CD28-4-1BB-CD3ζ | None | I/II; NCT01454596 | National Cancer Institute |
| ErbB | Head and neck cancer | CAR: CD28-CD3ζ | ErbB CAR coexpressed with 4αβ chimeric cytokine receptor to enable ex vivo expansion of engineered T cells using IL-4 | I; NCT01818323 | King’s College London |
| FAP | Malignant pleural mesothelioma | CAR: CD28-CD3ζ | None | I; NCT01722149 | University of Zurich |
| GD-2 | Neuroblastoma | CAR: CD28-OX40-CD3ζ | iCaspase9 safety switch/AP1903 dimerizing drug | I; NCT01822652 | Baylor College of Medicine |
| HER2 | HER2+ malignancies | CAR: CD28-CD3ζ | CD19 CAR-transduced EBV-specific CTLs expressing a dominant negative TGFβ receptor | I; NCT00889954 | Baylor College of Medicine |
| HER2 | Glioblastoma multiforme | CAR: CD28-CD3ζ | CMV-specific CTLs | I; NCT01109095 | Baylor College of Medicine |
| Ig Kappa Light Chain | Lymphoma; Myeloma; Leukemia | CAR: CD28-CD3ζ | None | I; NCT00881920 | Baylor College of Medicine |
| Mesothelin | Mesothelin-expressing cancers | CAR: CD28-CD3ζ | IL-2 | I/II: NCT01583686 | National Cancer Institute |
| Mesothelin | Metastatic pancreatic cancer | CAR: 4-1BB-CD3ζ | None | I; NCT01897415 | University of Pennsylvania |
| PSMA | Prostate cancer | CAR: CD28-CD3ζ | HSV thymidine kinase (used for imaging and suicide gene) | I; NCT01140373 | Memorial Sloan-Kettering Cancer Center |
ALL, Acute lymphoblastic leukemia; CLL, Chronic lymphocytic leukemia; NHL, non-Hodgkin lymphomas; EBV, Epstein–Barr virus; CTLs, Cytotoxic T lymphocytes; iCaspase9, inducible caspase 9; CMV, Cytomegalovirus; HSV, Herpes simplex virus; NP, Information not provided.
There is also great interest in developing CAR-T against other hematologic malignancies. The carbohydrate antigen Lewis Y (carbohydrate antigen) is being tested as a potential target in AML, MDS and multiple myeloma patients (clinicaltrials.gov NCT01716364). Specific targets for AML (175) and multiple myeloma(176) are in pre-clinical development.
The development of CARs for solid tumors has been challenging, in part because of the lack of extensive literature on specific surface markers expressed on malignant epithelial cells. Furthermore, it is not clear that many surface markers are exclusively expressed on tumor cells, and more often, targets with merely higher levels of expression on tumor than normal tissue have been selected as CAR targets. Although this approach has safety concerns (160, 177), it is possible that a therapeutic window will be found. For example, despite the death that rapidly ensued after administration of 1010 dose of Her2/neu CAR-T in a patient aggressively conditioned, new trials directed against the same antigen but starting with lower doses are underway (NCT00902044, NCT01109095).
T cell products that employ a safety check mechanism, whether based on transient expression of the CAR (such as RNA electroporation (125)) or a suicide gene encoded into the transduced cells, are an attractive method to initiate clinical testing. This may be necessary, for example for FAP-directed CAR-T cells (178, 179), GD2-directed CAR-T cells (180), or PSMA-directed CAR-T cells (clinicaltrials.gov NCT01140373), where pre-clinical testing or antibody-based testing in the clinic indicate some concerning potential tissue-directed toxicities. The choice of safety check mechanism may also affect the function of the CAR-T cells: transient expression of the CAR may sensitize the patient to the CAR (33); incorporating transient viral vectors or viral proteins may also be immunogenic. The humanized caspase9 suicide system is very attractive, and has been clinically tested in the setting of donor lymphocyte infusions after bone marrow transplant (181), but it is unclear whether all the transduced T cells are completely eliminated, should this be required. Alternatively, the synthetic biology of CAR-T cells may borrow more lessons from nature: that is, one way to increase specificity of the CAR-T cells is to separate T cell signal 1 (antigen) from signal 2 (costimulation). In one paper (182), this strategy was successfully implemented in in vitro and in mouse models, where the primary antigen-receptor had low affinity and delivered a weak (akin to a TCR signal), but a second engineered receptor that engaged separate antigen delivered the costimulatory signals (chimeric costimulatory receptor, CCR); only engagement of both engineered receptors generated a sustained T cell response.
The field of T cell engineering is now entering adolescence, and creative solutions to many of the current limitations are sure to emerge (Figure 8)). For example, one group of investigators hypothesized that in the setting of graft vs host disease following allogeneic bone marrow transplant, T cells could only cause damage to the tissues if they trafficked to those tissues; by reducing tissue trafficking with administration of a drug blocking CCR5 (maraviroc), GvHD was ameliorated in early studies (183), and confirmatory studies are underway. Incorporating chemokine receptors to alter tumor-specific T cell migration is also under investigation (77), as are mechanisms to improve T cell resistance to inhibitory signals and perhaps to avoid conditioning chemotherapy regimens (162).
Figure 8. Strategies for engineering effective genetically-modified T cells.
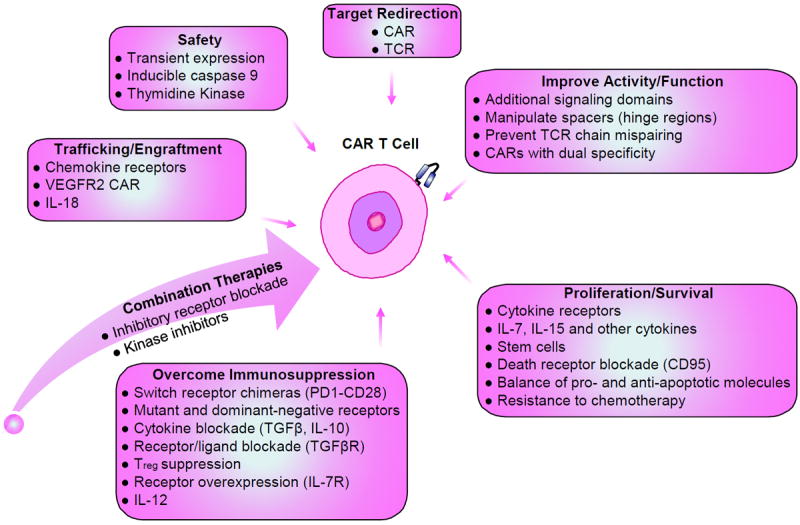
Various strategies can be undertaken to genetically modify T cells for adoptive therapy in an endeavor to enhance function and survival, proliferation, trafficking to tumor sites, and safety. Through genetic modification, these cells may also be armed to be efficacious in the immunosuppressive tumor microenvironment. Combination therapy can be used to improve the therapeutic efficacy of engineered T cells.
The future of CAR therapies looks bright, in part because early studies have shown that CAR-T cells are quite potent; strategies to expedite the discovery of suitable surface antigens, and generating the single-chain variable fragments to perform rapid throughput testing on tumors and on normal tissues to identify potential off-tumor reactivity, have the potential to make CAR-T cells more widely applicable.
Biomarkers for T cell therapies
The field of biomarkers has undergone dramatic evolution over the past few years, with an increased realization that relevant and meaningful measures of biological activity are unlikely to be generated simply on the basis of hypothesis testing, and that endeavors to interrogate for biological activity need to be supplemented by broader and hypothesis generating studies (184). This concept is particularly relevant for strategies that seek to therapeutically manipulate the immune system through immune modulation or immune activation, where the inherent multidimensional and integrated complexity of the immune system inevitably confounds scientific reductionism. A parallel concept is the implementation of uniform data reporting guidelines to support more transparent and systematic analysis of data from T cell therapy trials (185).
In a general sense, biomarkers provide information about the bioactivity of the tested therapy. Beyond this point, in early stage trials principal objectives of biomarker studies are to identify parameters that reveal specific information about the mechanism of action of the treatment and to provide proof of the tested principle being operative. In more advanced trials, the focused principle objective of biomarker studies is to identify and eventually validate biomarkers that correlate with the efficacy of the treatment and can potentially be pursued as surrogate endpoints for the treatment.
Unlike traditional strategies, where the treatment modality is an inert chemical compound, cell therapies are characterized by the fact that the “drug” is a biologically viable, dividing, and evolving entity, which interacts with and responds to the myriad complexities of the host biology. As a result, biomarker strategies for cell therapies must focus on studying not only the impact of the infused cells on patient biology, but also the infused cells themselves (186). A fundamental starting point for these studies is a thorough understanding of the properties of the manufactured cell product, obtained through product release, potency, and characterization assays.
Arguably, the field of biomarker studies for T cell therapy trials was ushered in by the seminal reports from the NCI group which demonstrated cancer regression in melanoma patients following adoptive transfer of bulk TIL-derived lymphocytes (187) and subsequently gene-engineered MART-1 specific T cells (98). These studies were among the first to examine persistence of infused cells, characterize their surface phenotype, and indirectly demonstrate in vivo functionality through cancer regression and autoimmunity.
Perhaps predictably, persistence of infused cells has been shown to correlate with cancer regression and durability of remission (188). Indeed, a major limitation for maximal efficacy of T cell therapy-based approaches has been thought to be the lack of robust long-term persistence of transferred cells, a limitation that now appears to have been overcome in at least some settings (32, 111). More controversy exists with regard to the phenotype and functional status of T cells optimally needed for anti-tumor immunity. Earlier work suggested that TIL cells cultured minimally were less differentiated, more diverse phenotypically, and had superior efficacy following transfer (189). Other work from the surgery branch of the NCI has indicated that naïve rather than memory cells were superior for adoptive transfer, demonstrating better transduction, more robust expansion, enhanced proliferative potential and telomere length, and were less susceptible to terminal differentiation (190), while the same group has shown that adoptive T cell treatment efficacy may be related to the persistence of T cells that are or can convert in vivo to memory cells (191). More recently, work principally from the Seattle group in primate models has suggested that central memory cells might be more effective in adoptive T cell transfer strategies (43). Even more recent and provocative work from the Munich group has suggested that there is a phenotypic plasticity within at least some naïve and memory T cell subsets (192). Together these disparate results highligt the potential folly of interrogating differentiation phenotypes of persisting cells as an essential element of biomarker studies. Direct assessment of the in vivo functionality of infused cells has been difficult to accomplish, at least in part due to the aforementioned relatively poor persistence of infused cells, although recent studies have demonstrated directly ex-vivo the ability of long-term persisting cells to recognize antigen-positive targets (111). Less direct measures of T cell bioactivity have included the measurement of systemic cytokine levels in patients post T cell infusion. Initial efforts in this area focused on specific cytokines directly associated with T cell effector functions; more recent and hypothesis agnostic efforts in the setting of potent clinical efficacy have revealed a broader profile of immune activation that might be an important element for ultimate efficacy of T cell-based immunotherapy strategies (32, 100, 111, 173).
The principal mechanism of action for T cells to effect anti-tumor activity is through direct engagement and cytolysis of target cells as well as production and secretion of soluble cytokine and chemokines which directly impact tumor cells and also orchestrate a more integrated anti-tumor inflammatory response. Accordingly, biomarker studies to interrogate T cell mechanism of action focus on detecting the presence and effector functionality of infused T cells. Although this objective is relatively straightforward to accomplish in cases where cancers are predominant in peripheral blood or bone marrow, it is a considerably more difficult challenge for cancers where disease is not readily accessible. Indeed, recent clinical data has provided compelling evidence to support the need to evaluate the T cell function at the site of disease (193, 194). Data principally accumulated in the context of adverse events demonstrate that infused T cells do in fact traffic throughout the body and home to sites where target antigen is expressed (144, 146), highlighting the critical need to develop innovative and sensitive approaches to enable the interrogation of tissues for T cell presence and functionality.
Approaches to monitor T cell persistence. In most adoptive T cell immunotherapy studies the total infused cell number corresponds to a small fraction of the total T cells, although lymphodepleting conditioning can skew this ratio at early timepoints post infusion. Homeostatic and antigen-driven expansion has been shown to drive high frequencies of infused T cells in patients at late times post infusion (32, 98, 111). Both molecular and flow-cytometry-based approaches have been developed to evaluate persistence and homing of infused T cells.
Quantitative PCR (Q-PCR)-based approaches have been developed in a number of cases to detect and quantify the persistence of infused T cells. Such approaches are feasible if the infused T cells have been genetically engineered prior to infusion, and typically afford for the ability to detect infused cells at frequencies as low as 0.01% of total cells, and provides important information about persistence, trafficking, and homing of infused T cells in patients. This approach is increasingly being applied in clinical studies to monitor T cell persistence see for example (32, 100, 111, 173), as well as to interrogate and demonstrate the contribution of infused cells to serious adverse events (SAE) (144). Recent technological advances in the ability to perform high throughput deep sequencing of CDR3 domains for TCR loci to detect and quantify individual TCR clonotypes in samples (195) opens up the potential to be able to obtain molecular signatures for individual clonotypes that persist in patients and correlate this signature to the original infusion product. A considerable limitation of molecular approaches is that they generate data from bulk populations of cells. The robust development of single cell multiplexed digital PCR-based approaches (196) opens up the exciting possibility for not only more sensitive detection of infused cells in samples, but also the ability to interrogate functionality of infused engineered cells at the single cell level.
Flow cytometric based approaches depend on the availability of antibody reagents to detect gene–engineered and infused cells. Such reagents have included MHC class I multimers (dextramers, tetramers, pentamers) which have been employed in a large number of clinical trials; more recently in the context of CAR trials idiotype-specific antibodies that recognize CAR-engineered cells have been employed to detect infused cells with high specificity (32, 100, 111). This approach typically requires that the frequency of antigen specific cells is at least 0.2% of the total CD3 population, although reports of considerably more sensitive detection have been reported (197), as well as the development of higher-throughput combinatorial strategies that increase sensitivity and reduce sample usage (198). One advantage of flow-cytometry based approaches is that they can be readily combined with stains for surface phenotypic or functional markers allowing for the generation of more integrated data sets.
Technical advancements in polychromatic flow-cytometry combined with an increased understanding of T cell biology have precipitated a number of flow-cytometry-based approaches to interrogate T cell function; specific markers and strategies are summarized in a recent review (186). The continued development of mass cytometry based platforms and algorithm driven hierarchical clustering approaches which allow for the ability to simultaneously interrogate very large numbers of T cell molecules including surface and intracellular proteins, phosphoproteins and RNA species (199, 200) has the potential to render traditional flow cytometry-based approaches to interrogate T cell functionality as a stand-alone platform obsolete.
As discussed above, the integrated complexity of the immune system mandates that part of the evaluation of T cell therapy focus on the impact of the treatment on the broader immune system. Although this relatively new concept has not yet been broadly implemented in clinical trials, one approach that has been implemented with some success has been to evaluate systemic cytokine levels in patients during treatment. In studies targeting leukemias using CAR-engineered T cells this strategy has revealed that engineered T cell activation and anti-tumor activity results in broad and potent cytokine-driven effects, including cytokine release syndrome (CRS) (32, 111), as well as macrophage activation syndrome (MAS) and hemophagocytic lymphohistiocytosis (HLH). Notably, the agnostic interrogation of cytokines in these trials unexpectedly identified IL-6 as a major cytokine induced by the CAR therapy, an observation which has resulted in the successful experimental deployment of the anti-IL-6 receptor antagonist antibody Tocilizumab to mitigate the observed cytokine induced toxicity (32), a treatment now being applied more systematically to counteract CRS (201).
Building T cell therapies into a pillar of medicine
Now that adoptive T cell therapy is showing such dramatic effects, the question becomes, how can we move past offering this as boutique medicine in major medical centers, and offer it in communities all over the world? There are several logistical issues that need to be addressed: shipping and tracking of autologous blood products, large-scale manufacturing of vectors and T cell products and validation of processes and immunologic assays necessary for quality control. This is likely to require adaptation of a variety of fields outside the scope of most physicians and immunologists, from blood banking, a field that has learned to manage cell collection and processing techniques in a standardized ways at the international level, to robotic manufacturing techniques used in high-throughput laboratories and systems such as building automobiles(202).
Understandably, some scientists have raised concerns about the complexity of this type of therapy; even performing multi-center studies with T cell products has been challenging at the academic level. Also, most regulatory health agencies are set up to handle the testing of drugs, which are manufactured centrally, the active ingredient is measured and controlled, and where one lot treats many patients. In contrast, the active ‘ingredient’ of a T cell product is challenging to define because it reflects a heterogeneous population of T cells; even the typical Phase I dose-escalation trial design is not necessarily the most appropriate method to test for safety of T cell products, given that the ‘dose’ is not static or even maximal immediately after infusion. Furthermore, each ‘lot’ produced can only be used for one patient, and thus far, T cell products have been manufactured locally. As these therapies move to Phase II or III studies to obtain an indication for use, it is not yet clear whether it will be possible to perform double-blind randomized controlled trials that are considered the gold standard in establishing a standard of care therapy.
Although these issues pose significant challenges, they are not necessarily insurmountable barriers; organ and bone marrow transplants are now considered fairly routine (203). As manufacturing of cell products becomes more automated, and as scientists better define the key components of what makes the most bioactive, optimal T cell product, it will become an absolute necessity to develop large-scale, centralized processes that can generate standardized quality-controlled T cell products. This endeavor will require scientists, physicians, and industry to work together in building the necessary infrastructure and adapting the current regulatory standards to reflect an entirely new pillar of medicine, comprised of personalized cell therapies (202).
References
- 1.Anderson WF. Prospects for human gene therapy. Science. 1984;226:401–9. doi: 10.1126/science.6093246. [DOI] [PubMed] [Google Scholar]
- 2.Friedmann T. A brief history of gene therapy. Nat Genet. 1992;2:93–8. doi: 10.1038/ng1092-93. [DOI] [PubMed] [Google Scholar]
- 3.Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, Bakker A, Roberts MR, June CH, Jalali S, Lin AA, Pennathur-Das R, Hege KM. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. 2000;96:785–93. [PubMed] [Google Scholar]
- 4.Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, Macken C, Richman DD, Christopherson C, June CH, Lazar R, Broad DF, Jalali S, Hege KM. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Ther. 2002;5:788–97. doi: 10.1006/mthe.2002.0611. [DOI] [PubMed] [Google Scholar]
- 5.Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, Vogel AN, Kalos M, Riley JL, Deeks SG, Mitsuyasu RT, Bernstein WB, Aronson NE, Levine BL, Bushman FD, June CH. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, Schneider T, Hofmann J, Kucherer C, Blau O, Blau IW, Hofmann WK, Thiel E. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360:692–8. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 7.Liang M, Kamata M, Chen KN, Pariente N, An DS, Chen IS. Inhibition of HIV-1 infection by a unique short hairpin RNA to chemokine receptor 5 delivered into macrophages through hematopoietic progenitor cell transduction. J Gene Med. 2010;12:255–65. doi: 10.1002/jgm.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, Guschin DY, Rupniewski I, Waite AJ, Carpenito C, Carroll RG, Orange JS, Urnov FD, Rebar EJ, Ando D, Gregory PD, Riley JL, Holmes MC, June CH. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–16. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–44. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 10.Louis CU, Straathof K, Bollard CM, Ennamuri S, Gerken C, Lopez TT, Huls MH, Sheehan A, Wu MF, Liu H, Gee A, Brenner MK, Rooney CM, Heslop HE, Gottschalk S. Adoptive transfer of EBV-specific T cells results in sustained clinical responses in patients with locoregional nasopharyngeal carcinoma. J Immunother. 2010;33:983–90. doi: 10.1097/CJI.0b013e3181f3cbf4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leen AM, Bollard CM, Mendizabal AM, Shpall EJ, Szabolcs P, Antin JH, Kapoor N, Pai SY, Rowley SD, Kebriaei P, Dey BR, Grilley BJ, Gee AP, Brenner MK, Rooney CM, Heslop HE. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood. 2013;121:5113–23. doi: 10.1182/blood-2013-02-486324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haque T, Wilkie GM, Jones MM, Higgins CD, Urquhart G, Wingate P, Burns D, McAulay K, Turner M, Bellamy C, Amlot PL, Kelly D, MacGilchrist A, Gandhi MK, Swerdlow AJ, Crawford DH. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial. Blood. 2007;110:1123–31. doi: 10.1182/blood-2006-12-063008. [DOI] [PubMed] [Google Scholar]
- 13.Barker JN, Doubrovina E, Sauter C, Jaroscak JJ, Perales MA, Doubrovin M, Prockop SE, Koehne G, O’Reilly RJ. Successful treatment of EBV-associated posttransplantation lymphoma after cord blood transplantation using third-party EBV-specific cytotoxic T lymphocytes. Blood. 2010;116:5045–9. doi: 10.1182/blood-2010-04-281873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leen AM, Myers GD, Sili U, Huls MH, Weiss H, Leung KS, Carrum G, Krance RA, Chang CC, Molldrem JJ, Gee AP, Brenner MK, Heslop HE, Rooney CM, Bollard CM. Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat Med. 2006;12:1160–6. doi: 10.1038/nm1475. [DOI] [PubMed] [Google Scholar]
- 15.Hanley PJ, Cruz CR, Savoldo B, Leen AM, Stanojevic M, Khalil M, Decker W, Molldrem JJ, Liu H, Gee AP, Rooney CM, Heslop HE, Dotti G, Brenner MK, Shpall EJ, Bollard CM. Functionally active virus-specific T cells that target CMV, adenovirus, and EBV can be expanded from naive T-cell populations in cord blood and will target a range of viral epitopes. Blood. 2009;114:1958–67. doi: 10.1182/blood-2009-03-213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leen AM, Christin A, Myers GD, Liu H, Cruz CR, Hanley PJ, Kennedy-Nasser AA, Leung KS, Gee AP, Krance RA, Brenner MK, Heslop HE, Rooney CM, Bollard CM. Cytotoxic T lymphocyte therapy with donor T cells prevents and treats adenovirus and Epstein-Barr virus infections after haploidentical and matched unrelated stem cell transplantation. Blood. 2009;114:4283–92. doi: 10.1182/blood-2009-07-232454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall SS. A commotion in the blood : life, death, and the immune system. xiv. New York: Henry Holt; 1997. p. 544. [Google Scholar]
- 18.Weiden PL, Flournoy N, Thomas ED, Prentice R, Fefer A, Buckner CD, Storb R. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N Engl J Med. 1979;300:1068–73. doi: 10.1056/NEJM197905103001902. [DOI] [PubMed] [Google Scholar]
- 19.Korngold R, Sprent J. Lethal graft-versus-host disease after bone marrow transplantation across minor histocompatibility barriers in mice. Prevention by removing mature T cells from marrow. J Exp Med. 1978;148:1687–98. doi: 10.1084/jem.148.6.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, Posati S, Rogaia D, Frassoni F, Aversa F, Martelli MF, Velardi A. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097–100. doi: 10.1126/science.1068440. [DOI] [PubMed] [Google Scholar]
- 21.Venstrom JM, Pittari G, Gooley TA, Chewning JH, Spellman S, Haagenson M, Gallagher MM, Malkki M, Petersdorf E, Dupont B, Hsu KC. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N Engl J Med. 2012;367:805–16. doi: 10.1056/NEJMoa1200503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porter DL, Levine BL, Bunin N, Stadtmauer EA, Luger SM, Goldstein S, Loren A, Phillips J, Nasta S, Perl A, Schuster S, Tsai D, Sohal A, Veloso E, Emerson S, June CH. A phase 1 trial of donor lymphocyte infusions expanded and activated ex vivo via CD3/CD28 costimulation. Blood. 2006;107:1325–31. doi: 10.1182/blood-2005-08-3373. [DOI] [PubMed] [Google Scholar]
- 23.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, Steinberg SM, White DE, Dudley ME. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yao X, Ahmadzadeh M, Lu YC, Liewehr DJ, Dudley ME, Liu F, Schrump DS, Steinberg SM, Rosenberg SA, Robbins PF. Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood. 2012;119:5688–96. doi: 10.1182/blood-2011-10-386482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, Surh CD, Rosenberg SA, Restifo NP. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–12. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wrzesinski C, Paulos CM, Kaiser A, Muranski P, Palmer DC, Gattinoni L, Yu Z, Rosenberg SA, Restifo NP. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother. 2010;33:1–7. doi: 10.1097/CJI.0b013e3181b88ffc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pagliara D, Savoldo B. Cytotoxic T lymphocytes for the treatment of viral infections and posttransplant lymphoproliferative disorders in transplant recipients. Curr Opin Infect Dis. 2012;25:431–7. doi: 10.1097/QCO.0b013e3283551dd3. [DOI] [PubMed] [Google Scholar]
- 32.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, Zhao Y, Kalos M, June CH. T Cells Expressing Chimeric Antigen Receptors Can Cause Anaphylaxis in Humans. CANCER IMMUNOLOGY RESEARCH. 2013;1 doi: 10.1158/2326-6066.CIR-13-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, 3rd, Armand P, Cutler C, Ho VT, Treister NS, Bienfang DC, Prasad S, Tzachanis D, Joyce RM, Avigan DE, Antin JH, Ritz J, Soiffer RJ. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. 2011;365:2055–66. doi: 10.1056/NEJMoa1108188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perna SK, De Angelis B, Pagliara D, Hasan ST, Zhang L, Mahendravada A, Heslop HE, Brenner MK, Rooney CM, Dotti G, Savoldo B. Interleukin 15 provides relief to CTLs from regulatory T cell-mediated inhibition: implications for adoptive T cell-based therapies for lymphoma. Clin Cancer Res. 2013;19:106–17. doi: 10.1158/1078-0432.CCR-12-2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu C, Lewis CM, Lou Y, Xu C, Peng W, Yang Y, Gelbard AH, Lizee G, Zhou D, Overwijk WW, Hwu P. Agonistic antibody to CD40 boosts the antitumor activity of adoptively transferred T cells in vivo. J Immunother. 2012;35:276–82. doi: 10.1097/CJI.0b013e31824e7f43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noji S, Hosoi A, Takeda K, Matsushita H, Morishita Y, Seto Y, Kakimi K. Targeting spatiotemporal expression of CD137 on tumor-infiltrating cytotoxic T lymphocytes as a novel strategy for agonistic antibody therapy. J Immunother. 2012;35:460–72. doi: 10.1097/CJI.0b013e31826092db. [DOI] [PubMed] [Google Scholar]
- 38.Donia M, Fagone P, Nicoletti F, Andersen RS, Hogdall E, Straten PT, Andersen MH, Svane IM. BRAF inhibition improves tumor recognition by the immune system: Potential implications for combinatorial therapies against melanoma involving adoptive T-cell transfer. Oncoimmunology. 2012;1:1476–83. doi: 10.4161/onci.21940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, Nakayama-Hosoya K, Iriguchi S, Uemura Y, Shimizu T, Takayama N, Yamada D, Nishimura K, Ohtaka M, Watanabe N, Takahashi S, Iwamoto A, Koseki H, Nakanishi M, Eto K, Nakauchi H. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114–26. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 40.Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, Fujii S, Koseki H, Kawamoto H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell. 2013;12:31–6. doi: 10.1016/j.stem.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 41.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, Yvon E, Weiss HL, Liu H, Rooney CM, Heslop HE, Brenner MK. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–70. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood. 2012;119:72–82. doi: 10.1182/blood-2011-07-366419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hinrichs CS, Borman ZA, Cassard L, Gattinoni L, Spolski R, Yu Z, Sanchez-Perez L, Muranski P, Kern SJ, Logun C, Palmer DC, Ji Y, Reger RN, Leonard WJ, Danner RL, Rosenberg SA, Restifo NP. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci U S A. 2009;106:17469–74. doi: 10.1073/pnas.0907448106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, Wang E, Douek DC, Price DA, June CH, Marincola FM, Roederer M, Restifo NP. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–7. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lugli E, Dominguez MH, Gattinoni L, Chattopadhyay PK, Bolton DL, Song K, Klatt NR, Brenchley JM, Vaccari M, Gostick E, Price DA, Waldmann TA, Restifo NP, Franchini G, Roederer M. Superior T memory stem cell persistence supports long-lived T cell memory. J Clin Invest. 2013;123:594–9. doi: 10.1172/JCI66327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res. 2011;17:6287–97. doi: 10.1158/1078-0432.CCR-11-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A. 2002;99:16168–73. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, Nuno MA, Richardson JE, Fan X, Ji J, Chu RM, Bender JG, Hawkins ES, Patil CG, Black KL, Yu JS. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother. 2013;62:125–35. doi: 10.1007/s00262-012-1319-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frohlich MW. Sipuleucel-T for the treatment of advanced prostate cancer. Semin Oncol. 2012;39:245–52. doi: 10.1053/j.seminoncol.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 52.Wheeler CJ, Black KL. DCVax-Brain and DC vaccines in the treatment of GBM. Expert Opin Investig Drugs. 2009;18:509–19. doi: 10.1517/13543780902841951. [DOI] [PubMed] [Google Scholar]
- 53.Fishman M. A changing world for DCvax: a PSMA loaded autologous dendritic cell vaccine for prostate cancer. Expert Opin Biol Ther. 2009;9:1565–75. doi: 10.1517/14712590903446921. [DOI] [PubMed] [Google Scholar]
- 54.June CH, Ledbetter JA, Linsley PS, Thompson CB. Role of the CD28 receptor in T-cell activation. Immunol Today. 1990;11:211–6. doi: 10.1016/0167-5699(90)90085-n. [DOI] [PubMed] [Google Scholar]
- 55.Levine BL, Ueda Y, Craighead N, Huang ML, June CH. CD28 ligands CD80 (B7-1) and CD86 (B7-2) induce long-term autocrine growth of CD4+ T cells and induce similar patterns of cytokine secretion in vitro. Int Immunol. 1995;7:891–904. doi: 10.1093/intimm/7.6.891. [DOI] [PubMed] [Google Scholar]
- 56.Levine BL, Bernstein WB, Connors M, Craighead N, Lindsten T, Thompson CB, June CH. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. J Immunol. 1997;159:5921–30. [PubMed] [Google Scholar]
- 57.Fowler DH, Odom J, Steinberg SM, Chow CK, Foley J, Kogan Y, Hou J, Gea-Banacloche J, Sportes C, Pavletic S, Leitman S, Read EJ, Carter C, Kolstad A, Fox R, Beatty GL, Vonderheide RH, Levine BL, June CH, Gress RE, Bishop MR. Phase I clinical trial of costimulated, IL-4 polarized donor CD4+ T cells as augmentation of allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2006;12:1150–60. doi: 10.1016/j.bbmt.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 58.Levine BL, Mosca JD, Riley JL, Carroll RG, Vahey MT, Jagodzinski LL, Wagner KF, Mayers DL, Burke DS, Weislow OS, St Louis DC, June CH. Antiviral effect and ex vivo CD4+ T cell proliferation in HIV-positive patients as a result of CD28 costimulation. Science. 1996;272:1939–43. doi: 10.1126/science.272.5270.1939. [DOI] [PubMed] [Google Scholar]
- 59.Riley JL, Carroll RG, Levine BL, Bernstein W, St Louis DC, Weislow OS, June CH. Intrinsic resistance to T cell infection with HIV type 1 induced by CD28 costimulation. J Immunol. 1997;158:5545–53. [PubMed] [Google Scholar]
- 60.Carroll RG, Riley JL, Levine BL, Feng Y, Kaushal S, Ritchey DW, Bernstein W, Weislow OS, Brown CR, Berger EA, June CH, St Louis DC. Differential regulation of HIV-1 fusion cofactor expression by CD28 costimulation of CD4+ T cells. Science. 1997;276:273–6. doi: 10.1126/science.276.5310.273. [DOI] [PubMed] [Google Scholar]
- 61.Renner C, Ohnesorge S, Held G, Bauer S, Jung W, Pfitzenmeier JP, Pfreundschuh M. T cells from patients with Hodgkin’s disease have a defective T-cell receptor zeta chain expression that is reversible by T-cell stimulation with CD3 and CD28. Blood. 1996;88:236–41. [PubMed] [Google Scholar]
- 62.Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, Rubin SC, Kaiser LR, June CH. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61:4766–72. [PubMed] [Google Scholar]
- 63.Bonyhadi M, Frohlich M, Rasmussen A, Ferrand C, Grosmaire L, Robinet E, Leis J, Maziarz RT, Tiberghien P, Berenson RJ. In vitro engagement of CD3 and CD28 corrects T cell defects in chronic lymphocytic leukemia. J Immunol. 2005;174:2366–75. doi: 10.4049/jimmunol.174.4.2366. [DOI] [PubMed] [Google Scholar]
- 64.Patten P, Devereux S, Buggins A, Bonyhadi M, Frohlich M, Berenson RJ. Effect of CD3/CD28 bead-activated and expanded T cells on leukemic B cells in chronic lymphocytic leukemia. J Immunol. 2005;174:6562–3. doi: 10.4049/jimmunol.174.11.6562. [DOI] [PubMed] [Google Scholar]
- 65.Maus MV, Thomas AK, Leonard DG, Allman D, Addya K, Schlienger K, Riley JL, June CH. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat Biotechnol. 2002;20:143–8. doi: 10.1038/nbt0202-143. [DOI] [PubMed] [Google Scholar]
- 66.Thomas AK, Maus MV, Shalaby WS, June CH, Riley JL. A cell-based artificial antigen-presenting cell coated with anti-CD3 and CD28 antibodies enables rapid expansion and long-term growth of CD4 T lymphocytes. Clin Immunol. 2002;105:259–72. doi: 10.1006/clim.2002.5277. [DOI] [PubMed] [Google Scholar]
- 67.Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC, Carroll RG, Riley JL, June CH. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol Ther. 2007;15:981–8. doi: 10.1038/mt.sj.6300134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maus MV, Thomas AK, Leonard DG, Allman D, Addya K, Schlienger K, Riley JL, June CH. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nature biotechnology. 2002;20:143–8. doi: 10.1038/nbt0202-143. [DOI] [PubMed] [Google Scholar]
- 69.Smith BD, Kasamon YL, Kowalski J, Gocke C, Murphy K, Miller CB, Garrett-Mayer E, Tsai HL, Qin L, Chia C, Biedrzycki B, Harding TC, Tu GH, Jones R, Hege K, Levitsky HI. K562/GM-CSF immunotherapy reduces tumor burden in chronic myeloid leukemia patients with residual disease on imatinib mesylate. Clin Cancer Res. 2010;16:338–47. doi: 10.1158/1078-0432.CCR-09-2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Broeren CP, Gray GS, Carreno BM, June CH. Costimulation light: activation of CD4+ T cells with CD80 or CD86 rather than anti-CD28 leads to a Th2 cytokine profile. J Immunol. 2000;165:6908–14. doi: 10.4049/jimmunol.165.12.6908. [DOI] [PubMed] [Google Scholar]
- 71.Godfrey WR, Ge YG, Spoden DJ, Levine BL, June CH, Blazar BR, Porter SB. In vitro-expanded human CD4(+)CD25(+) T-regulatory cells can markedly inhibit allogeneic dendritic cell-stimulated MLR cultures. Blood. 2004;104:453–61. doi: 10.1182/blood-2004-01-0151. [DOI] [PubMed] [Google Scholar]
- 72.Walker RE, Bechtel CM, Natarajan V, Baseler M, Hege KM, Metcalf JA, Stevens R, Hazen A, Blaese RM, Chen CC, Leitman SF, Palensky J, Wittes J, Davey RT, Jr, Falloon J, Polis MA, Kovacs JA, Broad DF, Levine BL, Roberts MR, Masur H, Lane HC. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood. 2000;96:467–74. [PubMed] [Google Scholar]
- 73.Levine BL, Humeau LM, Boyer J, MacGregor RR, Rebello T, Lu X, Binder GK, Slepushkin V, Lemiale F, Mascola JR, Bushman FD, Dropulic B, June CH. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci U S A. 2006;103:17372–7. doi: 10.1073/pnas.0608138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maier DA, Brennan AL, Jiang S, Binder-Scholl GK, Lee G, Plesa G, Zheng Z, Cotte J, Carpenito C, Wood T, Spratt SK, Ando D, Gregory P, Holmes MC, Perez EE, Riley JL, Carroll RG, June CH, Levine BL. Efficient clinical scale gene modification via zinc finger nuclease-targeted disruption of the HIV co-receptor CCR5. Hum Gene Ther. 2013;24:245–58. doi: 10.1089/hum.2012.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu K, Rosenberg SA. Transduction of an IL-2 gene into human melanoma-reactive lymphocytes results in their continued growth in the absence of exogenous IL-2 and maintenance of specific antitumor activity. J Immunol. 2001;167:6356–65. doi: 10.4049/jimmunol.167.11.6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kalbasi A, Shrimali RK, Chinnasamy D, Rosenberg SA. Prevention of interleukin-2 withdrawal-induced apoptosis in lymphocytes retrovirally cotransduced with genes encoding an antitumor T-cell receptor and an antiapoptotic protein. J Immunother. 2010;33:672–83. doi: 10.1097/CJI.0b013e3181e475cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33:780–8. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, Lee CC, Restifo NP, Schwarz SL, Cogdill AP, Bishop RJ, Kim H, Brewer CC, Rudy SF, VanWaes C, Davis JL, Mathur A, Ripley RT, Nathan DA, Laurencot CM, Rosenberg SA. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–46. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86:10024–8. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Uchida N, Cone RD, Freeman GJ, Mulligan RC, Cantor H. High efficiency gene transfer into murine T cell clones using a retroviral vector. J Immunol. 1986;136:1876–9. [PubMed] [Google Scholar]
- 81.Rosenberg SA, Aebersold P, Cornetta K, Kasid A, Morgan RA, Moen R, Karson EM, Lotze MT, Yang JC, Topalian SL, et al. Gene transfer into humans--immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323:570–8. doi: 10.1056/NEJM199008303230904. [DOI] [PubMed] [Google Scholar]
- 82.Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, Shearer G, Chang L, Chiang Y, Tolstoshev P, Greenblatt JJ, Rosenberg SA, Klein H, Berger M, Mullen CA, Ramsey WJ, Muul L, Morgan RA, Anderson WF. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science. 1995;270:475–80. doi: 10.1126/science.270.5235.475. [DOI] [PubMed] [Google Scholar]
- 83.Suerth JD, Schambach A, Baum C. Genetic modification of lymphocytes by retrovirus-based vectors. Curr Opin Immunol. 2012;24:598–608. doi: 10.1016/j.coi.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 84.Biasco L, Baricordi C, Aiuti A. Retroviral integrations in gene therapy trials. Mol Ther. 2012;20:709–16. doi: 10.1038/mt.2011.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bordignon C, Notarangelo LD, Nobili N, Ferrari G, Casorati G, Panina P, Mazzolari E, Maggioni D, Rossi C, Servida P, Ugazio AG, Mavilio F. Gene therapy in peripheral blood lymphocytes and bone marrow for ADA- immunodeficient patients. Science. 1995;270:470–5. doi: 10.1126/science.270.5235.470. [DOI] [PubMed] [Google Scholar]
- 86.Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, Morecki S, Andolfi G, Tabucchi A, Carlucci F, Marinello E, Cattaneo F, Vai S, Servida P, Miniero R, Roncarolo MG, Bordignon C. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–3. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 87.Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse E, Beldjord K, Asnafi V, MacIntyre E, Dal Cortivo L, Radford I, Brousse N, Sigaux F, Moshous D, Hauer J, Borkhardt A, Belohradsky BH, Wintergerst U, Velez MC, Leiva L, Sorensen R, Wulffraat N, Blanche S, Bushman FD, Fischer A, Cavazzana-Calvo M. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–42. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, Glimm H, Kuhlcke K, Schilz A, Kunkel H, Naundorf S, Brinkmann A, Deichmann A, Fischer M, Ball C, Pilz I, Dunbar C, Du Y, Jenkins NA, Copeland NG, Luthi U, Hassan M, Thrasher AJ, Hoelzer D, von Kalle C, Seger R, Grez M. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–9. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- 89.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, Vidaud M, Abel U, Dal-Cortivo L, Caccavelli L, Mahlaoui N, Kiermer V, Mittelstaedt D, Bellesme C, Lahlou N, Lefrere F, Blanche S, Audit M, Payen E, Leboulch P, l’Homme B, Bougneres P, Von Kalle C, Fischer A, Cavazzana-Calvo M, Aubourg P. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–23. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 90.Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, Down J, Denaro M, Brady T, Westerman K, Cavallesco R, Gillet-Legrand B, Caccavelli L, Sgarra R, Maouche-Chretien L, Bernaudin F, Girot R, Dorazio R, Mulder GJ, Polack A, Bank A, Soulier J, Larghero J, Kabbara N, Dalle B, Gourmel B, Socie G, Chretien S, Cartier N, Aubourg P, Fischer A, Cornetta K, Galacteros F, Beuzard Y, Gluckman E, Bushman F, Hacein-Bey-Abina S, Leboulch P. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 2010;467:318–22. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mavilio F, Pellegrini G, Ferrari S, Di Nunzio F, Di Iorio E, Recchia A, Maruggi G, Ferrari G, Provasi E, Bonini C, Capurro S, Conti A, Magnoni C, Giannetti A, De Luca M. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. 2006;12:1397–402. doi: 10.1038/nm1504. [DOI] [PubMed] [Google Scholar]
- 92.Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, Brugman MH, Pike-Overzet K, Chatters SJ, de Ridder D, Gilmour KC, Adams S, Thornhill SI, Parsley KL, Staal FJ, Gale RE, Linch DC, Bayford J, Brown L, Quaye M, Kinnon C, Ancliff P, Webb DK, Schmidt M, von Kalle C, Gaspar HB, Thrasher AJ. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–50. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, Schmidt M, Kramer A, Schwable J, Glimm H, Koehl U, Preiss C, Ball C, Martin H, Gohring G, Schwarzwaelder K, Hofmann WK, Karakaya K, Tchatchou S, Yang R, Reinecke P, Kuhlcke K, Schlegelberger B, Thrasher AJ, Hoelzer D, Seger R, von Kalle C, Grez M. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 94.Baum C, von Kalle C. Gene therapy targeting hematopoietic cells: better not leave it to chance. Acta Haematol. 2003;110:107–9. doi: 10.1159/000072459. [DOI] [PubMed] [Google Scholar]
- 95.Kustikova OS, Schiedlmeier B, Brugman MH, Stahlhut M, Bartels S, Li Z, Baum C. Cell-intrinsic and vector-related properties cooperate to determine the incidence and consequences of insertional mutagenesis. Mol Ther. 2009;17:1537–47. doi: 10.1038/mt.2009.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Newrzela S, Al-Ghaili N, Heinrich T, Petkova M, Hartmann S, Rengstl B, Kumar A, Jack HM, Gerdes S, Roeder I, Hansmann ML, von Laer D. T-cell receptor diversity prevents T-cell lymphoma development. Leukemia. 2012;26:2499–507. doi: 10.1038/leu.2012.142. [DOI] [PubMed] [Google Scholar]
- 97.Cassani B, Montini E, Maruggi G, Ambrosi A, Mirolo M, Selleri S, Biral E, Frugnoli I, Hernandez-Trujillo V, Di Serio C, Roncarolo MG, Naldini L, Mavilio F, Aiuti A. Integration of retroviral vectors induces minor changes in the transcriptional activity of T cells from ADA-SCID patients treated with gene therapy. Blood. 2009;114:3546–56. doi: 10.1182/blood-2009-02-202085. [DOI] [PubMed] [Google Scholar]
- 98.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL. Tumor Regression in Patients With Metastatic Synovial Cell Sarcoma and Melanoma Using Genetically Engineered Lymphocytes Reactive With NY-ESO-1. Journal of Clinical Oncology. 2011;29:917. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, Borquez-Ojeda O, Qu J, Wasielewska T, He Q, Bernal Y, Rijo IV, Hedvat C, Kobos R, Curran K, Steinherz P, Jurcic J, Rosenblat T, Maslak P, Frattini M, Sadelain M. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Newrzela S, Cornils K, Li Z, Baum C, Brugman MH, Hartmann M, Meyer J, Hartmann S, Hansmann ML, Fehse B, von Laer D. Resistance of mature T cells to oncogene transformation. Blood. 2008;112:2278–86. doi: 10.1182/blood-2007-12-128751. [DOI] [PubMed] [Google Scholar]
- 102.Hsu C, Jones SA, Cohen CJ, Zheng Z, Kerstann K, Zhou J, Robbins PF, Peng PD, Shen X, Gomes TJ, Dunbar CE, Munroe DJ, Stewart C, Cornetta K, Wangsa D, Ried T, Rosenberg SA, Morgan RA. Cytokine-independent growth and clonal expansion of a primary human CD8+ T-cell clone following retroviral transduction with the IL-15 gene. Blood. 2007;109:5168–77. doi: 10.1182/blood-2006-06-029173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Newrzela S, Cornils K, Heinrich T, Schlager J, Yi JH, Lysenko O, Kimpel J, Fehse B, von Laer D. Retroviral insertional mutagenesis can contribute to immortalization of mature T lymphocytes. Mol Med. 2011;17:1223–32. doi: 10.2119/molmed.2010.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Recchia A, Bonini C, Magnani Z, Urbinati F, Sartori D, Muraro S, Tagliafico E, Bondanza A, Stanghellini MT, Bernardi M, Pescarollo A, Ciceri F, Bordignon C, Mavilio F. Retroviral vector integration deregulates gene expression but has no consequence on the biology and function of transplanted T cells. Proc Natl Acad Sci U S A. 2006;103:1457–62. doi: 10.1073/pnas.0507496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.June CH, Blazar BR, Riley JL. Engineering lymphocyte subsets: tools, trials and tribulations. Nat Rev Immunol. 2009;9:704–16. doi: 10.1038/nri2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–7. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 107.Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–9. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 108.Biffi A, Bartolomae CC, Cesana D, Cartier N, Aubourg P, Ranzani M, Cesani M, Benedicenti F, Plati T, Rubagotti E, Merella S, Capotondo A, Sgualdino J, Zanetti G, von Kalle C, Schmidt M, Naldini L, Montini E. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood. 2011;117:5332–9. doi: 10.1182/blood-2010-09-306761. [DOI] [PubMed] [Google Scholar]
- 109.Ellis J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum Gene Ther. 2005;16:1241–6. doi: 10.1089/hum.2005.16.1241. [DOI] [PubMed] [Google Scholar]
- 110.Wang GP, Levine BL, Binder GK, Berry CC, Malani N, McGarrity G, Tebas P, June CH, Bushman FD. Analysis of lentiviral vector integration in HIV+ study subjects receiving autologous infusions of gene modified CD4+ T cells. Mol Ther. 2009;17:844–50. doi: 10.1038/mt.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schroers R, Hildebrandt Y, Hasenkamp J, Glass B, Lieber A, Wulf G, Piesche M. Gene transfer into human T lymphocytes and natural killer cells by Ad5/F35 chimeric adenoviral vectors. Exp Hematol. 2004;32:536–46. doi: 10.1016/j.exphem.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 113.Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, Meechoovet HB, Bautista C, Chang WC, Ostberg JR, Jensen MC. Adoptive transfer of chimeric antigen receptor redirected cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15:825–33. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 114.Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436:221–6. doi: 10.1038/nature03691. [DOI] [PubMed] [Google Scholar]
- 115.Huang X, Wilber AC, Bao L, Tuong D, Tolar J, Orchard PJ, Levine BL, June CH, McIvor RS, Blazar BR, Zhou X. Stable gene transfer and expression in human primary T cells by the Sleeping Beauty transposon system. Blood. 2006;107:483–91. doi: 10.1182/blood-2005-05-2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Maiti SN, Huls H, Singh H, Dawson M, Figliola M, Olivares S, Rao P, Zhao YJ, Multani A, Yang G, Zhang L, Crossland D, Ang S, Torikai H, Rabinovich B, Lee DA, Kebriaei P, Hackett P, Champlin RE, Cooper LJ. Sleeping beauty system to redirect T-cell specificity for human applications. J Immunother. 2013;36:112–23. doi: 10.1097/CJI.0b013e3182811ce9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H, Hackett PB, Kohn DB, Shpall EJ, Champlin RE, Cooper LJ. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008;68:2961–71. doi: 10.1158/0008-5472.CAN-07-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, Rosenberg SA, Morgan RA. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther. 2006;13:151–9. doi: 10.1016/j.ymthe.2005.07.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yoon SH, Lee JM, Cho HI, Kim EK, Kim HS, Park MY, Kim TG. Adoptive immunotherapy using human peripheral blood lymphocytes transferred with RNA encoding Her-2/neu-specific chimeric immune receptor in ovarian cancer xenograft model. Cancer Gene Ther. 2009;16:489–97. doi: 10.1038/cgt.2008.98. [DOI] [PubMed] [Google Scholar]
- 120.Mitchell DA, Karikari I, Cui X, Xie W, Schmittling R, Sampson JH. Selective modification of antigen-specific T cells by RNA electroporation. Hum Gene Ther. 2008;19:511–21. doi: 10.1089/hum.2007.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rowley J, Monie A, Hung CF, Wu TC. Expression of IL-15RA or an IL-15/IL-15RA fusion on CD8+ T cells modifies adoptively transferred T-cell function in cis. Eur J Immunol. 2009;39:491–506. doi: 10.1002/eji.200838594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Birkholz K, Hombach A, Krug C, Reuter S, Kershaw M, Kampgen E, Schuler G, Abken H, Schaft N, Dorrie J. Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene Ther. 2009;16:596–604. doi: 10.1038/gt.2008.189. [DOI] [PubMed] [Google Scholar]
- 123.Wang W, Lin C, Lu D, Ning Z, Cox T, Melvin D, Wang X, Bradley A, Liu P. Chromosomal transposition of PiggyBac in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105:9290–5. doi: 10.1073/pnas.0801017105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rabinovich PM, Komarovskaya ME, Wrzesinski SH, Alderman JL, Budak-Alpdogan T, Karpikov A, Guo H, Flavell RA, Cheung NK, Weissman SM, Bahceci E. Chimeric receptor mRNA transfection as a tool to generate antineoplastic lymphocytes. Hum Gene Ther. 2009;20:51–61. doi: 10.1089/hum.2008.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, Chew A, Carroll RG, Scholler J, Levine BL, Albelda SM, June CH. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70:9053–61. doi: 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M, Carroll RG, June CH, Grupp SA. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther. 2011;22:1575–86. doi: 10.1089/hum.2011.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kirkwood JM, Lee S, Moschos SJ, Albertini MR, Michalak JC, Sander C, Whiteside T, Butterfield LH, Weiner L. Immunogenicity and antitumor effects of vaccination with peptide vaccine+/-granulocyte-monocyte colony-stimulating factor and/or IFN-alpha2b in advanced metastatic melanoma: Eastern Cooperative Oncology Group Phase II Trial E1696. Clin Cancer Res. 2009;15:1443–51. doi: 10.1158/1078-0432.CCR-08-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, Royal RE, Kammula U, Restifo NP, Hughes MS, Schwartzentruber D, Berman DM, Schwarz SL, Ngo LT, Mavroukakis SA, White DE, Steinberg SM. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005;175:6169–76. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 129.Kirkwood JM, Butterfield LH, Tarhini AA, Zarour H, Kalinski P, Ferrone S. Immunotherapy of cancer in 2012. CA Cancer J Clin. 2012;62:309–35. doi: 10.3322/caac.20132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, Kaiser AD, Pouw N, Debets R, Kieback E, Uckert W, Song JY, Haanen JB, Schumacher TN. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. 2010;16:565–70. doi: 10.1038/nm.2128. 1p following 70. [DOI] [PubMed] [Google Scholar]
- 131.Ueno T, Fujiwara M, Tomiyama H, Onodera M, Takiguchi M. Reconstitution of anti-HIV effector functions of primary human CD8 T lymphocytes by transfer of HIV-specific alphabeta TCR genes. Eur J Immunol. 2004;34:3379–88. doi: 10.1002/eji.200425568. [DOI] [PubMed] [Google Scholar]
- 132.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer research. 2006;66:8878–86. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Stanislawski T, Voss RH, Lotz C, Sadovnikova E, Willemsen RA, Kuball J, Ruppert T, Bolhuis RL, Melief CJ, Huber C, Stauss HJ, Theobald M. Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nature immunology. 2001;2:962–70. doi: 10.1038/ni1001-962. [DOI] [PubMed] [Google Scholar]
- 134.Voss RH, Kuball J, Engel R, Guillaume P, Romero P, Huber C, Theobald M. Redirection of T cells by delivering a transgenic mouse-derived MDM2 tumor antigen-specific TCR and its humanized derivative is governed by the CD8 coreceptor and affects natural human TCR expression. Immunol Res. 2006;34:67–87. doi: 10.1385/IR:34:1:67. [DOI] [PubMed] [Google Scholar]
- 135.Boulter JM, Glick M, Todorov PT, Baston E, Sami M, Rizkallah P, Jakobsen BK. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 2003;16:707–11. doi: 10.1093/protein/gzg087. [DOI] [PubMed] [Google Scholar]
- 136.Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67:3898–903. doi: 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C, Greenberg PD. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. 2007;109:2331–8. doi: 10.1182/blood-2006-05-023069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Thomas S, Xue SA, Cesco-Gaspere M, San Jose E, Hart DP, Wong V, Debets R, Alarcon B, Morris E, Stauss HJ. Targeting the Wilms tumor antigen 1 by TCR gene transfer: TCR variants improve tetramer binding but not the function of gene modified human T cells. J Immunol. 2007;179:5803–10. doi: 10.4049/jimmunol.179.9.5803. [DOI] [PubMed] [Google Scholar]
- 139.Voss RH, Willemsen RA, Kuball J, Grabowski M, Engel R, Intan RS, Guillaume P, Romero P, Huber C, Theobald M. Molecular design of the Calphabeta interface favors specific pairing of introduced TCRalphabeta in human T cells. J Immunol. 2008;180:391–401. doi: 10.4049/jimmunol.180.1.391. [DOI] [PubMed] [Google Scholar]
- 140.Sebestyen Z, Schooten E, Sals T, Zaldivar I, San Jose E, Alarcon B, Bobisse S, Rosato A, Szollosi J, Gratama JW, Willemsen RA, Debets R. Human TCR that incorporate CD3zeta induce highly preferred pairing between TCRalpha and beta chains following gene transfer. J Immunol. 2008;180:7736–46. doi: 10.4049/jimmunol.180.11.7736. [DOI] [PubMed] [Google Scholar]
- 141.Okamoto S, Mineno J, Ikeda H, Fujiwara H, Yasukawa M, Shiku H, Kato I. Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res. 2009;69:9003–11. doi: 10.1158/0008-5472.CAN-09-1450. [DOI] [PubMed] [Google Scholar]
- 142.Torikai H, Reik A, Soldner F, Warren EH, Yuen C, Zhou Y, Crossland DL, Huls H, Littman N, Zhang Z, Tykodi SS, Kebriaei P, Lee DA, Miller JC, Rebar EJ, Holmes MC, Jaenisch R, Champlin RE, Gregory PD, Cooper LJ. Towards eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. 2013 doi: 10.1182/blood-2013-03-478255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, Phan GQ, Hughes MS, Kammula US, Miller AD, Hessman CJ, Stewart AA, Restifo NP, Quezado MM, Alimchandani M, Rosenberg AZ, Nath A, Wang T, Bielekova B, Wuest SC, Akula N, McMahon FJ, Wilde S, Mosetter B, Schendel DJ, Laurencot CM, Rosenberg SA. Cancer Regression and Neurological Toxicity Following Anti-MAGE-A3 TCR Gene Therapy. J Immunother. 2013;36:133–51. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, Binder-Scholl GK, Smethurst DP, Gerry AB, Pumphrey NJ, Bennett AD, Brewer JE, Dukes J, Harper J, Tayton-Martin HK, Jakobsen BK, Hassan NJ, Kalos M, June CH. Cardiovascular toxicity and titin cross-reactivity of affinity enhanced T cells in myeloma and melanoma. Blood. 2013 doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, Hughes MS, Kammula US, Phan GQ, Lim RM, Wank SA, Restifo NP, Robbins PF, Laurencot CM, Rosenberg SA. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620–6. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cameron BJ, Gerry A, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer J, Gupta M, Plesa G, Bossi g, Vuidepot A, Powlesland A, Legg A, Adams KJ, Bennett A, umphrey N, Williams D, Binder-Scholl G, Kulikovskaya I, Levine B, Riley J, Varela-Rohena A, Stadtmauer E, Rapoport A, Linette G, June C, Hassan NJ, Kalos M, Jakobsen BK. Identification of titin as an alternative specificity for engineered T cells expressing an enhanced affinity MAGE A3 TCR. 2013 submitted for publication. [Google Scholar]
- 147.Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, Duerkopp N, Roberts IM, Pogosov GL, Ho WY, Ochsenreither S, Wolfl M, Bar M, Radich JP, Yee C, Greenberg PD. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med. 2013;5:174ra27. doi: 10.1126/scitranslmed.3004916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol. 2002;20:70–5. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 149.Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, King PD, Larson S, Weiss M, Riviere I, Sadelain M. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nature medicine. 2003;9:279–86. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- 150.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, Carroll RG, Riley JL, Pastan I, June CH. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106:3360–5. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. Journal of immunology. 2004;172:104–13. doi: 10.4049/jimmunol.172.1.104. [DOI] [PubMed] [Google Scholar]
- 152.Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Molecular therapy : the journal of the American Society of Gene Therapy. 2010;18:413–20. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Davila ML, Brentjens R, Wang X, Riviere I, Sadelain M. How do CARs work?: Early insights from recent clinical studies targeting CD19. Oncoimmunology. 2012;1:1577–83. doi: 10.4161/onci.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. Journal of immunology. 2010;184:6938–49. doi: 10.4049/jimmunol.0901766. [DOI] [PubMed] [Google Scholar]
- 155.Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, Dimitrov DS, Morgan RA, FitzGerald DJ, Barrett DM, Wayne AS, Mackall CL, Orentas RJ. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–74. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Budde LE, Till BG, Raubitschek AA, Forman SJ, Press OW. Mathematical modeling of chimeric TCR triggering predicts the magnitude of target lysis and its impairment by TCR downmodulation. J Immunol. 2010;184:4284–94. doi: 10.4049/jimmunol.0903701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Davis MM, Krogsgaard M, Huse M, Huppa J, Lillemeier BF, Li QJ. T cells as a self-referential, sensory organ. Annu Rev Immunol. 2007;25:681–95. doi: 10.1146/annurev.immunol.24.021605.090600. [DOI] [PubMed] [Google Scholar]
- 158.Guy CS, Vignali KM, Temirov J, Bettini ML, Overacre AE, Smeltzer M, Zhang H, Huppa JB, Tsai YH, Lobry C, Xie J, Dempsey PJ, Crawford HC, Aifantis I, Davis MM, Vignali DA. Distinct TCR signaling pathways drive proliferation and cytokine production in T cells. Nat Immunol. 2013;14:262–70. doi: 10.1038/ni.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–2. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 161.Davila ML, Kloss CC, Gunset G, Sadelain M. CD19 CAR-targeted T cells induce long-term remission and B Cell Aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLoS One. 2013;8:e61338. doi: 10.1371/journal.pone.0061338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–41. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–8. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 164.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–58. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 165.Melenhorst JJ, Levine BL. Innovation and opportunity for chimeric antigen receptor targeted T cells. Cytotherapy. 2013 doi: 10.1016/j.jcyt.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 166.Hosing C, Kebriaei P, Wierda W, Jena B, Cooper LJ, Shpall E. CARs in chronic lymphocytic leukemia -- ready to drive. Curr Hematol Malig Rep. 2013;8:60–70. doi: 10.1007/s11899-012-0145-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Brentjens RJ, Curran KJ. Novel cellular therapies for leukemia: CAR-modified T cells targeted to the CD19 antigen. Hematology Am Soc Hematol Educ Program. 2012;2012:143–51. doi: 10.1182/asheducation-2012.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.June C, Rosenberg SA, Sadelain M, Weber JS. T-cell therapy at the threshold. Nat Biotechnol. 2012;30:611–4. doi: 10.1038/nbt.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kohn DB, Dotti G, Brentjens R, Savoldo B, Jensen M, Cooper LJ, June CH, Rosenberg S, Sadelain M, Heslop HE. CARs on track in the clinic. Mol Ther. 2011;19:432–8. doi: 10.1038/mt.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, Yang JC, Kammula US, Devillier L, Carpenter R, Nathan DA, Morgan RA, Laurencot C, Rosenberg SA. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–20. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, Forman SJ. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–56. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg D, Jurcic J, Heaney M, Heller G, Frattini M, Sadelain M. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–28. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ellison J. 2013 http://www.seattlepi.com/national/article/T-cell-leukemia-cure-shows-early-positive-4657999.php.
- 175.Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano Attianese GM, Cribioli E, Maltese F, Galimberti S, Lopez AF, Biondi A, Bonnet D, Biagi E. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161:389–401. doi: 10.1111/bjh.12282. [DOI] [PubMed] [Google Scholar]
- 176.Carpenter RO, Evbuomwan MO, Pittaluga S, Rose JJ, Raffeld M, Yang S, Gress RE, Hakim FT, Kochenderfer JN. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19:2048–60. doi: 10.1158/1078-0432.CCR-12-2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Morgan R, Yang J, Kitano M, Dudley M, Laurencot C, Rosenberg S. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Molecular Therapy. 2010;18:843–51. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, Liu H, Wang LL, Rowley DR, Pfizenmaier K, Gottschalk S. Antitumor Effects of Chimeric Receptor Engineered Human T Cells Directed to Tumor Stroma. Mol Ther. 2013 doi: 10.1038/mt.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, Rosenberg SA. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. 2013;210:1125–35. doi: 10.1084/jem.20130110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, Mei Z, Rooney CM, Heslop HE, Brenner MK. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, Brenner MK. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–83. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–5. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Reshef R, Luger SM, Hexner EO, Loren AW, Frey NV, Nasta SD, Goldstein SC, Stadtmauer EA, Smith J, Bailey S, Mick R, Heitjan DF, Emerson SG, Hoxie JA, Vonderheide RH, Porter DL. Blockade of lymphocyte chemotaxis in visceral graft-versus-host disease. N Engl J Med. 2012;367:135–45. doi: 10.1056/NEJMoa1201248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kalos M. An integrative paradigm to impart quality to correlative science. J Transl Med. 2010;8:26. doi: 10.1186/1479-5876-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Britten CM, Janetzki S, Butterfield LH, Ferrari G, Gouttefangeas C, Huber C, Kalos M, Levitsky HI, Maecker HT, Melief CJ, O’Donnell-Tormey J, Odunsi K, Old LJ, Ottenhoff TH, Ottensmeier C, Pawelec G, Roederer M, Roep BO, Romero P, van der Burg SH, Walter S, Hoos A, Davis MM. T cell assays and MIATA: the essential minimum for maximum impact. Immunity. 2012;37:1–2. doi: 10.1016/j.immuni.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 186.Kalos M. Biomarkers in T cell therapy clinical trials. J Transl Med. 2011;9:138. doi: 10.1186/1479-5876-9-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, Huang J, Powell DJ, Jr, Rosenberg SA. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173:7125–30. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tran KQ, Zhou J, Durflinger KH, Langhan MM, Shelton TE, Wunderlich JR, Robbins PF, Rosenberg SA, Dudley ME. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J Immunother. 2008;31:742–51. doi: 10.1097/CJI.0b013e31818403d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, Klebanoff CA, Johnson LA, Kerkar SP, Yang S, Muranski P, Palmer DC, Scott CD, Morgan RA, Robbins PF, Rosenberg SA, Restifo NP. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. 2011;117:808–14. doi: 10.1182/blood-2010-05-286286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Powell DJ, Jr, Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. 2005;105:241–50. doi: 10.1182/blood-2004-06-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Stemberger C, Neuenhahn M, Gebhardt FE, Schiemann M, Buchholz VR, Busch DH. Stem cell-like plasticity of naive and distinct memory CD8+ T cell subsets. Semin Immunol. 2009;21:62–8. doi: 10.1016/j.smim.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 193.Melenhorst JJ, Scheinberg P, Chattopadhyay PK, Gostick E, Ladell K, Roederer M, Hensel NF, Douek DC, Barrett AJ, Price DA. High avidity myeloid leukemia-associated antigen-specific CD8+ T cells preferentially reside in the bone marrow. Blood. 2009;113:2238–44. doi: 10.1182/blood-2008-04-151969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, Rubin SC, Coukos G. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 195.Robins H, Desmarais C, Matthis J, Livingston R, Andriesen J, Reijonen H, Carlson C, Nepom G, Yee C, Cerosaletti K. Ultra-sensitive detection of rare T cell clones. J Immunol Methods. 2012;375:14–9. doi: 10.1016/j.jim.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.White AK, VanInsberghe M, Petriv OI, Hamidi M, Sikorski D, Marra MA, Piret J, Aparicio S, Hansen CL. High-throughput microfluidic single-cell RT-qPCR. Proc Natl Acad Sci U S A. 2011;108:13999–4004. doi: 10.1073/pnas.1019446108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lamers CH, Willemsen R, van Elzakker P, van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, Oosterwijk E, Sleijfer S, Debets R, Gratama JW. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood. 2011;117:72–82. doi: 10.1182/blood-2010-07-294520. [DOI] [PubMed] [Google Scholar]
- 198.Hadrup SR, Bakker AH, Shu CJ, Andersen RS, van Veluw J, Hombrink P, Castermans E, Thor Straten P, Blank C, Haanen JB, Heemskerk MH, Schumacher TN. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat Methods. 2009;6:520–6. doi: 10.1038/nmeth.1345. [DOI] [PubMed] [Google Scholar]
- 199.Bodenmiller B, Zunder ER, Finck R, Chen TJ, Savig ES, Bruggner RV, Simonds EF, Bendall SC, Sachs K, Krutzik PO, Nolan GP. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat Biotechnol. 2012;30:858–67. doi: 10.1038/nbt.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe’er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31:545–52. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, Nichols KE, Suppa EK, Kalos M, Berg RA, Fitzgerald JC, Aplenc R, Gore L, Grupp SA. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121:5154–7. doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Levine BL, June CH. Perspective: assembly line immunotherapy. Nature. 2013;498:S17. doi: 10.1038/498S17a. [DOI] [PubMed] [Google Scholar]
- 203.http://www.wbmt.org/fileadmin/pdf/01_General/Distribution_1_Mio_transplants.pdf. 2013. one millionth stem cell transplant.
- 204.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA, Group B-S. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12:671–84. doi: 10.1038/nrc3322. [DOI] [PMC free article] [PubMed] [Google Scholar]