

Abstract
Tryptophan dioxygenase (TDO) and indole-amine 2,3-dioxygenase (IDO) are two heme-containing enzymes which catalyze the conversion of L-tryptophan to N-formylkynurenine (NFK). In mammals, TDO is mostly expressed in liver and is involved in controlling homeostatic serum tryptophan concentrations, whereas IDO is ubiquitous and is involved in modulating immune responses. Previous studies suggested that the first step of the dioxygenase reaction involves the deprotonation of the indoleamine group of the substrate by an evolutionarily conserved distal histidine residue in TDO and the heme-bound dioxygen in IDO. Here, we used classical molecular dynamics and hybrid quantum mechanical/molecular mechanical methods to evaluate the base-catalyzed mechanism. Our data suggest that the deprotonation of the indoleamine group of the substrate by either histidine in TDO or heme-bound dioxygen in IDO is not energetically favorable. Instead, the dioxygenase reaction can be initiated by a direct attack of heme-bound dioxygen on the C2=C3 bond of the indole ring, leading to a protein-stabilized 2,3-alkylperoxide transition state and a ferryl epoxide intermediate, which subsequently recombine to generate NFK. The novel sequential two-step oxygen addition mechanism is fully supported by our recent resonance Raman data that allowed identification of the ferryl intermediate (Lewis-Ballester et al. in Proc Natl Acad Sci USA 106:17371–17376, 2009). The results reveal the subtle differences between the TDO and IDO reactions and highlight the importance of protein matrix in modulating stereoelectronic factors for oxygen activation and the stabilization of both transition and intermediate states.
Keywords: Tryptophan dioxygenase, Indoleamine 2, 3-dioxygenase, Molecular dynamics, Quantum mechanics/molecular mechanics, Tryptophan dioxygenation
Introduction
Human tryptophan dioxygenase (hTDO) and human indoleamine 2,3-dioxygenase (hIDO) are the only two heme-containing enzymes in humans that catalyze the oxidative ring cleavage reaction of L-Trp to N-formylkynurenine (NFK), the initial and rate-limiting step of the kynurenine pathway [2, 3]. Tryptophan dioxygenase (TDO) is mostly expressed in the liver; it has been found in bacteria, insects, and other mammals in addition to humans [2, 4, 5]. Indoleamine 2,3-dioxygenase (IDO), on the other hand, is only found in mammals and is ubiquitously expressed in all tissues [3, 6]. As indicated by their names, other than L-Trp, IDO is capable of oxidizing various indoleamine derivatives, such as serotonin, tryptamine, and melatonin [6, 7], whereas TDO is specific for Trp [2, 7].
The crystallographic structures of two bacterial isoforms of TDO (xcTDO from Xanthomonas campestris [4] and rmTDO from Ralstonia metallidurans [8]) have been reported. They show that [8] the enzyme is tetrameric, with one heme per monomer. In the L-Trp-bound xcTDO [Protein Data Bank (PDB) ID 2NW8), the substrate binds to the distal heme pocket, in the vicinity of the ligand binding site; the substrate is stabilized by hydrogen-bonding interactions with the surrounding protein matrix via its indoleamine, carboxylate, and ammonium groups, as well as by hydrophobic interactions via the aromatic indole ring. Crystallographic data of hIDO [9], on the other hand, show that the enzyme is monomeric, but exhibits high structure-based homology with TDO, with most of the critical residues involving substrate–protein interactions conserved.
Recently, it was discovered that IDO promotes immune escape in cancer [10]; in addition, inhibition of IDO was shown to cooperate with cytotoxic agents to promote regression of established tumors [11]. The recognition of hIDO as a potent anticancer drug target has triggered intense research on the two dioxygenases at the molecular and physiological levels [10, 11]. Despite the efforts, the mechanism by which Trp is oxidized by relatively inert dioxygen in TDO and IDO remains elusive. Since their discovery more than four decades ago [6, 12], several scenarios have been postulated to account for the dioxygenase activity [2, 13, 14]. Pathway A in Scheme 1 illustrates the widely accepted based-catalyzed mechanism first proposed by Hamilton [15]. In this scenario, the reaction is initiated by the deprotonation of the indoleamine group of L-Trp by an active-site base, which is followed by electrophilic addition of the heme-bound dioxygen to the C2=C3 bond of Trp, leading to the heme iron-bound 3-indolenylperoxo intermediate, which subsequently converts to the product, NFK, by means of a dioxetane reaction or Criegee rearrangement [2, 15, 16]. In line with this hypothesis, a 3-hydroperoxyindolenine intermediate has been identified in the photosensitized oxidation reaction of L-Trp in an enzyme-free system; although the specificity is much lower, the reaction was shown to yield the same NFK product via a dioxetane intermediate [17].
Scheme 1.
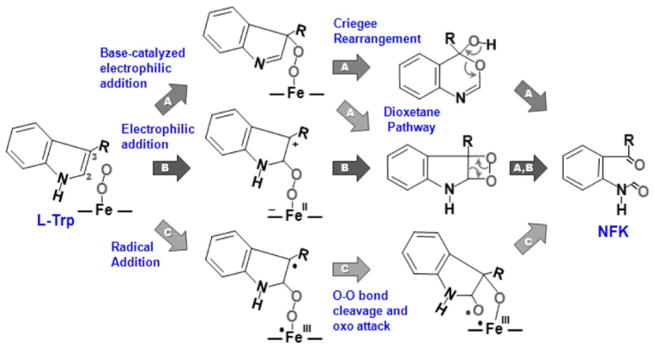
Proposed reaction mechanisms for Trp dioxygenation performed by tryptophan dioxygenase and indoleamine 2,3-dioxygenase
On the basis of the idea that the reaction was initiated by the deprotonation of Trp, significant efforts have been devoted to identifying the active-site base in each enzyme. By using CO as a structure probe, earlier resonance Raman and mutagenesis studies [14, 18] suggested that the active-site base that deprotonates the indoleamine group of the substrate is H55 in xcTDO (H76 in hTDO), whereas that in hIDO is the heme-bound dioxygen [18]. However, recent experimental results showed that hIDO is able to oxygenate 1-methyltryptophan [19, 20], casting doubt on the base-catalyzed mechanism. In addition, recent density functional theory (DFT) calculations, based on a heme–O2–indole model system in a vacuum, suggested that a concerted proton transfer from indoleamine to the heme-bound dioxygen and electrophilic addition of the dioxygen to the C2=C3 bond exhibited a very high activation barrier owing to a highly distorted transition state [13]; instead, the data suggested that the reaction could be initiated by a direct electrophilic or radical addition of the terminal atom of the dioxygen to the C2 position of the indole ring without the deprotonation of the indoleamine group (see pathways B and C in Scheme 1). The addition reaction leads to a heme-bound alkylperoxide with a cation or radical at the C3 position, respectively, which could then rearrange to produce NFK. It is noted that an electrophilic addition mechanism similar to pathway B, but with the dioxygen attached to the C3 position, leaving a cation at the C2 atom, was also shown to be energetically feasible (not shown in Scheme 1). It is also important to note that since the DFT calculations were conducted for a vacuum, without the consideration of the protein matrix and solvent effects, additional studies are required to further validate the dioxygenase chemistry.
Disregarding the mechanism, it is generally believed that during the TDO and IDO reactions two oxygen atoms are simultaneously inserted into the substrate, thereby setting the dioxygenase reaction apart from the reactions carried out by monooxygenases, such as cytochromes P450. However, recently, our resonance Raman data revealed a ferryl intermediate populated during the reaction of hIDO, demonstrating a novel sequential two-step oxygen insertion mechanism. In agreement with the spectroscopic data, our preliminary quantum mechanical/molecular mechanical (QM/MM) calculations showed that the dioxygenase reactions of both IDO and TDO are initiated by radical addition of the heme-bound dioxygen to the C2=C3 bond of the indole ring, leading to a ferryl and indole epoxide intermediate, via an alkylperoxo transition state [1].
To uncover the mechanistic details, here we present a thorough evaluation of the ferryl-based mechanism by using state-of-the-art classical molecular dynamics (MD) and QM/MM simulations of the xcTDO and hIDO reactions. Specifically, we have systematically examined the feasibility of proton transfer during the initial step of the reaction, demonstrated that the initial deprotonation of the substrate is not required, and that this process is highly unfavorable for both enzymes. In addition, we have characterized the electronic and structural properties of the ternary complex, the peoxo transition state, and the ferryl intermediate populated along the reaction coordinate of each enzyme in different spin states, and analyzed the impact of substrate–ligand and substrate–protein interactions on the catalytic mechanism. Lastly, we have quantitatively analyzed the importance of the protein matrix in stabilizing the transition and intermediate states of the reaction by examining an enzyme-free model complex in a vacuum, as compared with the enzyme system.
Computational methods
TDO starting structure
The crystal structure of ferrous L-Trp-bound xcTDO (PDB ID 2NW8) [4] contains two subunits: the first 15 amino acids in one subunit form a short helix that penetrates into the other subunit, forming part of the substrate-binding site. For the computational studies, a one-subunit model was built by using subunit A without residues 1–35, but with residues 21–35 of subunit B added into it (it is noted that residues 1–20 are missing in the crystal structure). An O2 molecule was added to the heme iron in silico to generate the ternary complex.
IDO starting structure
For hIDO, only the crystal structures of the ferric monomeric enzyme with either 4-phenylimidazole or cyanide coordinated to the heme iron are available. In these structures, two molecules of 2-(N-cyclohexylamino)ethanesulfonic acid, a component of the crystallization buffer, were co-crystallized. To build the hIDO model, the 4-phenylimidazole-bound structure (PDB ID 2D0T) [9], with 4-phenylimidazole and 2-(N-cyclohexylamino)ethanesulfonic acid removed, was used as a base structure; an O2 molecule was added to the heme iron in silico. After a short relaxation period, a 5-ns MD simulation was performed. Five snapshots were selected for flexible docking of L-Trp into hIDO–O2 by using the Autodock program [21]. One hundred docking runs were performed for each structure selected. The docking results were clustered into two possible conformations, which were subsequently used for the MD and QM/MM simulations.
MD simulations
All classical simulations were performed by using the PMEMD module of the Amber10 package [22], with the Amber99SB force field parameters [23] for all residues. The starting structures were immersed in a preequilibrated octahedral box of TIP3P water molecules. SHAKE was used to keep bonds involving hydrogen atoms at their equilibrium lengths [24], which allowed a 2-fs time step to be employed for the integration of Newton’s equations. All simulations were performed at 1 atm and 300 K, and these conditions were maintained with the Berendsen barostat and thermostat [25]. Periodic boundary conditions and Ewald sums (grid spacing of 1 Å) were used to treat long-range electrostatic interactions; a 12-Å cutoff was used for computing direct interactions. The heme parameters used in these simulations were developed and thoroughly tested by our group in previous works [26–31]. For each ternary complex [xcTDO–O2–L-Trp, hIDO–O2–L-Trp (conformation 1, Cf1), and hIDO–O2–L-Trp (conformation 2, Cf2)], 5-ns-long MD simulations in explicit water were performed and carefully analyzed. After the MD simulations, the systems were slowly cooled to 0 K to obtain the initial structures for the QM/MM simulations.
QM/MM simulations
All optimizations were performed with a conjugate gradient algorithm, at the DFT level by using the SIESTA code with our QM/MM implementation [32]. The quantum and the molecular mechanics subsystems are combined through a hybrid Hamiltonian introducing a modification of the Hartree potential and a QM/MM coupling term. The protein (or classical) environment affects the electronic density in a self-consistent fashion owing to the addition of the classical point charge potential to the Hartree potential. The coupling term has two main contributions representing the electrostatic interaction between the electrons and nuclei, defining the QM charge density with the classical point charge and an additional term corresponding to the van der Waals interactions between the atoms in the quantum and classical regions through a 6–12 Lennard-Jones potential. For all atoms, basis sets of double zeta plus polarization quality were employed with cutoff and energy shift values of 150 Ry and 25 meV. All calculations were performed using the generalized gradient approximation functional proposed by Perdew, Burke, and Ernzerhof (PBE) [33]. Only residues located within 10 Å from the heme reactive center were allowed to move freely in the QM/MM runs. The interface between the QM and MM portions of the system was treated with the scaled position link atom method. The SIESTA code showed excellent performance for medium-sized and large systems, and was proven to be appropriate for biomolecules, and specifically for heme models [34–36]. For all systems the spin-unrestricted approximation was used, unless otherwise stated. In hIDO, the QM subsystem included the heme group (without side chains), the O2, the substrate, L-Trp, and the imidazole group of the proximal His. For xcTDO, the imidazole group of His55 was also included. The rest of the protein unit and the water molecules were treated classically. Further technical details about the QM/MM implementation can be found elsewhere [32, 36, 37]. Basis quality effects were tested by computing the energy barriers and ΔE using a better-quality basis set; with cutoff and energy shift values of 300 Ry and 0.5 meV, which significantly increases the computational cost.
Since obtaining accurate free-energy profiles requires extensive sampling, which is computationally very expensive and difficult to achieve at the DFT QM/MM level, we resorted to computing potential energy profiles by using restrained energy minimizations along the reaction path that connects reactant and product states [32, 38, 39]. For this approach, an additional term, V(ξ) = k(ξ − ξ0)2, was added to the potential energy, where k is an adjustable force constant (set to be 200 kcal/mol Å2 here) and ξ0 is a reference value, which was varied stepwise with an interval of 0.1 Å, along the reaction coordinate. By variation of ξ0, the system is forced to follow the energy minimum reaction path along the given coordinate ξ. To avoid possible hysteresis problems in the reaction coordinate scans due to accidental changes in the MM part of the system, a distance cutoff of 10 Å was used, which only allows MM atoms that close to the QM active site to move during the reaction coordinate scan. This method has been widely used by our group in several works and has been proven to successfully avoid the problem mentioned [32, 40].
It is important to note that the use of coordinate driving can give misleading results, especially when only one initial structure is used. To minimize this possibility, detailed analysis of the reactive structures by MD simulations was performed and more than one conformation was selected in cases where significant movements of the reactive atoms were involved, as described later for each case.
For the model studies in a vacuum, the same SIESTA (PBE) calculations as described above were used. The in vacuum B3LYP-based calculations were performed using the Gaussian 98 program [41], using 6–31G* basis sets for hydrogen, carbon, nitrogen and oxygen atoms. Hay–Wadt’s effective core potentials and basis sets were applied for iron.
Results and discussion
QM/MM simulation of the ternary complex of xcTDO
To start the study of the TDO reaction mechanism, we first performed 5-ns-long MD simulations of the ternary complex (Fe2+–O2–L-Trp) of xcTDO built by adding O2 in silico to a monomeric model of the L-Trp-bound deoxy derivative of xcTDO (see “Computational methods”), corresponding to the starting point of the chemical reaction. The resulting structure was subsequently optimized by QM/MM protocols. As shown in Fig. 1a, the interactions between the substrate and the surrounding amino acid residues, such as R117, Y113, H55, F51, and the propionate-7 group of the heme, found in the crystal structure of xcTDO are conserved during the MD simulation and in the optimized structure. The overall root mean square deviation calculated from the Cα trace of the optimized structure with respect to the reference crystal structure is only 1.4 Å, indicating that binding of O2 to the L-Trp-bound enzyme does not significantly perturb its global structure.
Fig. 1.

QM/MM optimized structure of the ternary complexes of tryptophan dioxygenase from Xanthomonas campestris (xcTDO) and human indoleamine 2,3-dioxygenase (hIDO). For hIDO, only Cf1A is shown here. The expanded views in c highlight the hydrogen-bonding network between the Fe–O–O moiety, the substrate, and the surrounding protein matrix in xcTDO (top) and hIDO (bottom)
The geometrical parameters of the optimized structure are summarized in Table 1. The data show that the Fe–O–O moiety in the ternary complex of xcTDO lies parallel to the C2=C3 bond of the indole ring, which sits perpendicular to the heme plane. In addition, the terminal oxygen atom (Ot) of the dioxygen forms hydrogen bonds with both the ammonium group of L-Trp and the amine group of G125, whereas the proximal oxygen atom (Op) forms only one hydrogen bond with the amine group of G125 (Fig. 1c, top). The close proximity of the heme-bound O2 to the aromatic indole ring is consistent with prior resonance Raman data showing a hydrophobic environment surround the heme-bound ligand in hTDO [14].
Table 1.
Parameters for the optimized structures of the ternary complexes of tryptophan dioxygenase from Xanthomonas campestris (xcTDO) and human indoleamine 2,3-dioxygenase (hIDO) obtained from quantum mechanical/molecular mechanical simulations
| Parameter | xcTDO | hIDO
|
||
|---|---|---|---|---|
| Cf1A | Cf1B | Cf2 | ||
| d Fe–Op | 1.772 | 1.858 | 1.833 | 1.763 |
| d Op–Ot | 1.313 | 1.312 | 1.312 | 1.314 |
| ∠ Fe–Op–Ot | 124.1 | 121.0 | 120.0 | 120.0 |
| d Fe–Nprox | 2.134 | 2.108 | 2.091 | 2.110 |
| d HTrpNH–Op | – | 3.497 | 2.147 | 2.254 |
| d HTrpNH–Ot | – | 3.611 | 2.651 | 2.157 |
| d C2,Trp–Op | 3.393 | 3.394 | 3.295 | – |
| d C3,Trp–Ot | 3.288 | 3.588 | 4.801 | – |
| d Nε,H55–HTrpNH | 1.673 | – | – | – |
| ∠ NTrpNH–HTrpNH–Nε,H55 | 163.1 | – | – | – |
| ∠ NTrpNH–HTrpNH–Op | – | 92.2 | 133.0 | 149.7 |
The distances (d) and angles (∠) are in angstroms and degrees, respectively. The subscripts p and t for the oxygen atoms refer to the proximal and terminal atoms, respectively
C2,Trp, C3,Trp, HTrpNH, and NTrpNH are the C2 and C3 atoms and the indoleamine hydrogen and nitrogen atoms of L-Trp, respectively. Nprox and Nε,H55 are the nitrogen atom of the proximal His ligand of the heme and the Nε atom of the H55 residue (in xcTDO), respectively
QM/MM simulation of the ternary complex of hIDO
Given that no substrate-bound crystal structure is available for hIDO, we used the substrate-free structure as a base model and obtained putative structures of the ternary complex by a combined MD–docking approach (see “Computational methods”). Starting from each of five equilibrated structures of the oxy complex, we performed 100 flexible L-Trp docking simulations, which resulted in two major clusters of conformations, Cf1 and Cf2, with probabilities of 40 and 35%, respectively. The L-Trp binding energies of Cf1 and Cf2 are similar, with the binding energy of the former being slightly higher than that of the latter (7.0–8.2 vs. 6.0–7.0 kcal/mol). As a control, we docked L-Trp into the equilibrated MD structure derived from the crystal structure of the L-Trp-bound xcTDO (but with the L-Trp deleted). All the structures obtained are consistent with the reference crystal structure, with a binding energy of approximately 8 kcal/mol, confirming the reliability of our methods. For each conformation, we performed a 5-ns-long MD simulation in explicit water. For the subsequent QM/MM optimization, two snapshots of Cf1, referred to as Cf1A and Cf1B, were selected owing to the high conformational freedom of the active site, whereas only one snapshot of Cf2 was selected.
Intriguingly, Cf1A (Fig. 1b) exhibits a structure similar to that of the ternary complex of xcTDO (Fig. 1a). Although the hydrogen bond between the indoleamine and H55 is lost, as the latter is substituted by S167 in hIDO, all the rest of the critical hydrogen-bonding interactions between the heme-bound dioxygen, substrate, heme pro-pionate-7 group, and the surrounding protein matrix appear to be preserved. It is important to note that G125 in xcTDO is replaced by A264 in hIDO; however, it does not disturb the hydrogen-bonding interactions between its amine group and both atoms of the heme-bound dioxygen.
In Cf1B, the indole ring of L-Trp tilts slightly away from the heme normal; in addition, the Fe–O–O moiety rotates approximately 90° with respect to that in Cf1A, with its terminal (Ot) and proximal (Op) oxygen atoms forming hydrogen bonds with A264 and the indoleamine group of L-Trp, respectively (Fig. 2b). Cf2, on the other hand, displays a clear distinct structure, with L-Trp slightly displaced outside the active site (Fig. 2a). The indole ring of L-Trp lies almost parallel to the heme plane; instead of orienting between the side chains of F163 and F226, like in Cf1A, the indole ring moves closer to F226. Furthermore, both atoms of heme-bound dioxygen form hydrogen bonds with the indoleamine group of L-Trp as well as the amine group of A264 (Fig. 2b).
Fig. 2.

QM/MM optimized structure of the ternary complexes of hIDO. a The structure of Cf2. b The differences in the hydrogen-bonding network for Cf1A, Cf1B, and Cf2
In addition to the differences in the active-site structures, the major global structural difference between Cf1 and Cf2 lies in the heme conformation and the relative positioning of the distal helix (containing F163/F164) with respect to the proximal helix (containing the heme ligand, H346). It is noted that Cf1A and Cf1B exhibit almost identical global structures; hence, they are referred to as Cf1 here. In Cf2, the heme is in the same orientation as that in the starting crystal structure, but the distance between the proximal and distal helices (defined by the distance between Cα of H346 and Cα of F163) increases from 13.2 to 14.6 Å as compared with that observed in the Trp-free crystallographic structure. In Cf1, the distance increases further to 15.6 Å, to accommodate the indole ring of the substrate located further inside the active site; in addition, the substrate forces pyrrole rings C and D of the heme to shift down from the heme plane.
The overall root mean square deviation calculated from the Cα trace of Cf1 and Cf2 with respect to the reference crystal structure is approximately 1.4 Å. The structural regions showing high deviations are localized in the region involving direct interaction with the substrate, including (1) the side chain of R231, which moves from solvent into the active site to form hydrogen bonds with the carboxylate group of the substrate, (2) the side chains of F163, F164, and Y126, which interact with the aromatic indole ring of the substrate, and (3) the loop region containing S263 (which forms a hydrogen bond with the heme propionate-7 group of the heme) and A264 (whose amine group forms an hydrogen bond with the heme-bound dioxygen). These data support a partial induced-fit mechanism, proposed for xcTDO [4].
The data presented here for the ternary complex of hIDO are in good agreement with prior experimental data [1, 14, 42]. The structural flexibility of the ternary complex of hIDO revealed in this work is consistent with its resonance Raman spectrum showing a significantly broader Fe–O2 stretching mode (νFe–O2) as compared with that of hTDO. It also agrees well with its much broader substrate selectivity and its ease of autooxidation [2] (as the high degree of conformational freedom presumably increases the solvent accessibility of the distal heme pocket, which has been shown to be an important structural factor leading to autooxidation of O2-bound heme proteins [43]). The dynamic nature of the hydrogen-bonding interactions between the heme-bound dioxygen and its surrounding environment in hIDO is consistent with the unusual activation of the typically silent νO–O mode in its resonance Raman spectrum that was not observed in hTDO, and the relatively low νFe–O2 frequency, as compared with that of hTDO [1]. The data also agree well with a more positive polar ligand environment in the CO derivative of hIDO, as compared with that in hTDO [14, 44]. The disturbance of the heme conformation observed in Cf1A of hIDO, on the other hand, is consistent with the enhancement of the asymmetric in-plane and out-of-plane heme modes in the resonance Raman spectrum of hIDO, but not in hTDO [1]. Finally, the slightly shorter iron proximal His bond length in IDO compared with TDO (2.108 vs. 2.134 Å) revealed in our data (Table 1) is consistent with its relatively higher Fe–His stretching (νFe–His) frequency (236 vs. 229 cm−1) [14]. The νFe–His frequencies of both IDO and TDO are significantly higher than that found in typical globins (approximately 220 cm−1), indicating an imidazolate character of the proximal His ligand, which may provide an appropriate electronic push to promote the dioxygenase reaction.
It has been shown that in heme proteins carrying out oxygen reactions, such as cytochromes P450, different mechanisms can be operative depending on the spin state of the system (a property sometimes referred to as multi-state reactivity) [45]. As such, we compared the energetic parameters, Mulliken charges, and spin populations of the ternary complex of xcTDO and hIDO, in their singlet, triplet, and quintuplet states. The data are summarized in Table 2. For hIDO, we decided to focus our analysis on Cf1A, since the structural data discussed above clearly suggest that this is the reactive conformation. The data show that the singlet and triplet states are very close in energy for both enzymes, whereas the quintuplet states are significantly higher in energy. As expected, in the quintuplet state, the O2 is only weakly bonded to the iron, as indicated by an Fe–Op distance of 2.325 Å in hIDO and 2.101 Å xcTDO, and a spin density of approximately 1.3 and 1.5, respectively, closer to that expected for a triplet state in the free O2 molecule. The O2 molecule, however, is still interacting with the porphyrin, as indicated by the significant amount of charge transfer.
Table 2.
Energetic parameters, Mulliken charges, and spin populations of the ternary complexes of xcTDO and hIDO in the singlet (S), triplet (T), and quintuplet (Q) spin states
| xcTDO
|
hIDO
|
|||||
|---|---|---|---|---|---|---|
| S | T | Q | S | T | Q | |
| ΔEspin | 0 | 1.2 | 17.8 | 0 | 0.6 | 11.2 |
| Mulliken charge | ||||||
| Fe | +0.774 | +0.81 | +1.009 | +0.820 | +0.812 | +0.941 |
| O2 | −0.326 | −0.371 | −0.407 | −0.335 | −0.377 | −0.368 |
| L-Trp | −0.218 | −0.282 | −0.277 | −0.069 | −0.099 | −0.152 |
| Heme + Hprox | +0.284 | +0.414 | +0.466 | +0.407 | +0.470 | +0.498 |
| H55 | +0.252 | +0.238 | +0.219 | – | – | – |
| Spin population | ||||||
| Fe | −0.036 | +1.102 | +2.567 | +0.941 | +1.070 | +2.548 |
| O2 | +0.026 | +0.827 | +1.333 | −0.875 | +1.052 | +1.513 |
| L-Trp | +0.012 | +0.114 | +0.143 | −0.029 | +0.005 | +0.006 |
| Heme + Hprox | −0.032 | +1.066 | +2.520 | +0.907 | +0.947 | +2.480 |
ΔEspin (kcal/mol) is the difference in energy with respect to the S spin state
Hprox corresponds to the proximal His ligand
In general, in both enzymes, the Mulliken charges on the various functional groups listed in Table 2 are not sensitive to the spin state. In hIDO, the positive and negative Mulliken charges on the heme iron and O2, respectively, highlight the superoxide character of the heme-bound O2, consistent with the relatively long O–O bond length (1.312 Å, see Table 1), with respect to that of free dioxygen (approximately 1.24 Å), and the experimentally observed νO–O frequency at 1,137 cm−1 [1]. Similar superoxide character of the heme-bound O2 is also evident in xcTDO. The L-Trp in hIDO is slightly negatively charged, plausibly due to electron transfer from the heme-bound superoxide. In contrast, the L-Trp in xcTDO exhibits much higher negative electron density, due to electron transfer from H55, highlighting the strong interaction between the two species.
In hIDO, in the singlet state, the spin densities of the ferric heme iron and superoxide ligand are of similar magnitude and are antiparallel (as indicated by their opposite signs), implying that the two moieties are anti-ferromagnetically coupled, characteristic for an open-shell singlet state expected for these types of oxy complexes [26, 38]. Although both hIDO and xcTDO calculations were performed in the unrestricted DFT approximation, in xcTDO, the spin densities are all very low, indicating that the optimized system can be considered as a closed-shell singlet state. This is probably due to the strong hydrogen-bond interaction between the bound O2 and the charged NH3+ group of L-Trp (Ot–H distance of 1.813 Å), which positions the O2 very close to the heme, leading to a much shorter Fe–O bond than the bonds normally observed for heme–O2 complexes [26, 38]. In the triplet state, as expected, the spin densities on the iron and superoxide are also of similar magnitude, but are now parallel.
Evaluation of the deprotonation reaction of indoleamine group of the substrate
As discussed in “Introduction,” it has been suggested that the first step of the dioxygenase reaction involves the deprotonation of the indoleamine group of the substrate by either H55 in xcTDO or heme-bound dioxygen in hIDO. As a first step to evaluate the feasibility of the deprotonation reaction, we computed the proton affinity of TrpN−, as compared with three model systems in a vacuum: (1) His, (2) Op of a heme-bound dioxygen, and (3) Ot of a heme-bound dioxygen. As summarized in Table 3, the results show that the proton affinity of TrpN− is significantly higher than that of His. This is not surprising given that the pKa of indoleamine is 20.95 in dimethyl sulfoxide and 16.2 in water, whereas that of His is close to 6 [46, 47]. The data also show that the proton affinity of His is similar to that of the heme-bound dioxygen, with the Ot exhibiting a higher basicity as compared with Op. Taken together, the data suggest that the proton transfer from L-Trp to His or either atom of the heme-bound dioxygen is an endergonic process, which is in agreement with the fact that heme-bound dioxygen is typically not a good base, unless it is reduced to its peroxo derivative [45, 48]. The data suggest that unless there is significant stabilization provided by the surrounding protein matrix (for the deprotonated form of Trp and the protonated form of His in xcTDO, or dioxygen in hIDO) to overcome the unfavorable ΔE, the deproto-nation of L-Trp is not an energetically feasible process.
Table 3.
Proton affinities and energy required for proton transfer from Trp to His or heme-bound dioxygen
| Model system | Proton affinity | ΔE | |
|---|---|---|---|
| TrpN− ⇒ TrpNH | 268.0 | 0 | |
|
|
228.0 | 40 | |
| Heme–O2 ⇒ heme–Op–OtH+ | 234.4 | 34.4 | |
| Heme–O2 ⇒ heme–(OpH+)–Ot | 214.4 | 54.4 |
ΔE is the difference between the proton affinity of the corresponding compound and the proton affinity of TrpN−. The values are in kilo-calories per mole
To evaluate the effect of the protein matrix in xcTDO, we computed the energy profile associated with the postulated proton transfer reaction along the reaction coordinate defined by d(NTrp–H)–d(H–NH55) with QM/MM methods. Here NTrp and NH55 are the indoleamine nitrogen atom and the Nε atom of the substrate and H55, respectively. The QM region includes L-Trp, heme, dioxygen, proximal His ligand, and H55 imidazole groups. This approach allows the reaction to proceed via the energetically lowest passage, either a proton transfer or a proton-coupled electron transfer process (i.e., a net hydrogen atom transfer), in both the singlet and the triplet states. As shown in Fig. 3a, the energy profile resulting from the singlet state shows that the energy monotonically increases as the reaction proceeds along the reaction coordinate. The proton transfer does not yield a stable state, as no minimum was observed following the proton transfer; in addition, reoptimization of the product state led to its spontaneous return to the reactant state. Mulliken population analysis shows that the resulting charge on NH55-H+ is +0.756, whereas that on is −0.569, indicating that the reaction corresponds to a proton transfer reaction and not a proton-coupled electron transfer process, and that some charge transfer from TrpN− to NH55-H+ is present. The remaining negative charge is distributed in the O2–heme–Hisprox moiety, with a charge of −0.194. The data hence suggest that the negative charge on the deprotonated L-Trp can be delocalized in the O2–heme–Hisprox moiety, as well as the protonated H55, plausibly owing to the strong hydrogen-bonding network connecting them.
Fig. 3.
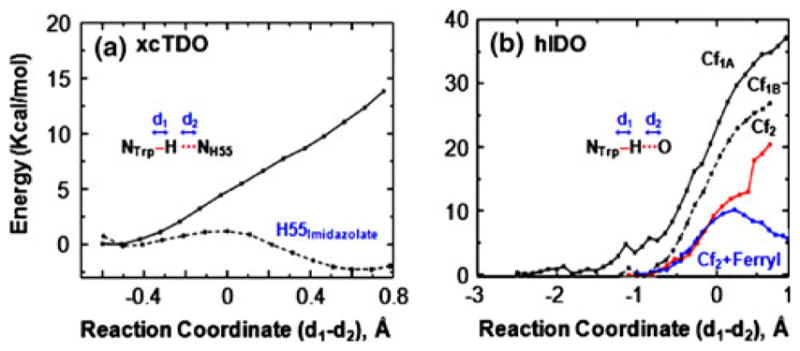
Energy profile for proton transfer from L-Trp to H55 in xcTDO (a) and to the heme-bound dioxygen in hIDO (b)
Similar results were observed in the triplet state reaction (data not shown). As a control to demonstrate that the results were not an artifact of our computational method, we calculated the energy profile for the proton transfer from L-Trp to H55 in the imidazolate state (the pKa of neutral imidazole is 18.6 in dimethyl sulfoxide) [46] As expected, the proton is readily transferred with a low activation barrier (less than 2 kcal/mol), and the product is more stable than the reactant by approximately 2 kcal/mol (Fig. 3a). It is noteworthy that although the deprotonation of the indoleamine group of the substrate does not appear to be essential for the dioxygenase reaction, the hydrogen bond between the indoleamine group of the substrate and H55 in xcTDO (or H76 in hTDO) could still be a critical factor in controlling substrate binding as well as catalysis, on the basis of two observations: (1) the substitution of the indoleamine with N-methyl in the substrate significantly diminishes dioxygenase activity in TDO [49] and (2) the mutation of H55 (in xcTDO) [50, 51] or H76 (in hTDO) [42] to Ala or Ser caused a significant increase in the Km value and a decrease in the kcat value.
The proton transfer from the indoleamine group of the substrate to the heme-bound dioxygen was also evaluated in hIDO. The reaction coordinate was defined as d(NTrp–H)–d(H–O). Here O is Op or Ot of the dioxygen for Cf1A/Cf1B or Cf2, respectively, which were selected on the basis of the shorter distance to the indoleamine group. Figure 3b shows the energy profile associated with each conformation of hIDO in the singlet state. In Cf1A and Cf1B, the energy monotonically increases as the proton approaches Op, which reaches approximately 40 and 25 kcal/mol, respectively, as d(NTrp–H) and d(H–Op) approach approximately 2.0 and 1.0 Å, respectively. In Cf2, the energy rise is slightly more modest as the reaction proceeds, consistent with the observation that Ot has a higher basicity than Op in the model systems listed in Table 3. The energy appears to reach a plateau at approximately 13 kcal/mol, when d(NTrp–H) and d(H–O) are 1.48 and 1.09 Å, respectively. However, no energy minimum was observed; in addition, reoptimization of the structure leads to its return to the initial state. Attempts to move the reaction further along the reaction coordinate caused an additional increase in energy to more than 20 kcal/mol. The energy profiles resulting from the triplet states, like those of the singlet states, show that the proton transfer does not yield stable states. Mulliken population analysis suggests that the proton transfer reaction is coupled with electron transfer, leading to a ferric hydroperoxo and a neutral L-Trp radical.
As a control, like in the previous case, we computed the energy profile associated with proton transfer from the indoleamine of L-Trp to the reactive ferryl (Fe4+=O2−) species in Cf2. The results show the formation of a stable product state, with a low activation barrier of approximately 10 kcal/mol, although the reaction is endergonic by 5 kcal/mol. Mulliken population analysis of the product revealed that the proton transfer is also coupled with electron transfer, leaving a neutral L-Trp radical behind. Taken together, our data suggest that proton transfer from the indoleamine group of the substrate to either H55 in xcTDO or heme-bound dioxygen in hIDO is not energetically feasible.
Previous studies carried out by Batabyal and Yeh [42] showed that the mutations of H76 to Ser and Ala in hTDO led to approximately 70-fold and tenfold lower kcat, respectively; combined with resonance Raman data of the CO derivatives, the authors proposed that the dioxygenase reactions carried out by hTDO and hIDO are initiated by the deprotonation of the indoleamine group of L-Trp by the H76 residue and the heme-bound dioxygen, respectively. In contrast with this earlier hypothesis, our current data showed that the deprotonation of the indoleamine by either H55 in xcTDO (i.e., H76 in hTDO) or heme-bound dioxygen in hIDO is not energetically feasible, and that the deprotonation of the indoleamine is not essential for the initiation of the dioxygenase reaction carried out by either enzyme. Taken together, the data suggest that the lower kcat observed for the H76 mutants is a result of perturbed Trp orientation with respect to the heme-bound dioxygen, which is a critical factor for the reaction (vide infra), owing to the disruption of the hydrogen bond between H76 and the indolamine group of Trp. In addition, the CO complexes of IDO and TDO are plausibly not an accurate model for their reactive oxy derivatives, as suggested by QM and QM/MM simulation studies of several other heme proteins, revealing important differences in the structural and electronic properties of the CO complexes compared with those of the O2 adducts [27, 52].
Direct oxygen attack as the first step in the dioxygenase reaction
To examine if the heme-bound O2 could directly attack either the C2 or the C3 atom of the C2=C3 bond of L-Trp, we computed the energy profile along the reaction coordinate defined by d(Ot–C2,Trp). For hIDO the calculation was performed for Cf1A only, as it is similar to the ternary complex of xcTDO, and its Ot is best positioned for the oxygen attack reaction.
The results show that in both enzymes the heme-bound superoxide in the active ternary complex can be readily inserted into the C2=C3 bond of the indole ring, giving rise to a 2-alkylperoxo transition state, which spontaneously converts to a 2,3-alkyl epoxide and a compound II type of ferryl intermediate, via a homolytic O–O bond cleavage reaction as schematically illustrated in Scheme 2, and consistent with our previous experimental observations [1]. As shown in Fig. 4a, the energy profiles are similar for both proteins, although in xcTDO a much sharper energy drop is associated with the O–O bond cleavage reaction, leading to the ferryl epoxide intermediate. The data resulting from the energetic, Mulliken, and spin population analyses of the reaction are summarized in Table 4.
Scheme 2.
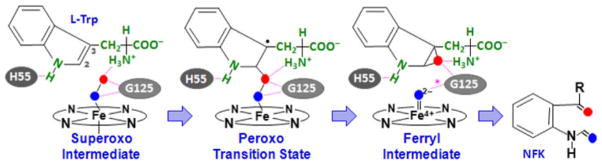
Proposed ferryl-based dioxygenase mechanism. The scheme is based on the reaction involving tryptophan dioxygenase from Xanthomonas campestris; a similar mechanism is applicable to human indoleamine 2,3-dioxygenase (hIDO), except that in hIDO H55 is replaced by S167, which is incapable of forming a hydrogen bond with the indole amine group of the substrate, G125 is replaced by A264, and the hydrogen bond indicated by the asterisk in the ferryl intermediate is absent
Fig. 4.

Energy profile associated with the radical attack of the heme-bound dioxygen on the C2=C3 bond of L-Trp in xcTDO and hIDO along the singlet reaction coordinate (a) and the structures of the associated transition and intermediate states (b, c). The O–O bond lengths of the 2-alkylperoxo transition state (1.51 and 1.42 Å for xcTDO and hIDO, respectively) are elongated as compared with those in the superoxo species
Table 4.
Energetic parameters, Mulliken charges, and spin populations of the transition states and products in the S and T spin states
| xcTDO
|
hIDO
|
||||||
|---|---|---|---|---|---|---|---|
| S | T | SS | T | ||||
| Eact | +11.2 (5.3) | +8.1 (7.6) | +15.5 | +13.8 | |||
| ΔE | −12.1 (−12.6) | −21.7 (−21.9) | −9.2 | −16.2 | |||
| ΔEspin (product) | +10.8 | 0 | +7.7 | 0 | |||
| Mulliken charge
|
||||||||
|---|---|---|---|---|---|---|---|---|
| TS | P | TS | P | TS | P | TS | P | |
| Fe | +0.830 | +0.791 | +0.840 | +0.797 | +0.837 | +0.799 | +0.853 | +0.806 |
| O2 | −0.488 | – | −0.534 | – | −0.519 | – | −0.546 | – |
| 2,3-Alkyl epoxide | – | −0.318 | – | −0.317 | – | +0.063 | – | +0.064 |
| Ferryl-Hprox | – | +0.060 | – | +0.058 | – | −0.067 | – | −0.068 |
| H55 | +0.198 | +0.258 | +0.273 | +0.256 | – | – | – | – |
| Spin population
|
||||||||
|---|---|---|---|---|---|---|---|---|
| TS | P | TS | P | TS | P | TS | P | |
| Fe | −0.006 | +0.001 | +0.914 | +1.255 | +0.891 | +0.001 | +1.005 | +1.252 |
| O2 | +0.002 | – | +0.434 | – | −0.124 | – | +0.274 | – |
| 2,3-Alkyl epoxide | – | +0.002 | – | +0.007 | – | +0.001 | – | +0.006 |
| Ferryl-Hprox | – | +0.009 | – | +1.996 | – | +0.000 | – | +1.998 |
Ea and ΔE (kcal/mol) are the activation energies and total reaction energies associated with the Fe3+–O2− + L-Trp → Fe4+=O2− + 2,3-alkyl epoxide reaction along the singlet or triplet reaction coordinate. Values in parentheses for xcTDO correspond to those obtained with a larger basis. ΔEspin (product) is the energy of the product, 2,3-alkyl epoxide + ferryl-Hprox, in the two spin states, obtained by using that of the triplet state as a reference
TS transition state, P product state, Hprox proximal His ligand
Conformational analysis along the reaction coordinate shows that in the active ternary complex of xcTDO (Fig. 1a, c), the Fe–O–O moiety lies perpendicular to the heme plane and parallel to the indole ring, providing optimum alignment of the O–O bond, with respect to the C2=C3 bond, for the insertion of the dioxygen into the C2 position of the indole ring, yielding the 2-alkylperoxo transition state. The optimized structures of the 2,3-alkyl-epoxo transition state and the ferryl 2,3-alkyl epoxide intermediate state, shown in Fig. 4b, combined with that of the ternary complex, show that, along the reaction coordinate, the indole ring of the substrate is held in position by H55, via a hydrogen bond; in addition, the two atoms of dioxygen form hydrogen bonds with the amino group of the substrate, as well as the amine group of the G125 residue, highlighting the importance of stereoelectronic factors in the dioxygenase reaction.
Owing to the absence of H55 in hIDO, the indole ring of L-Trp exhibits higher conformational freedom as compared with TDO, leading to a more flexible ternary complex (Fig. 2b). Consequently, in xcTDO all the hydrogen-bonding interactions stabilizing the heme-bound O2 are conserved during the superoxo → peroxo‡ → ferryl transition, whereas in hIDO the hydrogen bond between Op and A264 is lost in the ferryl intermediate, owing to a reorientation of the indole ring (Fig. 4c). This high mobility, observed only in hIDO, is likely to disfavor the subsequent attack of the ferryl oxygen on the 2,3-alkyl epoxide, accounting for the experimental observation that the ferryl intermediate can only be accumulated to detection level during the IDO reaction, and not during the TDO reaction [1].
To examine if the spin state of the system affects the reaction, we also examined the reactions in the triplet state. The data (Table 4) show that the dioxygenase reaction is only slightly perturbed by the spin state. The data also confirm that in both enzymes, the O–O bond cleavage is homolytic, regardless of the spin state, as indicated by the almost neutral Mulliken charges on the ferryl–Hprox moiety (approximately +0.06 and −0.07 for xcTDO and hIDO, respectively). Mulliken charges of the O2 moiety also show an increase in the negative charge in the transition state as compared with that in the ternary complex (Table 4), supporting the superoxide character of the transition state. The data listed in Table 4 also reveal that the strong His55–Trp interaction is maintained along the reaction coordinate in TDO, as evidenced by the charge transfer. Finally, in both enzymes, the ferryl epoxide intermediate is slightly more stable in the triplet state, consistent with the fact that a compound II type of ferryl intermediate in hemeproteins, such as catalases or peroxidases, typically displays a triplet ground state [53, 54].
To test if the type of basis set used for the calculation influences the results, we also computed the energy barriers and ΔE for the xcTDO reaction by using a better-quality basis set. As listed in Table 4, similar results were obtained, although smaller energy barriers were observed for both the singlet and the triplet reactions. The data confirm the reliability of our calculations with the double zeta plus polarization basis set, which were accomplished with a much lower computational cost.
Effect of protein matrix
To further evaluate how the protein matrix influences the dioxygenase reaction, we computed the same reaction carried out by a enzyme-free model complex in a vacuum, by using the ternary complex, 2-alkylperoxo transition state, and ferryl epoxide intermediate structures obtained from the QM/MM simulation of the xcTDO reaction described already, but with the protein moiety deleted. The results show that, in both the singlet and the triplet states, the in vacuum enzyme-free reaction follows the same trend as the enzyme reaction. The ternary complex displays a singlet ground state, whereas the ferryl epoxide intermediate exhibits a triplet ground state. The activation barriers for the singlet and triplet reactions are 23.0 and 11.1 kcal/mol, respectively, with respect to 11.2 and 8.1 kcal/mol barriers observed in the xcTDO enzyme reactions (Table 4), indicating that the xcTDO protein matrix significantly stabilizes the transition state, especially in the singlet state. The origin of transition-state stabilization in the enzyme possibly arises in part owing to stabilization of the significant negative charge on the oxygen moiety in the peroxo state, due to strong hydrogen bonds with protein backbone amides. In this context, the intrinsically more polarized oxygen in the triplet state may account for the different amount of protein stabilization. Finally, the in vacuum simulation results also show that the enzyme stabilizes the ferryl epoxide intermediate by approximately 3.5 and 5.7 kcal/mol for the singlet and triplet states.
It is important to note that a similar epoxidation mechanism of the dioxygenation has been investigated by B3LYP–DFT calculations based on a model heme-indole system in a vacuum by Chung et al. [13]. In that work, the reaction was found to be initiated by oxygen attack on the C2 or C3 atom of the indole ring, yielding a heme-bound alkylperoxo intermediate, with an activation barrier of approximately 12.0–22.7 kcal/mol and ΔE of 7.0–21.5 kcal/mol (depending on the spin state of the system). The reaction is followed by an O–O bond cleavage reaction, leading to the ferryl epoxide transition state, with an energy of approximately 11.0–16.0 kcal/mol [13]. On the basis of the above-mentioned values, the effective activation barrier for the ferryl epoxide formation (starting from the oxy complex) was estimated to be at least 23.0 kcal/mol, which was judged to be too high to compete with the alternative radical and electrophilic addition pathways illustrated in Scheme 1. Contrary to the B3LYP-DFT results, the QM/MM data presented here as well as the resonance Raman data [1] support the ferryl-based mechanism (Scheme 2), highlighting the importance of protein matrix in modulating the reactions of IDO and TDO. To examine if the observed differences are a result of the basis sets and functional used, we also computed the energies of the reaction in a vacuum at the protein-optimized geometries by using B3LYP calculations and Gaussian basis sets. The activation barriers were found to be 16.1 and 6 kcal/mol for the singlet and triplet reactions, respectively, which were only slightly lower than those computed at the SIESTA/PBE level.
In addition to stabilizing the ferryl epoxide intermediate, the protein matrix is presumably critical for substrate positioning, as the B3LYP–DFT results reported by Chung et al. [13] indicate that the indole ring of the substrate lies parallel to the heme plane, whereas our QM/MM results show that the indole ring adopts a perpendicular orientation (Figs. 1, 4), which constrains the C2=C3 bond parallel to the O=O bond, facilitating the oxygen attack and the subsequent O–O bond cleavage reaction.
It is noteworthy that similar a ferryl epoxide intermediate has recently been proposed for the dioxygenase reaction carried out by a non-heme dioxygenase, apocarotenoid oxygenase, on the basis of B3LYP–DFT calculations [55]. In apocarotenoid oxygenase, the active site consists of a ferrous iron atom coordinated by three His ligands, and possibly also a water molecule. As in xcTDO and hIDO, the reaction starts with dioxygen coordination to the ferrous iron, which is followed by the oxygen attack on a C=C bond of the substrate, leading to dioxygenation and subsequent C–C bond cleavage, via either a dioxetane or an epoxide mechanism, with similar activation barriers (approximately 15.9–17.6 kcal/mol), with the dioxetane mechanism being slightly favored when the water molecule is coordinated to the iron. The consecutive two-step oxygen insertion mechanism also resembles that of another non-heme dioxygenase, homoprotocatechuate 2,3-dioxygenase [56], although the detailed mechanisms significantly differ between these two types of dioxygenases.
Conclusions
Taken together, the QM/MM data reported here are consistent with our previous experimental observation [1], supporting the ferryl-based mechanism illustrated in Scheme 2. In addition to our previous calculation, in this work we provides a systematic evaluation of the feasibility of proton transfer as the initial step of the dioxygenase reaction. Our results show that the proton transfer from indoleamine to either H55 in xcTDO or heme-bound dioxygen in hIDO is not likely to be the first step of the reaction, as in either case it is not energetically favorable. Our new data, obtained by detailed analysis of the electronic and structural properties of the reactant, transition state, and ferryl intermediate associated with the first step of the reaction in two spin states, show that heme-bound dioxygen exhibits superoxide character; it attacks the C2 atom of the C2=C3 bond, which gives rise to a metastable ferryl and 2,3-alkyl epoxide intermediate, via a 2-alkylperoxo transition state. The subsequent attack of the ferryl oxygen on the 2,3-alkyl epoxide can lead to NFK, via a mechanism which remains to be elucidated. Our results also reveal the unique multiconformational nature of the ternary complex of hIDO as compared with xcTDO, in particular the flexible nature of the substrate as well as the heme with respect to the protein matrix surrounding them, which plausibly raises the activation barrier for the subsequent insertion of the ferryl oxygen into the indole 2,3-epoxide, accounting for the transient accumulation of the ferryl intermediate in hIDO, but not in hTDO [1]. The new data also demonstrate the importance of the protein matrix in positioning the substrate in a favorable regio-orientation with respect to the heme-bound dioxygen, thereby activating the dioxygen for its insertion into the substrate. We also showed that the protein matrix plays a key role in stabilizing the 2-alkylperoxo transition state and the ferryl epoxide intermediate, thereby lowering the activation barrier for the reaction.
In summary, our data provide a detailed microscopic picture of the dioxygenase chemistry carried out by the two physiologically important enzymes, and introduce a paradigm shift in our common perception of heme dioxygenase chemistry. They reveal the importance of stereoelectronic factors in TDO and IDO, as well as the subtle mechanistic differences between the two enzymes. Recently, hIDO has attracted a great deal of attention as a result of the recognition of its potential as a therapeutic target for cancer [11]. Our data offer a starting point for additional computational and structural investigations, which are anticipated to provide valuable insights for the development of anticancer drugs specifically targeting hIDO.
Acknowledgments
This work was partially supported by grants from Universidad de Buenos Aires 08-X625 to M.A.M. and 08-X074 to D.A.E., ANPCYT 07-1650 to M.A.M. and 06-25667 to D.A.E., Conicet PIP 01207 and a Guggenheim Foundation grant awarded to D.A.E, and NIH Molecular Biophysics Training Grant GM008572 to A.L.-B. D.A.E., and M.A.M. are members of CONICET, L.C. holds a CONICET Ph.D. fellowship. Computer power was provided by the Centro de Computacion de Alto Rendimiento (CECAR) at Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires. We thank Pau Arroyo Mañez for useful discussions.
Abbreviations
- Cf1
Conformation 1
- Cf2
Conformation 1
- DFT
Density functional theory
- hIDO
Human indoleamine 2,3-dioxygenase
- hTDO
Human tryptophan dioxygenase
- IDO
Indoleamine 2,3-dioxygenase
- MD
Molecular dynamics
- MM
Molecular mechanical
- NFK
N-Formylkynurenine
- PDB
Protein Data Bank
- PBE
Generalized gradient approximation functional proposed by Perdew, Burke, and Ernzerhof
- QM
Quantum mechanical
- TDO
Tryptophan dioxygenase
Contributor Information
Luciana Capece, Departamento de Química Inorgánica, Analítica y Química Física/INQUIMAE-CONICET, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Ciudad Universitaria, Pabellón 2, C1428EHA Buenos Aires, Argentina.
Ariel Lewis-Ballester, Department of Physiology and Biophysics, Albert Einstein College of Medicine, 1300 Morris Park Avenue, New York, NY 10461, USA.
Dipanwita Batabyal, Department of Physiology and Biophysics, Albert Einstein College of Medicine, 1300 Morris Park Avenue, New York, NY 10461, USA.
Natali Di Russo, Departamento de Química Inorgánica, Analítica y Química Física/INQUIMAE-CONICET, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Ciudad Universitaria, Pabellón 2, C1428EHA Buenos Aires, Argentina.
Syun-Ru Yeh, Email: syeh@aecom.yu.edu, Department of Physiology and Biophysics, Albert Einstein College of Medicine, 1300 Morris Park Avenue, New York, NY 10461, USA.
Dario A. Estrin, Departamento de Química Inorgánica, Analítica y Química Física/INQUIMAE-CONICET, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Ciudad Universitaria, Pabellón 2, C1428EHA Buenos Aires, Argentina
Marcelo A. Marti, Email: marcelo@qi.fcen.uba.ar, Departamento de Química Inorgánica, Analítica y Química Física/INQUIMAE-CONICET, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Ciudad Universitaria, Pabellón 2, C1428EHA Buenos Aires, Argentina. Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Ciudad Universitaria, Pabellón 2, C1428EHA Buenos Aires, Argentina
References
- 1.Lewis-Ballester A, Batabyal D, Egawa T, Lu C, Lin Y, Marti MA, Capece L, Estrin DA, Yeh SR. Proc Natl Acad Sci USA. 2009;106:17371–17376. doi: 10.1073/pnas.0906655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sono M, Roach MP, Coulter ED, Dawson JH. Chem Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 3.Hayaishi O. J Biochem. 1976;79:13–21. doi: 10.1093/oxfordjournals.jbchem.a131115. [DOI] [PubMed] [Google Scholar]
- 4.Forouhar F, Anderson JLR, Mowat CG, Vorobiev SM, Hussain A, Abashidze M, Bruckmann C, Thackray SJ, Seetharaman J, Tucker T, Xiao R, Ma L-C, Zhao L, Acton TB, Montelione GT, Chapman SK, Tong L. Proc Natl Acad Sci USA. 2007;104:473–478. doi: 10.1073/pnas.0610007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greengard O, Feigelson P. J Biol Chem. 1962;237:1903–1907. [PubMed] [Google Scholar]
- 6.Yamamoto S, Hayaishi O. J Biol Chem. 1967;242:5260–5266. [PubMed] [Google Scholar]
- 7.Shimizu T, Nomiyama S, Hirata F, Hayaishi O. J Biol Chem. 1978;253:4700–4706. [PubMed] [Google Scholar]
- 8.Zhang Y, Kang SA, Mukherjee T, Bale S, Crane BR, Begley TP, Ealick SE. Biochemistry. 2007;46:145–155. doi: 10.1021/bi0620095. [DOI] [PubMed] [Google Scholar]
- 9.Sugimoto H, Oda S-i, Otsuki T, Hino T, Yoshida T, Shiro Y. Proc Natl Acad Sci USA. 2006;103:2611–2616. doi: 10.1073/pnas.0508996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, Extermann M, Mellor AL, Munn DH, Antonia SJ. Int J Cancer. 2002;101:151–155. doi: 10.1002/ijc.10645. [DOI] [PubMed] [Google Scholar]
- 11.Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Nat Med. 2005;11:312–319. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 12.Koyake Y, Masayama I. Z Physiol Chem. 1936;243:237–244. [Google Scholar]
- 13.Chung LW, Li X, Sugimoto H, Shiro Y, Morokuma K. J Am Chem Soc. 2008;130:12299–12309. doi: 10.1021/ja803107w. [DOI] [PubMed] [Google Scholar]
- 14.Batabyal D, Yeh SR. J Am Chem Soc. 2007;129:15690–15701. doi: 10.1021/ja076186k. [DOI] [PubMed] [Google Scholar]
- 15.Hamilton GA. Adv Enzymol Relat Areas Mol Biol. 1969;32:55–96. doi: 10.1002/9780470122778.ch3. [DOI] [PubMed] [Google Scholar]
- 16.Leeds JM, Brown PJ, McGeehan GM, Brown FK, Wiseman JS. J Biol Chem. 1993;268:17781–17786. [PubMed] [Google Scholar]
- 17.Ronsein GE, Oliveira MCB, Miyamoto S, Medeiros MHG, Di Mascio P. Chem Res Toxicol. 2008;21:1271–1283. doi: 10.1021/tx800026g. [DOI] [PubMed] [Google Scholar]
- 18.Terentis AC, Thomas SR, Takikawa O, Littlejohn TK, Truscott RJW, Armstrong RS, Yeh S-R, Stocker R. J Biol Chem. 2002;277:15788–15794. doi: 10.1074/jbc.M200457200. [DOI] [PubMed] [Google Scholar]
- 19.Chauhan N, Thackray SJ, Rafice SA, Eaton G, Lee M, Efimov I, Basran J, Jenkins PR, Mowat CG, Chapman SK, Raven EL. J Am Chem Soc. 2009;131:4186–4187. doi: 10.1021/ja808326g. [DOI] [PubMed] [Google Scholar]
- 20.Lu C, Lin Y, Yeh S-R. J Am Chem Soc. 2009;131:12866–12867. doi: 10.1021/ja9029768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 22.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, DeBolt S, Ferguson D, Seibel G, Kollman P. Comput Phys Commun. 1995;91:1–41. [Google Scholar]
- 23.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 25.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 26.Marti MA, Crespo A, Capece L, Boechi L, Bikiel DE, Scherlis DA, Estrin DA. J Inorg Biochem. 2006;100:761–770. doi: 10.1016/j.jinorgbio.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 27.Capece L, Estrin DA, Marti MA. Biochemistry. 2008;47:9416–9427. doi: 10.1021/bi800682k. [DOI] [PubMed] [Google Scholar]
- 28.Bidon-Chanal A, Martí MA, Estrín DA, Luque FJ. NATO science for peace and security series A: chemistry and biology. Springer; Netherlands: 2009. Self-organization of molecular systems; pp. 33–47. [Google Scholar]
- 29.Boechi L, Martà MA, Milani M, Bolognesi M, Luque FJ, Estrin DA. Proteins Struct Funct Genet. 2008;73:372–379. doi: 10.1002/prot.22072. [DOI] [PubMed] [Google Scholar]
- 30.Capece L, Marti MA, Bidon-Chanal A, Nadra A, Luque FJ, Estrin DA. Proteins Struct Funct Bioinform. 2008;75:885–894. doi: 10.1002/prot.22297. [DOI] [PubMed] [Google Scholar]
- 31.Nadra AD, Martà MA, Pesce A, Bolognesi M, Estrin DA. Proteins Struct Funct Genet. 2008;71:695–705. doi: 10.1002/prot.21814. [DOI] [PubMed] [Google Scholar]
- 32.Crespo A, Scherlis DA, Marti MA, Ordejon P, Roitberg AE, Estrin DA. J Phys Chem B. 2003;107:13728–13736. [Google Scholar]
- 33.Perdew JP, Burke K, Ernzerhof M. Phys Rev Lett. 1996;77:3865. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 34.Capece L, Marti MA, Crespo A, Doctorovich F, Estrin DA. J Am Chem Soc. 2006;128:12455–12461. doi: 10.1021/ja0620033. [DOI] [PubMed] [Google Scholar]
- 35.Marti MA, Crespo A, Bari SE, Doctorovich FA, Estrin DA. J Phys Chem B. 2004;108:18073–18080. [Google Scholar]
- 36.Marti MA, Capece L, Crespo A, Doctorovich F, Estrin DA. J Am Chem Soc. 2005;127:7721–7728. doi: 10.1021/ja042870c. [DOI] [PubMed] [Google Scholar]
- 37.Crespo A, Marti MA, Kalko SG, Morreale A, Orozco M, Gelpi JL, Luque FJ, Estrin DA. J Am Chem Soc. 2005;127:4433–4444. doi: 10.1021/ja0450004. [DOI] [PubMed] [Google Scholar]
- 38.Bikiel DE, Boechi L, Capece L, Crespo A, De Biase PM, Di Lella S, González Lebrero MC, Martí MA, Nadra AD, Perissinotti LL, Scherlis DA, Estrin DA. Phys Chem Chem Phys. 2006;8:5611–5628. doi: 10.1039/b611741b. [DOI] [PubMed] [Google Scholar]
- 39.Perissinotti LL, Marti MA, Doctorovich F, Luque FJ, Estrin DA. Biochemistry. 2008;47:9793–9802. doi: 10.1021/bi801104c. [DOI] [PubMed] [Google Scholar]
- 40.Crespo A, Marti MA, Roitberg AE, Amzel LM, Estrin DA. J Am Chem Soc. 2006;128:12817–12828. doi: 10.1021/ja062876x. [DOI] [PubMed] [Google Scholar]
- 41.Frisch MJ, GWT, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Stratmann RE, Jr, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Gonzalez C, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA. Gaussian 98. Gaussian; Pittsburgh: 1998. [Google Scholar]
- 42.Batabyal D, Yeh S-R. J Am Chem Soc. 2009;131:3260–3270. doi: 10.1021/ja807969a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brantley RE, Jr, Smerdon SJ, Wilkinson AJ, Singleton EW, Olson JS. J Biol Chem. 1993;268:6995–7010. [PubMed] [Google Scholar]
- 44.Samelson-Jones BJ, Yeh SR. Biochemistry. 2006;45:8527–8538. doi: 10.1021/bi060143j. [DOI] [PubMed] [Google Scholar]
- 45.Meunier B, de Visser SP, Shaik S. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 46.Bordwell FG. Acc Chem Res. 1988;21:456–463. [Google Scholar]
- 47.Lide DR, editor. Handbook of chemistry and physics. 82. CRC Press; Boca Raton: 2001–2002. [Google Scholar]
- 48.Guallar V, Harris DL, Batista VS, Miller WH. J Am Chem Soc. 2002;124:1430–1437. doi: 10.1021/ja016474v. [DOI] [PubMed] [Google Scholar]
- 49.Saito I, Matsuura T, Nakagawa M, Hino T. Acc Chem Res. 1977;10:346–352. [Google Scholar]
- 50.Thackray SJ, Bruckmann C, Anderson JLR, Campbell LP, Xiao R, Zhao L, Mowat CG, Forouhar F, Tong L, Chapman SK. Biochemistry. 2008;47:10677–10684. doi: 10.1021/bi801202a. [DOI] [PubMed] [Google Scholar]
- 51.Chauhan N, Basran J, Efimov I, Svistunenko DA, Seward HE, Moody PCE, Raven EL. Biochemistry. 2008;47:4761–4769. doi: 10.1021/bi702405a. [DOI] [PubMed] [Google Scholar]
- 52.Martí MA, Bikiel DE, Crespo A, Nardini M, Bolognesi M, Estrin DA. Proteins Struct Funct Bioinform. 2005;62:641–648. doi: 10.1002/prot.20822. [DOI] [PubMed] [Google Scholar]
- 53.Rovira C, Fita I. J Phys Chem B. 2003;107:5300–5305. [Google Scholar]
- 54.Bathelt CM, Zurek J, Mulholland AJ, Harvey JN. J Am Chem Soc. 2005;127:12900–12908. doi: 10.1021/ja0520924. [DOI] [PubMed] [Google Scholar]
- 55.Borowski T, Blomberg MR, Siegbahn PE. Chemistry (Weinheim) 2008;14:2264–2276. doi: 10.1002/chem.200701344. [DOI] [PubMed] [Google Scholar]
- 56.Kovaleva EG, Lipscomb JD. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]