

Abstract
Glycosylation is an important post-translational modification that influences many biological processes critical for development, normal physiologic function, and diseases. Unfortunately, progress towards understanding the roles of glycans in biology has been slow due to the challenges of studying glycans and the proteins that interact with them. Glycan microarrays provide a high-throughput approach for the rapid analysis of carbohydrate-macromolecule interactions. Protocols detailed here are intended to help laboratories with basic familiarity of DNA or protein microarrays to begin printing and performing assays using glycan microarrays. Basic and advanced data processing are also detailed, along with strategies for improving reproducibility of data collected with glycan arrays.
Keywords: Glycosylation, Microarray, Neoglycoconjugate, Carbohydrate-Dependent Binding, Serum Antibody Profile
INTRODUCTION
DNA microarray technology revolutionized our ability to probe expression of thousands of genes during a single high-throughput experiment. Extension of microarray technology to proteins similarly advanced the field of proteomics. More recently, application of microarray technology to study carbohydrate-binding molecules has impacted the field of glycobiology. The importance of glycosylation on the interactions of molecules has long been recognized. However, the tools available for studying glycobiology have lagged behind other fields. Glycan microarrays, which display many different carbohydrates or glycans on a solid support in a spatially-defined arrangement, provide a versatile tool for studying carbohydrate-mediated binding that addresses many of the technical limitations that have traditionally slowed glycobiology (Liang et al., 2008; Liu et al., 2009; Oyelaran and Gildersleeve, 2009; Song and Pohl, 2009). This chapter details protocols for printing glycan microarrays, assays for studying glycan-binding properties of various samples, and key aspects of data processing related to these high-throughput experiments. The protocols focus on the construction of neoglycoprotein/glycoprotein arrays. Many of the considerations and technical challenges, however, are relevant to other glycan array formats and protein arrays.
BASIC PROTOCOL 1: PRODUCTION OF GLYCAN MICROARRAYS
Production of high quality arrays is critical for a successful glycan array experiment. This printing protocol describes attachment of glycoproteins and neoglycoproteins to an epoxidecoated slide. These protocols will allow a laboratory equipped for microarray processing to adapt existing protocols for DNA or protein microarrays to glycan microarrays. The protocols require a robotic array printer and fluorescence scanner, which are widely used for printing and processing DNA and/or protein microarrays. Additionally, the following protocol describes quality control checks for evaluating array slides.
Materials
Neoglycoproteins and/or glycoproteins (see comments below regarding sources)
Printing Buffer (see recipe)
384-well V-bottom sample plates with lids (X6004, Genetix)
Aluminum plate seals (07-200-683, Costar)
Robotic Microarray Printer (MicroGridII, Genomic Solutions)
Pins (Stealth Microspotting SMP3 Pins, Arrayit)
Epoxide Coated Slides (SuperEpoxy 2 Premium Microarray Substrates, Arrayit)
Hygrometer
Neoglycoproteins
Acquisition of a diverse set of glycans and carbohydrates is a key step for construction of a glycan array. The focus of this protocol is on the production of neoglycoprotein arrays. Neoglycoproteins are non-naturally occurring conjugates formed by covalently linking a glycan to a carrier protein, such as albumin. A variety of BSA and HSA conjugates are commercially-available (some sources include Dextra Labs, IsoSep AB, Glycorex, and Glycotech), and a number of other neoglycoproteins can be obtained in a single step by reductive amination of BSA with oligosaccharides containing a free reducing end (Gildersleeve et al., 2008; Roy et al., 1984). Using commercial sources and simple conjugations, one can obtain approximately 50-100 neoglycoproteins at present.
Prepare Sample Plates Containing Glycoproteins and Neoglycoproteins
Dilute glycoproteins and neoglycoproteins in printing buffer to a concentration of 125 μg/mL. The concentration of albumin conjugates can be determined by measuring UV absorption (280 nm) and comparing the results to a titration curve generated from pure albumin for conjugates with functional groups that do not absorb in the same region. Alternatively, concentration can be determined using the Bradford assay.
Determine the layout of the sample plates, which must be coordinated with the number and configuration of pins that will be used for printing. See Figure 1 for details.
Place 15 μL of solution into the appropriate well(s) of a 384-well V-bottom sample plate with lid. Depending on the number of samples and setup of the sample plate, multiple sample plates may be needed.
Seal plate with an aluminum seal to prevent evaporation and cross-over contamination. 5. Store sealed plates at −20 °C in a non-defrosting freezer. Minimize freeze-thawing of samples.
Immediately before printing and prior to removing the aluminum seal, thaw samples and spin plates down for 5 minutes at 200 × g.
Figure 1. Coordination of slide layout, configuration of sample plate, and pin positioning.
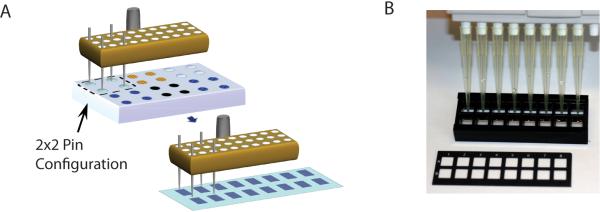
(A) An example is shown with four pins loaded in the pin tool. Pins are lowered into the wells of a source plate (e.g. 384 well plate), allowing the pins to fill with neoglycoprotein solution. The robot then moves the pin head to the slides to print arrays of spots. 16 complete arrays are printed on each slide. The spacing of the pins and the array layout should match the spacing of the 16-well slide module. (B) Slides are fitted with a 16-well module to form 16 separated wells. The wells have the same spacing as is normally found on 96-well plates, and a multichannel pipettor can be used to add solutions to the wells. The slide shown in the picture has a mask that subdivides the slide into 16 areas; however, this is shown for illustrative purposes, and slides without masks can also be used.
NOTES:
A concentration of 125 μg/mL was selected to saturate the printed surface with glycoprotein or neoglycoproteins. Concentration of protein in the printing buffers should be adjusted to achieve saturation if either the composition of the printing buffer or the array surface changes.
Include appropriate controls such as unmodified BSA and HSA (negative controls) and BSA conjugated with Cy3 or Alexa Fluor 647. These controls will provide standards for scanning and aid in aligning images during data processing
Sample plates may be reused for different print batches provided that they are sealed with aluminum foil and frozen at −20° C between uses. Before reusing the sample plate, check for evaporation from the printing buffer that may occur during printing. Samples may lose 2-5 μL of water during printing. Add Milli-Q water to bring the sample volume up to 15 μL, and mix sample by repipetting. Sample plates handled in this way have been used for up to 6 months.
The maximum number of spots that can be printed within a well depends on a number of parameters, including the spot size, the pitch, the location and layout of pins, and the type of arrayer and software. Using the listed parameters, arrayer, and a 16 well/grid format, 440 spots can be printed in each well.
Program Robotic Printer
The programming methods and parameters are specific to the printer and software. Some guidelines are described below, based on our use of a MicroGrid II array printer and BioRobotics TAS Application Suite software.
-
7.
Determine the configuration of multiple array copies to be printed onto the slide. Position multiple copies of the array so that the arrays are aligned with the wells of slide modules positioned on top of the slide. For instance, printing 16 arrays in an 8×2 configuration conveniently allows for an 8-well multichannel pipette to be used with commercially available slide modules. (Figure 1)
-
8.
Program the arrayer to print 2-4 adjacent spots for each component.
-
9.
Program the robotic printer to print spots at an appropriate separation. The distance between the centers of adjacent spots (referred to as the pitch) is typically twice the desired diameter of microarray spots.
-
10.
Optimize printer settings (dwell time, velocity, and number of prespots) to achieve uniform spots of the desired size.
NOTE: The amount of material transferred during printing depends on the pin's weight, composition, and geometry as well as the slide's surface properties. Thus, for each combination of pins and slides, printer settings should be optimized to obtain uniform spots of the desired size. As a starting point, our preferred settings for printing are described in Table 1. The troubleshooting section discusses strategies for refining printing parameters.
Table 1.
Recommended Printing Parameters
| Step | Parameter | Description | Recommended Value |
|---|---|---|---|
| Sample Loading | Pin depth | Distance pin is submerged into source solution | 5 mm |
| Speed | Speed pin is lowered into the source solution | 4 mm/s | |
| Dwell time | Time pin held in source solution before wiggles or dips | 1 second | |
| Number of wiggles | Times pin moved laterally while submerged | 0 | |
| Number of dips | Times pin moved vertically while submerged | 2 | |
| Dip height | Distance pin is further submerged | 2 mm | |
| Inter-dip delay | Time pin kept submerged between dips | 1 second | |
| Lid Handling | Specifies when lid is replaced | Immediately | |
| Pre-Spotting | Number of pre-spots | Times a freshly loaded pin is spotted onto a slide before it is used to print slides that will be used for experiments | 18 |
| Delay before pre-spotting | Time pin is momentarily suspended above slide before descending to contact the slide | 0.5 second | |
| Target height | Distance tool is lowered to bring pin into contact with slide | 1.5 mm | |
| Speed | Speed that tool is lowered to bring pin into contact with slide | 4 mm/s | |
| Surface Height | Thickness of slide | 1 mm | |
| Dwell time | Time tool is held in place at its target height | 0.250 second | |
| Multiple Strikes | Number of times the pin is tapped onto the slide to form each spot | 1 | |
| Pitch | Distance pin moves laterally or vertically between pre-spots | 0.45 mm | |
| Printing | Number of print slides | High-print quality slides that will be used for experiments | 12 |
| Delay before spotting | Time pin is momentarily suspended above slide before contact | 0 second | |
| Target height | Distance tool is lowered as pin contacts slide | 1 mm | |
| Speed | Speed that tool is lowered as pin contacts slide | 4 mm/s | |
| Dwell time | Time tool is held in place at its target height | 0 second | |
| Multiple Strikes | Number of times the pin is tapped onto the slide to form each spot | 1 | |
| Number of spots/sample | Number of spots per samples | 2-4 spots | |
| Pitch | Distance pin moves laterally or vertically between spots | 0.2 mm | |
| Washes | Dip Height | Distance pins submerged into recirculating baths | 5 mm |
| Bath Dwell Time | Time pins are held in bath | 3 seconds | |
| MWS Washes | Times wash in Main Wash Station (MWS) is repeated | 2 | |
| Fill Time | Time that water is pumped into MWS | 3 seconds | |
| Drain Time | Time that vacuum is turned on to drain MWS | 8 seconds | |
| Number of Wash Cycles | Number of times the entire wash cycle is repeated | 2 |
Setup and Initiate Robotic Printer
-
11.
Check pins under microscope for debris or damage. Follow manufacturer's guidelines for cleaning pins by sonication and/or blowing with particle-free compressed gas. (See Figure 2 and Video 1.)
-
12.
Load pins into tool. Number of pins and configuration of pins depends on the desired distance between arrays and the number of arrays to be printed on each slide. See Figure 1 for further explanation and example configuration.
-
13.
Humidify printing chamber to a relative humidity of 50-60%. Maintain humidity within this range throughout printing.
-
14.
Thaw for 5 minutes at room temperature, and centrifuge (200 × g for 5 minutes) sample plate to collect sample at the bottom of the wells. Remove aluminum plate seal and discard.
-
15.
Load pre-spotting and array slides such that the slide's printed surface faces upward. (See Video 2.)
-
16.
Load the first sample plate into the arrayer. In order to minimize evaporation, program arrayer to replace the plate lid between printing samples. (See Video 3.)
-
17.
Start the print run.
-
18.
During printing, periodically check humidity and that pins are moving freely within the tool. Maintain humidity with the microarrayer at 50-60% by adjusting the microarrayer's humidifier. Pin sticking is an occasional problem caused by moist pins or debris on the tool. If a pin sticks persistently, use forceps to move the pin until it slide freely.
-
19.
After the first sample plate is complete, load the second sample plate. Even if the arrayer can load several plates at once, plates should be loaded individually so that only the sample plate currently being printed is in the arrayer. Minimize the time that sample plates are in the arrayer in order to reduce evaporation. Promptly re-seal plates with a new aluminum plate seal, and return them to the freezer. Alternatively, plates may be refrigerated at 4 °C for up to 3 days if multiple slide batches are to be printed in succession.
-
20.
After the print run completes, inspect slides using a microscope for smearing, merged spots, missing spots, and other defects. (Figure 3) Keep a record of any defects, and save an electronic image of the arrays, if possible.
-
21.
Store slides at −20 °C in a non-defrosting freezer until use. Sealing slides in a bag containing Drierite helps prevent condensation of water vapor onto the slides. Slides stored in this manner can be used for up to 6 months. Longer time frames have not been tested.
Figure 2. Inspection of pins prior to printing.

Magnified views of the pins are shown. The full length of the pin should be inspected for any debris that may clog the channel. A clean pin is free of debris throughout its channel and near the pin tip.
Figure 3. Microscope images of printed arrays.

The figure shows magnified portions of arrays after printing but prior to an assay. (A) High quality printing produces uniform spots evenly spaced on the glass surface, which is free of debris. (B) Small spots may be due to a partially clogged pin or low volume of sample in the pin due to poor pin loading or excessive pre-spotting. (C) Debris on the surface may have no or high signal, depending on its fluorescence. (D) Missing spots are typically due to pin sticking.
BASIC PROTOCOL 2: GLYCAN ARRAY PROFILING OF CARBOHYDRATE-BINDING PROPERTIES
Many different glycan-binding proteins (GBPs) can be studied using glycan microarrays. Some of the most commonly studied samples include lectins, monoclonal antibodies, and serum antibodies. The general steps of the array assay are similar for each of these, but specific conditions and reagents should be tailored to the particular sample. A key consideration is matching the sample with an appropriate detection method. A variety of methods have been used to detect sample binding, but the most common approach relies on either direct or indirect tagging to produce a fluorescent signal. Lectins can be labeled with a fluorophore for direct detection or can be labeled with a tag, such as biotin, followed by detection with an appropriate fluorophore-labeled secondary reagent, such as Cy3-labeled streptavidin. Monoclonal and serum antibodies are typically detected with a fluorophore-labeled secondary antibody specific for the primary antibody's species and isotype of interest.
NOTES:
Selection of an appropriate fluorophore is a key consideration. In addition to matching the appropriate excitation and emission properties for your scanner, degradation and photobleaching should be considered. In our experience, Cy3, Alexa555, and DyeLight fluorophores work well, but Cy5 is not recommended due to high levels of photobleaching and degradation.
Technical parameters of the assay, such as blocking buffers, incubation times, number of washes, and choice of secondary antibodies, should be optimized for new samples similar to optimization of ELISA and Western blotting.
Materials
Glycan arrays (Basic Protocol 1)
Slide module (ProPlate, Invitrogen)
Adhesive seals for slide module (ProPlate, Invitrogen)
Immunoglobulin-free Bovine Serum Albumin (BSA) (A3059, Sigma)
Immunoglobulin-free Human Serum Albumin (HSA) (A8763, Sigma)
Phosphate Buffered Saline (PBS)
PBST Array Wash Buffer (see recipe)
Reference Pooled Serum Sample
Secondary Antibody and/or Cy3-conjugate Streptavidin (JacksonImmunoResearch)
NOTES:
Prepare all solutions fresh on the day of the experiment.
All BSA and HSA must be globulin-free to avoid high levels of background binding. Albumin that has not been sufficiently purified contains globulins that will interfere in the assay. Other sources of albumin purified by gel electrophoresis may be substituted after confirming that they do not contain contaminating immunoglobulins that bind to the array.
Assemble Slide Module and Microarray
Remove glycan arrays from storage at −20 °C.
Inspect glycan arrays for smudging and other defects under microscope. For highest quality results, use only arrays that are defect free. Arrays with defects can be used in certain circumstances, depending on the location and extent of the defects. For example, they are often used for optimizing assay parameters and confirming results. Place the slide atop the slide module such that the printed surface faces the slide module and the wells align with the printed arrays on the slide.
Secure the slide module in place according to the manufacturer's instructions.
Inspect the microarray under a microscope to verify that none of the array components have been smeared.
Block glycan microarray
-
5.
Prepare a fresh solution of blocking buffer (3% BSA in PBS).
-
6.
Gently pipette blocking buffer into each well of the slide module. (For a 16-well slide module, add 200 μL of blocking buffer.) Use care to pipette solution against the side wall of slide module, rather than dropping the liquid directly on the array spots, to prevent smearing of the printed array surface.
NOTE: Rapid or forceful pipetting of blocking buffer may smear the glycan microarray or cause spreading of the array spots. These can appear as “comets” or spots with long tails. Once the unprinted array surface has been blocked, the printed array features are protected from smearing.
-
7.
Seal the slide module with an adhesive strip to prevent evaporation.
-
8.
Incubate for 2 hours at room temperature for lectins and monoclonal antibodies, or overnight at 4 °C for assays involving serum.
-
9.
Remove blocking solution and wash array immediately prior to addition of samples (step 13) so as not to dry out the arrays while preparing samples. Wash the arrays six times with sufficient volume of PBST Array Wash buffer to cover the bottom of the well (e.g. 200 μL/well for a 16/well module). Discard wash solution by inverting and vigorously tapping the slide module over absorbent paper towels to remove as much wash buffer as possible.
Incubate sample with glycan microarray
-
10.
Prepare samples in the sample dilution buffer appropriate for the type of sample being analyzed according to Table.
NOTES:
-
a.
The optimal concentration depends on the particular sample. Most plant lectins and many monoclonal antibodies give strong signals in the 1-10 μg/mL range.
-
b.
Many lectins require metals, such as calcium or manganese, for full activity.
-
c.
Serum antibodies from certain individuals and animals can react with BSA and/or HSA. Both BSA and HSA should be included in the buffer to avoid binding of these antibody populations with BSA and HSA-conjugates on the array surface.
-
11.
Prepare a reference sample that serves as a positive control and will be used later in data analysis. Pooled serum from multiple donors can provide a reference when analyzing serum samples. To minimize freeze-thaw cycles for the reference sample, divide a single pooled sample into 100-200 aliquots. Use a freshly thawed sample for each experiment. Lectins, either individually or pooled, can provide a similar reference when analyzing lectins. A combination of the following four biotinylated lectins will provide signals for a broad range of glycans on arrays: wheat germ agglutinin (WGA, 10 μg/mL), concanavalin A (conA, 10 μg/mL), Bauhinia purpurea lectin (BPL, 10 μg/mL), and Ricinus communis agglutinin (RCA120, 10 μg/mL).
-
12.
Pipette enough sample to cover the bottom of a well. For a 16-well module, 100 μL/well of sample solution is recommended for best reproducibility. However, the volume can be reduced to 50 μL/well if required to conserve sample.
NOTE: For best results, analyze each sample in duplicate, utilizing wells on two different slides printed with different pins.
-
13.
Seal wells with adhesive seals to prevent evaporation.
-
14.
Place arrays in orbital shaker at 100 RPM during incubation. See Error! Reference source not found. for recommended incubation conditions.
Incubate with fluorophore-labeled secondary reagent (If necessary)
-
15.
In cases where samples have been fluorophore-labeled prior to incubation, skip to step 19. Otherwise, thoroughly wash away unbound samples with PBST Array Wash Buffer (200 μL/well for a 16 well module). Let the solution stand for two minutes before discarding wash. Repeat for a total of 3 washes.
-
16.
Dilute the secondary reagent in the appropriate buffer according to Error! Reference source not found..
-
17.
Add secondary reagent solution to the well (100 μL/well for a 16 well module).
-
18.
Seal wells and incubate according to Error! Reference source not found..
NOTE: Prevent photo-bleaching of fluorescently labeled samples by covering arrays with aluminum foil to block light during incubation.
Final Wash Prior to Scanning
-
19.
Wash wells with PBST wash buffer (200 μL/well for a 16 well module). Repeat for a total of 7 washes.
-
20.
Gently remove the slide module from the glass slide according to the manufacturer's instructions. Handle the array by its edges, and use caution to not smear the printed surface.
-
21.
Submerge the glass slides in a bath of PBST wash buffer for 5 minutes. Position the glass slides so that the printed surface faces up.
-
22.
Dry the glass slides using centrifugation as follows: Place the glass slides into 50 mL conical tubes and orient the slides so that they are perpendicular to the arm of the centrifuge. Centrifuge at 200 × g for 5 minutes.
-
23.
Carefully decant any liquid from the centrifuge tube so as not to re-wet the array's printed surface.
-
24.
Remove the slides from centrifuge tubes.
-
25.
Clean the non-printed back side of the glass slides using Kimwipes.
-
26.
Protect the slide from light by using a covered slide holder until ready for scanning. Scan promptly.
NOTE: Scanning immediately following the final wash is recommended. However, little signal loss has been reported over hours to days when the slide is stored properly in the dark at −20 °C (Gu et al., 2006).
BASIC PROTOCOL 3: SCANNING AND DATA ANALYSIS OF GLYCAN MICROARRAYS
Processed glycan microarrays are imaged using a fluorescence scanner with resolution of 10 μm or finer. Quality images show small, uniform spots of varying intensity depending on the amount of sample bound to each neoglycoconjugate or glycoprotein on the array (see Figure 4). Specialized software, such as GenePix Pro 6.0 (Molecular Devices, Sunnyvale, CA), measures the average and/or median pixel intensity for each spot and can be used to subtract background pixel intensity. Background-corrected pixel intensities can be used to measure sample binding in most cases. Further processing of pixel intensities is needed in certain cases. Protocols below include increasing the dynamic range of the array measurements and normalizing array data using a reference sample. These advanced techniques may be useful when comparing samples with largely different affinities for an array component or when analyzing samples on different arrays.
Figure 4. Scans of processed arrays.
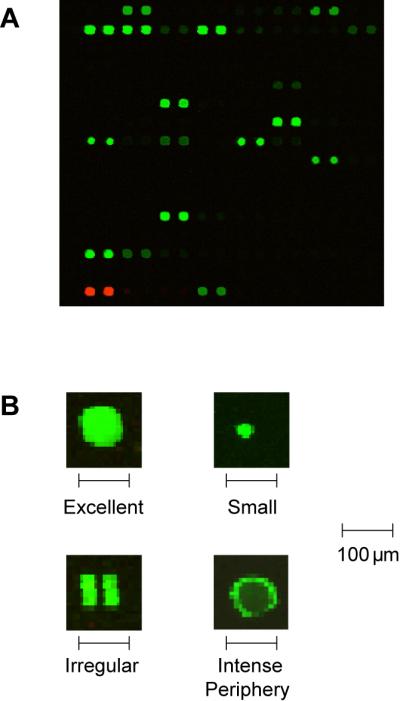
Examples of images scanned using a fluorescence scanner. (A) High quality results show circular spots of varying intensity but uniform size. (B) Under higher magnification, high quality spots have homogeneous intensity throughout the spot, and duplicate spots have nearly identical intensity. Printing and processing problems can result in variations in intensity across individual spots, irregular spot morphology, or missing spots.
Materials
Arrays
Fluorescence scanner (GenePix 4000A, Molecular Devices, Sunnyvale, CA)
Image processing software (GenePix Pro 6.0, Molecular Devices, Sunnyvale, CA)
Microsoft Excel
Basic Scanning and Data Processing
Set fluorescence scanner to a resolution of 10 μm or finer.
Scan using a laser power and photomultiplier tube (PMT) voltage sufficiently high to maximize image quality of array features with low amounts of bound sample without saturating high signals.
Load a GAL file, which contains information regarding where the array features (spots) are supposed to be based on the printing parameters. This will produce a grid of spot boundaries for each array on the slide. Using the software, adjust the positions of the grids to align with the actual spots in the image. Positive controls, such as Cy3-BSA help confirm alignment. Additional refinement of the spot positions and resizing of the spot boundaries can be done manually for each spot on the array, or can be carried out using object recognition algorithms of image processing software. Any bad spots (e.g. spots obscured by lint or fluorescent streaks) should be flagged using the software.
Use image processing software to measure median pixel intensity (MPI) of individual array features, neighboring background pixel intensity (BPI), and MPI-BPI for each array feature.
Transfer or export the data to Excel.
Determine an appropriate minimum value of pixel intensity (floor value), and set all values below the floor value equal to the floor value.
Calculate the final signal value for each array component by averaging the signal from each of the corresponding spots.
NOTES:
Optimal intensity measurement is highly dependent on accurate identification of spots. The positioning and size of each spot boundary should be checked carefully prior to processing.
Use of a floor value is not essential but is very helpful. In certain cases, the background can be equal or somewhat higher than the intensity within a spot. After background correction, the median values for these spots will be negative or zero, which can disrupt many processing steps during subsequent analyses of the data. Therefore, we always set values below 1 equal to 1. Use of higher floor values can also be beneficial. Spot intensities that are only slightly above the background intensity level are highly variable. By using a higher floor value, one can deemphasize differences resulting from experimental noise. A floor value of 0.5-1.0 times the background intensity is recommended.
Alternate Protocol 1
Advanced processing and methods
Extend Dynamic Range of Pixel Intensity Measurements (optional)
If a single scan cannot be obtained that provides good signal from low intensity spots without saturating high intensity spots, two scans can be combined to increase dynamic range according to a method described by Lyng et al. (Lyng et al., 2004). This method is most effective when there are a number of signals on the array, such as with serum antibodies.
First, obtain the primary scan at a higher PMT voltage in order to obtain good signal from low intensity spots.
Perform secondary scan with lower PMT voltage such that no pixels or spots are saturated.
Obtain the raw pixel intensities for these scans as described for the basic scanning protocol. (Steps 3-6 of Basic Protocol 3)
For the entire slide, identify all array features with intensities that are approximately 30-50% of saturation. For each of these middle intensity features, calculate the ratio of pixel intensity measured at the higher PMT voltage to the pixel intensity measured at the lower PMT voltage. This will give a ratio for each of the middle intensity features. Next find the median value of these ratios. This is the correction factor (CF).
For only the features that are saturated or nearly saturated in the primary scan, use the correction factor to calculate the adjusted intensity based on the feature's intensity in the secondary scan. The adjusted intensity is the intensity in the secondary scan multiplied by the correction factor. For all other spots, use the values from the primary, higher PMT voltage scan.
Compute the average signal intensity from duplicate spots on a slide.
Log transform (base 2) the signal intensities to help compare signals over a large range.
Alternate Protocol 2
Normalize Slide using Reference Sample (optional)
Determine the median signal intensity of the reference sample over the entire array, excluding any control samples (such as Cy3) that do not bind sample and do not vary due to minor processing differences.
Calculate the ratio of the reference sample's median signal calculated in the preceding step to a standard value (for example, we use a value of 10,000). This ratio is the normalization factor (NF).
Multiply all signal intensities on the slide by the normalization factor. However, if the signals have been log transformed, normalize the data by subtracting the log transform (base 2) of the normalization factor.
For values that were previously set to the floor, reset them to an equivalent floor on the log base 2 scale. Both slides should have the same floor value after normalization.
Calculate the final data value as the average of data from the two slides.
Quality Control
Compute the coefficient of variance for the spots corresponding to each array component. For components with a coefficient of variance greater than 30%, re-inspect the scanned images for smearing, lint, and other abnormalities.
Data for the reference sample(s) should be continually accumulated and evaluated for consistency. Compare data for the reference sample(s) on a given slide to previous data obtained for that reference sample. Significant changes in binding data can help detect a variety of problems, such as sample degradation, storage problems, printing problems, and analysis problems.
REAGENTS AND SOLUTIONS
10x Phosphate-Buffered Saline (PBS)
14.4 g Na2HPO4
2.0 g KCl 2.4 g K2HPO4
Milli-Q water to 1 L
PBST Array Wash Buffer
1X phosphate-buffered saline (PBS), pH=7.4
0.05% Tween-20 (v/v) (Sigma-Aldrich, St. Louis, MO)
Printing Buffer
1X phosphate-buffered saline (PBS), pH = 7.4
2.5% glycerol (v/v)
0.006% Triton-X 100 (Sigma-Aldrich, St. Louis, MO)
COMMENTARY
Background Information
Glycan microarrays are emerging as a powerful tool for studying the interactions between a broad range of glycans and carbohydrate-binding molecules (Liang et al., 2008; Liu et al., 2009; Oyelaran and Gildersleeve, 2009; Song and Pohl, 2009). These arrays are beginning to provide new insights into the many biological processes influenced by glycosylation. So far, the applications of glycan microarrays include characterizing binding properties of monoclonal antibodies and lectins [for some recent examples, see (Blixt et al., 2008a; Manimala et al., 2006; Manimala et al., 2007; Schallus et al., 2008; Song et al., 2009)], defining substrate specificities of glycosyltransferases (Ban and Mrksich, 2008; Park and Shin, 2007), as well as measuring antibody titers against tumors (Wang et al., 2008), pathogens (Blixt et al., 2008b; Kamena et al., 2008; Parthasarathy et al., 2008) and autoantigens (Freedman et al., 2009; Seow et al., 2009). As glycan microarrays become more widely accessible, their application will likely be extended to new problems.
A key issue for the construction of a glycan microarray is obtaining a diverse collection of carbohydrates to populate the array. It has been estimated that there are about 7,000 glycan determinants in the human glycome (Cummings, 2009), but only a tiny fraction of these are readily accessible in homogenous form. At present, even the largest glycan arrays contain only several hundred glycans. The focus of this protocol is on the construction of neoglycoprotein microarrays. The neoglycoprotein approach was chosen for several reasons. First, neoglycoproteins have been used for many years to study carbohydrate-protein interaction (Wong, 1995). Second, a variety of carbohydrate-BSA and HSA conjugates are commercially-available. Many others can be prepared in a single step from readily accessible oligosaccharides (Gildersleeve et al., 2008; Roy et al., 1984). Therefore, moderately sized neoglycoprotein arrays can be produced by many laboratories. Third, neoglycoproteins are multivalent probes that can be used in a variety of other experiments such as ELISAs, Western blots, and as multivalent inhibitors. This is very useful for confirming results observed on the array and for carrying out other follow up experiments. It should be noted that a number of other glycan array formats have been developed (Culf et al., 2006). Examples of other strategies include immobilization of neoglycolipids (Liu et al., 2007), fluorous-tagged oligosaccharides (Pohl, 2008), nucleophile-linked oligosaccharides (Ratner et al., 2004) (Blixt et al., 2004), and non-covalent adsorption of large polysaccharides (Wang et al., 2002). Methods for accessing larger glycan libraries and making protein conjugates and neoglycoconjugates have been reviewed (Seeberger, 2008).
In addition to variations in glycan structure, the array format is well-suited for varying the mode of glycan presentation. Many carbohydrate-binding proteins achieve high avidity interactions via formation of multivalent complexes. Features of presentation such as spacing and orientation of glycans affect the ability to form a multivalent interaction. Microarrays containing neoglycoconjugates can easily vary glycan density on the array. For example, glycan density can be varied by modulating the average number of glycans attached to each molecule of carrier protein (Oyelaran et al., 2009).
Critical Parameters
Methods and parameters used for imaging and processing data can significantly affect results. Small adjustments in the size and position of boundaries used to define array spots can have a substantial effect on results. Parameters for finding spots (such as limits on resizing features) can be easily overlooked details, yet can significantly impact results. Setting limits on the resizing features, such as no less than 70% and no more than 150% of the expected diameter, can improve automatic object recognition. Manual inspection of spot recognition is highly recommended. Although manual adjustment of objects is needed occasionally, it is important to do so with caution and consistency; the same standards for measuring pixel intensity and background should be applied to all components on the array.
Troubleshooting
Printing Problems
Printing problems are the most likely cause of irregular spot morphology or high variability in the data. To help find printing problems early, slides should be viewed under a microscope after printing and prior to blocking. Table 4 summarizes causes and remedies for several common printing problems.
Table 4.
Troubleshooting guide for Glycan Microarray Printing and Processing
| Problem | Possible Cause | Solution |
|---|---|---|
| Missing Spots | a. Pin Sticking | a. Dry Pins and Tool Thoroughly |
| b. Clogged Pin | b. Sonicate Pins | |
| c. Damaged Pin | c. Replace Pin | |
| Irregular Spot | a. Inadequate Prespotting | a. Change Prespotting |
| Morphology / Merged Spots | b. For “donuts”, be sure Triton X 100 is in the print buffer | |
| c. For small spots, increase neoglycoprotein concentration in source plate | ||
| High Background | a. Globulins in BSA or HSA | a. Block only with globulin-free albumin |
| b. Inadequate washes | b. Increase number of washes | |
| c. Poor Blocking | c. Increase concentration of BSA, HSA, or tween during blocking | |
| d. Dirty Slide Module | d. Replace Slide Module | |
| Low or No Signal | a. Inadequate incubation time | a. Increase time or temperature of incubation |
| b. Low concentration of GBP | b. Increase concentration of GBP | |
| c. Photo bleaching | c. Protect fluorescent dyes from light during incubation | |
| d. Protein is inactive or lacks carbohydrate binding activity | ||
| e. Appropriate glycan ligand is not present on the array | ||
| High Variability | a. Batch-to-batch printing variations | a. Analyze groups of samples on arrays from the same printing batch |
The first step in evaluating printing problems is to determine if the problems are associated with a particular pin or sample. If the problems are restricted to a single pin, that pin should be cleaned or replaced. Clogged or damaged pins typically lead to missing spots, although irregular spot morphology can occur. Missing or irregular spots are also indications of sample-specific problems, such as insufficient loading of sample into pins. Small pre-spots and a decrease in spot size as the run progresses can occur when volume of sample in the sample plates is low.
When problems appear more widespread, consider adjusting printing parameters to either increase or decrease the amount of sample deposited with each pin contact. Pre-spotting reduces the amount of sample deposited by the first pin strikes after loading. Increasing the number of pre-spots may eliminate some merged spots. Prolonging the contact time (i.e., increased dwell time) between pins and slide will make spots larger. Also check the concentration of neoglycoprotein and Triton X-100 in the print solution. Intense signal at the periphery of spots (known as “donuts” and shown in panel B of Figure 4) may result when Triton X-100 is not included in the print buffer (Deng et al., 2006).
Deterioration or contamination of the sample plates are another cause of poor quality slides. Many print problems can be corrected by replacing the sample plates. However, this is not always practical when samples must be conserved. Adding water to the samples to replace losses from evaporation and mixing samples by re-pipetting can also improve print quality.
High Background
High background can be caused by insufficient blocking, high sample concentration, or slide contamination. The blocking solution may contain contaminants, such as immunoglobulins, that bind to some array components. Alternatively, the blocking solution may not be effective at blocking. Problems with the blocking solution can be found by analyzing binding of a secondary antibody to a blocked slide that has not been incubated with any sample. If the secondary antibody alone binds to a blocked slide, consider increasing the concentration of blocking solution, changing, or adding another blocking agent. It is important to note that many common blocking agents for biological experiments, such as gelatin, casein, milk, and animal serum, contain glycoproteins and/or carbohydrates which may inhibit binding to glycans on the array.
High background can also be reduced by lowering the concentration of sample, increasing washes, or using a fresh slide module. Contamination of the sample plate (in particular carry-over from insufficiently washed pins) may increase background of samples printed after high-signal samples.
Low or No Signal
An absence of signal can be due to a variety of causes. First, the glycan array may not possess the correct carbohydrate ligand for recognition, or it may not present that ligand in an appropriate context for binding. Second, the carbohydrate binding entity being tested may denature or lose activity during purification, during storage, or as a result of freezing and thawing prior to the assay. Third, the secondary reagent used for detection may be inactive or unable to recognize the sample of interest. Finally, one should keep in mind that not every protein evaluated on a glycan array is capable of binding to carbohydrates.
When a sample shows low or no signal, troubleshooting begins by analyzing a positive control (such as monoclonal antibodies, sera, or lectins known to bind glycans on the array) to rule out technical problems with the array. Problems with fluorophore tags can produce low signal for positive controls. Since fluorophore-labeled reagents fade over time, they require periodic replacement. Adjust concentration of the secondary reagent or switching to a more stable dye such as Cy3 or an Alexa dye if the fluorophore requires frequent replacement.
If positive controls give high signal, low concentration of sample or suboptimal incubation conditions likely causes the sample's low signal. Check that the sample has been properly mixed, and increase the concentration of sample. Also, check that the incubation conditions (especially, temperature, length, and metal and salt concentrations) are appropriate. Some lectins require metals for binding.
Anticipated Results
Characteristics of High Quality Images
Processing and scanning of array samples produces images of multiple, well-defined spots, as shown in Figure 4. Spot intensity correlates with the amount of sample present. Slides should have few, if any, saturated pixels. High quality images have few missing spots and are free of any smears or debris. Spots should be uniformly sized. Merged, irregularly shaped, large, or small spots indicate printing problems. Although spots may differ in intensity, each individual spot should be homogeneous in intensity. Spots with brighter intensity at the periphery (referred to as “donuts”) are also indications of immobilization irregularities.
Expected Variation in Array Data
Variability in glycan microarray data should be carefully monitored to ensure reliable results. The first check on variability is comparing the signal from each of the spots for each array component. The median absolute difference between components final binding value on the two slides should be ~20% in absolute signal, which corresponds to ± ~0.3 on a log base 2 scale.
Another check on variability is to compare array values for the same sample (e.g. a reference sample) analyzed at several different times. It is highly recommended to include a reference sample on each slide and to continually compare newly acquired data for the reference sample to previously obtained data for that sample. The median standard deviation of each array component should be less than or equal to 35%, even across different arrays and different batches of slides. Array components whose signals vary substantially more or drift overtime could indicate printing or storage problems.
Time Considerations
Time Considerations for Printing
Depending on the number of components that are included on the array, printing can be completed in a few hours to 1.5 days. If printing slides will take more than a day, the arrayer should be kept on overnight with the array slides secure in place at room temperature. The vacuum must stay on to keep the slides aligned in position. The sample plates, however, should be promptly returned to the refrigerator or freezer in order to minimize evaporation.
When multiple batches are to be printed one after the other (for example, three batches of 12 slides printed over 5 days), the sample plates should be resealed with aluminum seals and stored at 4 °C between printing runs. This is done to minimize the number of freeze-thaw cycles. More than three print runs is not recommended without replacing water due to evaporation loss.
Time Considerations for Assays
There are no natural pause points when performing binding assays with the arrays. Once the slides have been blocked, it is recommended that samples be processed without interruption. The assays can be performed in a single day. Depending on the number of washes and lengths of incubation periods, the assay generally takes 5-8 hours. If needed, the processed slides can be stored for several hours to days at −20 °C in the dark prior to scanning(Gu et al., 2006).
Supplementary Material
Table 2.
Recommended buffers and starting dilutions of samples for incubation on glycan microarray
| Sample Type | Incubation Buffer | Concentration | Conditions |
|---|---|---|---|
| Lectin | 1% BSA in PBST | 1-50 μg/mL | RT for 2 hrs |
| Monoclonal Antibody | 3% BSA in PBST | 1-50 μg/mL | 37 °C while gently shaken for 2-4 h |
| Serum Antibodies | 3% BSA in PBST | 1:50 to 1:200 | 37 °C while gently shaken for 4 h |
Table 3.
Recommended Secondary Reagents and Conditions
| Sample Type | Secondary Label | Buffer | Concentration | Conditions |
|---|---|---|---|---|
| Biotinylated Lectin | Cy3-Streptavidin | 1% BSA in PBS buffer | 2 μg/mL | RT for 2 hrs |
| Monoclonal Antibody | Monoclonal antibody specific for species and isotype of 1° Ab | 3% BSA in PBS | 2 μg/mL | 37 °C while gently shaken for 2 h |
| Human Serum Antibodies | Monoclonal anti-human IgG, IgM, and/or IgA | 3% HSA + 1% BSA in PBS | 2 μg/mL | 37 °C while gently shaken for 2 h |
Literature Cited
- Ban L, Mrksich M. On-chip synthesis and label-free assays of oligosaccharide arrays. Angew Chem Int Ed Engl. 2008;47:3396–9. doi: 10.1002/anie.200704998. [DOI] [PubMed] [Google Scholar]
- Blixt O, Han S, Liao L, Zeng Y, Hoffmann J, Futakawa S, Paulson JC. Sialoside analogue arrays for rapid identification of high affinity siglec ligands. J Am Chem Soc. 2008a;130:6680–1. doi: 10.1021/ja801052g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blixt O, Head S, Mondala T, Scanlan C, Huflejt ME, Alvarez R, Bryan MC, Fazio F, Calarese D, Stevens J, et al. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc Natl Acad Sci U S A. 2004;101:17033–8. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blixt O, Hoffmann J, Svenson S, Norberg T. Pathogen specific carbohydrate antigen microarrays: a chip for detection of Salmonella O-antigen specific antibodies. Glycoconj J. 2008b;25:27–36. doi: 10.1007/s10719-007-9045-0. [DOI] [PubMed] [Google Scholar]
- Culf AS, Cuperlovic-Culf M, Ouellette RJ. Carbohydrate microarrays: survey of fabrication techniques. OMICS. 2006;10:289–310. doi: 10.1089/omi.2006.10.289. [DOI] [PubMed] [Google Scholar]
- Cummings RD. The repertoire of glycan determinants in the human glycome. Mol Biosyst. 2009;5:1087–104. doi: 10.1039/b907931a. [DOI] [PubMed] [Google Scholar]
- Deng Y, Zhu XY, Kienlen T, Guo A. Transport at the air/water interface is the reason for rings in protein microarrays. J Am Chem Soc. 2006;128:2768–9. doi: 10.1021/ja057669w. [DOI] [PubMed] [Google Scholar]
- Freedman MS, Laks J, Dotan N, Altstock RT, Dukler A, Sindic CJ. Anti-alpha-glucose-based glycan IgM antibodies predict relapse activity in multiple sclerosis after the first neurological event. Mult Scler. 2009;15:422–30. doi: 10.1177/1352458508101944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gildersleeve JC, Oyelaran O, Simpson JT, Allred B. Improved procedure for direct coupling of carbohydrates to proteins via reductive amination. Bioconjug Chem. 2008;19:1485–90. doi: 10.1021/bc800153t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Q, Sivanandam TM, Kim CA. Signal stability of Cy3 and Cy5 on antibody microarrays. Proteome Sci. 2006;4:21. doi: 10.1186/1477-5956-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamena F, Tamborrini M, Liu X, Kwon YU, Thompson F, Pluschke G, Seeberger PH. Synthetic GPI array to study antitoxic malaria response. Nat Chem Biol. 2008;4:238–40. doi: 10.1038/nchembio.75. [DOI] [PubMed] [Google Scholar]
- Liang PH, Wu CY, Greenberg WA, Wong CH. Glycan arrays: biological and medical applications. Curr Opin Chem Biol. 2008;12:86–92. doi: 10.1016/j.cbpa.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Feizi T, Campanero-Rhodes MA, Childs RA, Zhang Y, Mulloy B, Evans PG, Osborn HM, Otto D, Crocker PR, et al. Neoglycolipid probes prepared via oxime ligation for microarray analysis of oligosaccharide-protein interactions. Chem Biol. 2007;14:847–59. doi: 10.1016/j.chembiol.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Liu Y, Palma AS, Feizi T. Carbohydrate microarrays: key developments in glycobiology. Biol Chem. 2009;390:647–56. doi: 10.1515/BC.2009.071. [DOI] [PubMed] [Google Scholar]
- Lyng H, Badiee A, Svendsrud DH, Hovig E, Myklebost O, Stokke T. Profound influence of microarray scanner characteristics on gene expression ratios: analysis and procedure for correction. BMC Genomics. 2004;5:10. doi: 10.1186/1471-2164-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manimala JC, Roach TA, Li Z, Gildersleeve JC. High-throughput carbohydrate microarray analysis of 24 lectins. Angew Chem Int Ed Engl. 2006;45:3607–10. doi: 10.1002/anie.200600591. [DOI] [PubMed] [Google Scholar]
- Manimala JC, Roach TA, Li Z, Gildersleeve JC. High-throughput carbohydrate microarray profiling of 27 antibodies demonstrates widespread specificity problems. Glycobiology. 2007;17:17C–23C. doi: 10.1093/glycob/cwm047. [DOI] [PubMed] [Google Scholar]
- Oyelaran O, Gildersleeve JC. Glycan arrays: recent advances and future challenges. Curr Opin Chem Biol. 2009;13:406–13. doi: 10.1016/j.cbpa.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyelaran O, Li Q, Farnsworth D, Gildersleeve JC. Microarrays with varying carbohydrate density reveal distinct subpopulations of serum antibodies. J Proteome Res. 2009;8:3529–38. doi: 10.1021/pr9002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Shin I. Carbohydrate microarrays for assaying galactosyltransferase activity. Org Lett. 2007;9:1675–8. doi: 10.1021/ol070250l. [DOI] [PubMed] [Google Scholar]
- Parthasarathy N, Saksena R, Kovac P, Deshazer D, Peacock SJ, Wuthiekanun V, Heine HS, Friedlander AM, Cote CK, Welkos SL, et al. Application of carbohydrate microarray technology for the detection of Burkholderia pseudomallei, Bacillus anthracis and Francisella tularensis antibodies. Carbohydr Res. 2008 doi: 10.1016/j.carres.2008.05.021. [DOI] [PubMed] [Google Scholar]
- Pohl NL. Fluorous tags catching on microarrays. Angew Chem Int Ed Engl. 2008;47:3868–70. doi: 10.1002/anie.200704801. [DOI] [PubMed] [Google Scholar]
- Ratner DM, Adams EW, Su J, O'Keefe BR, Mrksich M, Seeberger PH. Probing protein-carbohydrate interactions with microarrays of synthetic oligosaccharides. Chembiochem. 2004;5:379–82. doi: 10.1002/cbic.200300804. [DOI] [PubMed] [Google Scholar]
- Roy R, Katzenellenbogen E, Jennings HJ. Improved procedures for the conjugation of oligosaccharides to protein by reductive amination. Can J Biochem Cell Biol. 1984;62:270–5. doi: 10.1139/o84-037. [DOI] [PubMed] [Google Scholar]
- Schallus T, Jaeckh C, Feher K, Palma AS, Liu Y, Simpson JC, Mackeen M, Stier G, Gibson TJ, Feizi T, et al. Malectin: a novel carbohydrate-binding protein of the endoplasmic reticulum and a candidate player in the early steps of protein N-glycosylation. Mol Biol Cell. 2008;19:3404–14. doi: 10.1091/mbc.E08-04-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeberger PH. Automated oligosaccharide synthesis. Chem Soc Rev. 2008;37:19–28. doi: 10.1039/b511197h. [DOI] [PubMed] [Google Scholar]
- Seow CH, Stempak JM, Xu W, Lan H, Griffiths AM, Greenberg GR, Steinhart AH, Dotan N, Silverberg MS. Novel anti-glycan antibodies related to inflammatory bowel disease diagnosis and phenotype. Am J Gastroenterol. 2009;104:1426–34. doi: 10.1038/ajg.2009.79. [DOI] [PubMed] [Google Scholar]
- Song EH, Pohl NL. Carbohydrate arrays: recent developments in fabrication and detection methods with applications. Curr Opin Chem Biol. 2009;13:626–32. doi: 10.1016/j.cbpa.2009.09.021. [DOI] [PubMed] [Google Scholar]
- Song X, Xia B, Stowell SR, Lasanajak Y, Smith DF, Cummings RD. Novel fluorescent glycan microarray strategy reveals ligands for galectins. Chem Biol. 2009;16:36–47. doi: 10.1016/j.chembiol.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CC, Huang YL, Ren CT, Lin CW, Hung JT, Yu JC, Yu AL, Wu CY, Wong CH. Glycan microarray of Globo H and related structures for quantitative analysis of breast cancer. Proc Natl Acad Sci U S A. 2008;105:11661–6. doi: 10.1073/pnas.0804923105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Liu S, Trummer BJ, Deng C, Wang A. Carbohydrate microarrays for the recognition of cross-reactive molecular markers of microbes and host cells. Nat Biotechnol. 2002;20:275–81. doi: 10.1038/nbt0302-275. [DOI] [PubMed] [Google Scholar]
- Wong SY. Neoglycoconjugates and their applications in glycobiology. Curr Opin Struct Biol. 1995;5:599–604. doi: 10.1016/0959-440x(95)80050-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.