

Abstract
Fas is a transmembrane cell surface protein recognized by Fas ligand (FasL). When FasL binds to Fas, the target cells undergo apoptosis. A soluble Fas molecule that lacks the transmembrane domain is produced from skipping of exon 6 encoding this region in alternative splicing procedure. The soluble Fas molecule has the opposite function of intact Fas molecule, protecting cells from apoptosis. Here we show that knockdown of hnRNP A1 promotes exon 6 skipping of Fas pre-mRNA, whereas overexpression of hnRNP A1 reduces exon 6 skipping. Based on the bioinformatics approach, we have hypothesized that hnRNP A1 functions through interrupting 5′ splice site selection of exon 5 by interacting with its potential binding site close to 5′ splice site of exon 5. Consistent with our hypothesis, we demonstrate that mutations of the hnRNP A1 binding site on exon 5 disrupted the effects of hnRNP A1 on exon 6 inclusion. RNA pull-down assay and then western blot analysis with hnRNP A1 antibody prove that hnRNP A1 contacts the potential binding site RNA sequence on exon 5 but not the mutant sequence. In addition, we show that the mutation of 5′ splice site on exon 5 to a less conserved sequence destructed the effects of hnRNP A1 on exon 6 inclusion. Therefore we conclude that hnRNP A1 interacts with exon 5 to promote distal exon 6 inclusion of Fas pre-mRNA. Our study reveals a novel alternative splicing mechanism of Fas pre-mRNA.
Keywords: Fas, Apoptotic, Anti-apoptotic, Pre-mRNA splicing, hnRNP A1, Exon 6, 5′ splice site
Introduction
The Fas (Apo-I) gene, also designated as CD95, induces apoptosis after interaction with its antibody [1, 2]. Fas is a cell surface protein belonging to the TNF receptor family [1, 3]. The Fas protein consists of a signal sequence, an extracellular domain comprising three cysteine-rich sub-domains characteristic of TNFR superfamily, a transmembrane domain, and an intracellular domain. Exon 6 of Fas pre-mRNA encodes the transmembrane domain [4]. Skipping of Fas exon 6 causes a production of a soluble form, in which the transmembrane domain is missing. This soluble isoform blocks apoptosis induced by Fas antibody (Fig. 1a).
Fig. 1.
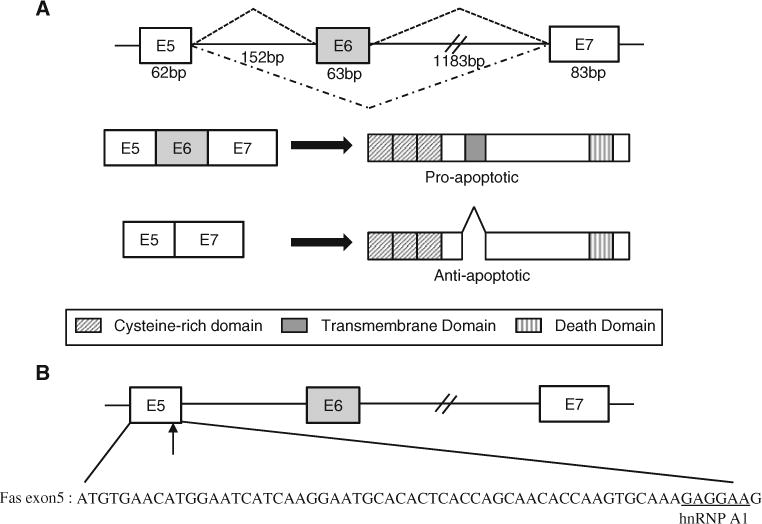
a Alternative splicing of exon 6 produces anti-apoptotic and pro-apoptotic Fas protein. b The sequence of exon 5 is shown. Potential binding sites of hnRNP A1 on Fas exon 5 are underlined. Exons are shown with boxes, introns are shown with lines
Pre-mRNA splicing is one of major regulatory events of gene expression [5–8]. Pre-mRNA splicing requires splicing signals on pre-mRNA that include 5′ splice site, 3′ splice site, branch point and polypyrimidine tract [9]. Pre-mRNA splicing occurs in a large RNA–protein complex called spliceosome [10]. In the process of spliceosome assembly, U1, U2, U4/U5/U6 snRNPs as well as other proteins, including U2AF65 are recruited [11–13]. Chemical reactions of splicing include 5′ splice site cleavage, 3′ splice site cleavage and ligation of two exons [14–16]. Pre-mRNA splicing is positively regulated by serine–arginine rich (SR) proteins [9, 17]. SR proteins target RNA through RNA recognition motif (RRM) domain, whereas RS domain functions as activator [18–21].
Pre-mRNA splicing can be also negatively regulated by heterogeneous nuclear ribonucleoproteins (hnRNPs) [22–24]. hnRNPs inhibit splicing through site-specific binding with the target RNA [25]. hnRNPs recognize RNA through RRM [26]. hnRNPs also contain RGG boxes (repeats of Arg–Gly–Gly tripeptides), additional glycine-rich, acidic or proline-rich domains [13]. The modularity of the hnRNPs ensures structural variation that promotes functional diversity [27]. hnRNP A1 is one of hnRNP family members [28]. Relative concentrations of hnRNP A1 and ASF/SF2 regulate 5′ splice site selection. For example, an excess of hnRNP A1 favors distal 5′ splice site selection [29]. hnRNP A1 blocks spliceosomal assembly through inhibiting the recruitment of snRNPs, and through looping out the entire exons [30, 31]. hnRNP A1 regulates alternative splicing of a number of pre-mRNAs, including survival of motor neuron (SMN2), BRCA1 and its own [32, 33]. In addition to the inhibition of pre-mRNA splicing, hnRNP A1 stimulates pre-mRNA splicing as well as functions in the proofreading procedure of 3′ splice site [24, 34].
The mechanisms of Fas exon 6 splicing are shown only in a few cases. One of the regulators, RBM5 which is involved in 3′ splice site recognition of fas exon 6, inhibits the transition between prespliceosomal complexes to mature spliceosome [35]. Another regulation is that TIA-1 and PTB regulate fas exon 6 splicing through an antagonistic effect [36]. HuR protein also regulates Fas exon 6 splicing through exon definition [37].
Here we show that hnRNP A1 promotes Fas exon 6 inclusion by characterizing its effects using shRNA knockdown and overexpression. We identified exon 5 as the functional target of hnRNP A1 through mutagenesis and RNA–protein binding analysis. We demonstrate that a strong signal of 5′ splice site is required for the function of hnRNP A1 on exon 6 inclusion of Fas pre-mRNA.
Results
Knockdown of hnRNP A1 increases Fas exon 6 skipping
In order to ask if hnRNP A1 regulates alternative splicing of Fas exon 6, we applied a bioinformatics tool—Human Splicing Finder to locate the potential contact sites of hnRNP A1 on Fas pre-mRNA covering exon 5–7. Fas exon 5–7 has provided several possible binding sites for hnRNP A1. One of the most likely binding sites is located at 1 nt upstream from the 5′ splice site of exon 5 (Fig. 1b). Thus we predict that hnRNP A1 regulates alternative splicing of Fas exon 6 through binding to its contact sites. To test this possibility, we carried out several experiments and examined whether the expression level of hnRNP A1 affected alternative splicing of Fas exon 6.
In the first set of experiments, we knocked down hnRNP A1 mRNA using lentivirus based shRNA in cultured cells. RNA and protein were extracted after 3 days of infection with hnRNP A1 shRNA virus. RT-PCR analysis with hnRNP A1 specific primers and western blotting analysis with anti-hnRNP A1 antibody were performed to compare the hnRNP A1 expression level of shRNA treated cells with untreated cells. Both RT-PCR and western blotting results show that the expression of hnRNP A1 is significantly decreased (~95 %) in hnRNP A1 knocked down cells as compared with that in untreated cells and that in cells infected with non-silencing (NS) shRNA virus (Fig. 2a). To answer the question if knockdown of hnRNP A1 regulates alternative splicing of exon 6, we performed RT-PCR analysis in three different cell lines. The primer pair used in PCR was located in exon 5 and exon 7. The first cell line tested is MDA MB 231, a metastatic breast cancer cell line. We found that under normal conditions, only exon 6 included form is detectable whereas exon 6 skipped form is undetectable in MDA MB 231 cells (Fig. 2a, left panel). The results in Fig. 2a show that the knockdown of hnRNP A1 promotes exon 6 skipped forms significantly. The quantitation results show that exon 6 skipped form of total RNA is increased by ~20 % after treatment of hnRNP A1 shRNA (Fig. 2a, right panel). The second cell line we tested is HeLa cells. We have found that only exon 6 included forms appears in HeLa cells as in the MDA MB 231 cell line. As shown in Fig. 2b, hnRNP A1 knockdown induces increase of exon 6 skipped form (~11 %). We next asked if the effect of hnRNP A1 is unique to the cell lines which express only wild type (exon 6 included form) Fas. To answer this question, we tested the effect of hnRNP A1 in HCT 116 cells, that expresses both exon 6 included and skipped isoforms. The results in Fig. 2c show that exon 6 skipped form of total RNA is significantly increased (~ 40 %) whereas the treatment with non-silencing shRNA did not cause the alteration. Therefore we conclude that knockdown of hnRNP A1 promotes the production of exon 6 skipping form.
Fig. 2.
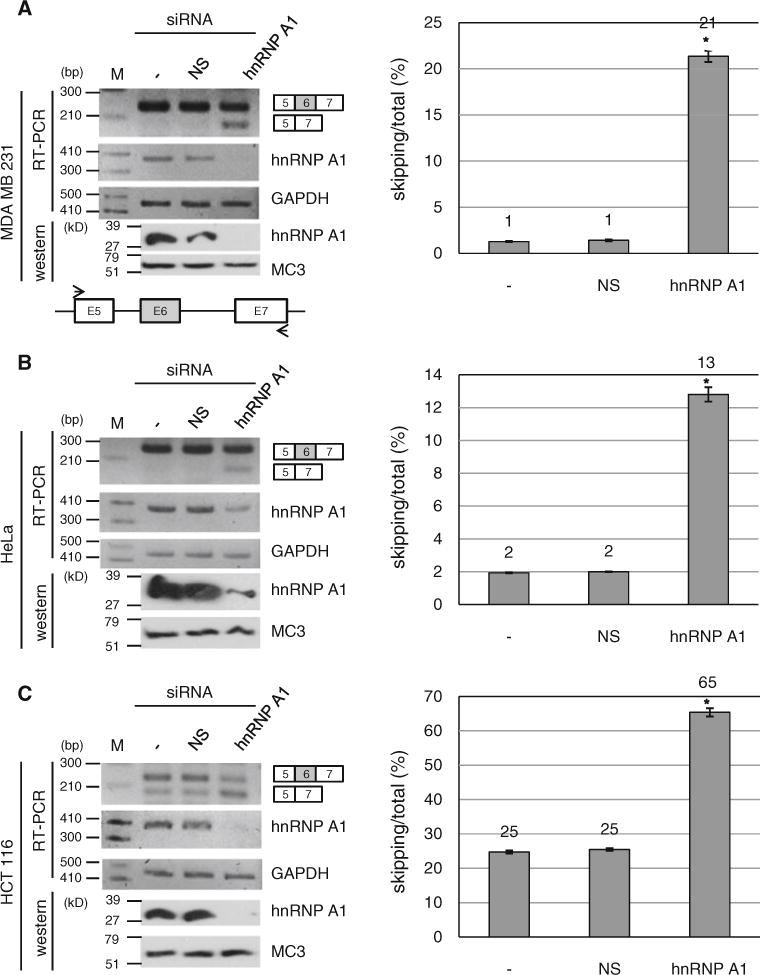
hnRNP A1 knockdown promotes exon 6 skipped form of Fas pre-mRNA. RT-PCR analysis for endogenous Fas exon 6 inclusion/skipping was performed with RNAs from non-treated cells, non-silencing shRNA virus infected cells and hnRNP A1 shRNA virus infected cells. Expression levels of hnRNP A1 are shown with RT-PCR and western analysis. GADPH and MC3 (monoclonal antibody for U2AF65) were used as controls for RT-PCR and western blotting. Primers are basepaired with exon 5 and exon 7 as shown with arrows. The quantitative results for exon 6 skipping of total RNA are shown at the right panel. MDA MB 231 cell line (a) HeLa cell line (b) and HCT 116 cell line (c) are used
Overexpression of hnRNP A1 reduces Fas exon 6 skipped form
In the second set of experiments, we overexpressed hnRNP A1 in the cell lines mentioned above together with a Fas minigene containing exon 5–7. We asked if overexpression of hnRNP A1 has the opposite effects on Fas exon 6 splicing as knockdown of hnRNP A1. The results in Fig. 3a show that hnRNP A1 overexpression induced a decrease of exon 6 skipped form of total RNA by ~ 17 % in MDA MB 231 cell line. However the expression with control plasmid (pcDNA3.1) did not cause the alteration of exon 6 splicing significantly. Similarly, the results in Fig. 3b and c show that overexpression of hnRNP A1 reduced exon 6 skipping of total RNA by ~12 and ~21 % in HeLa cells and HCT 116 cells independently. Therefore we conclude that hnRNP A1 overexpression reduced exon 6 skipped form of Fas pre-mRNA. Taken the results of Figs. 2 and 3 together, we conclude that hnRNP A1 promotes exon 6 inclusion of Fas pre-mRNA.
Fig. 3.
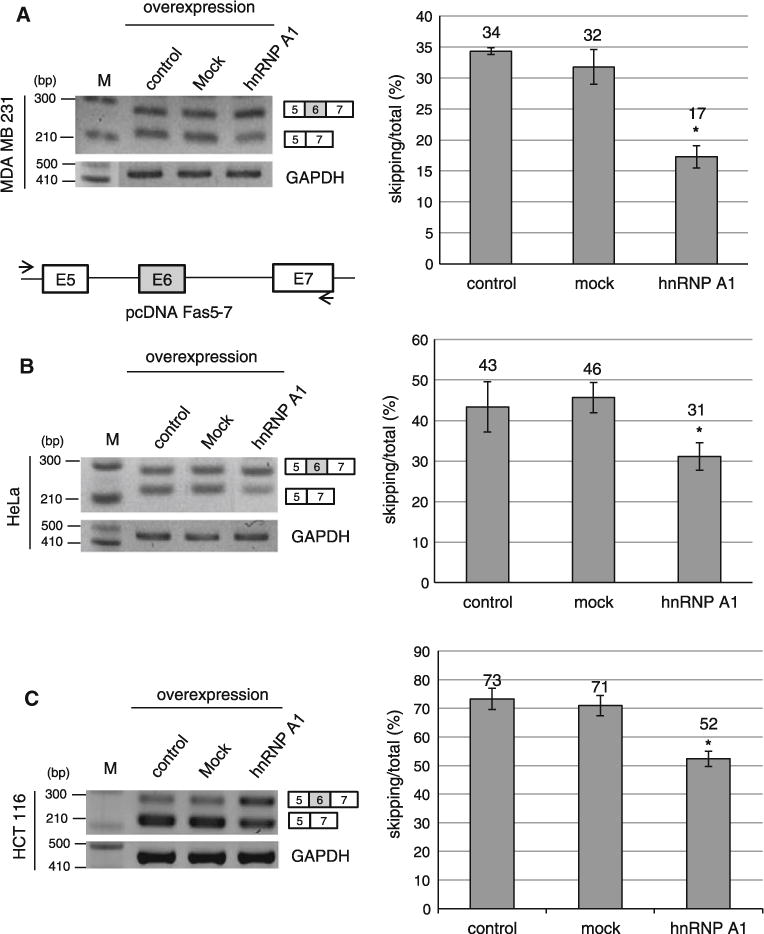
Overexpression of hnRNP A1 reduces exon 6 skipped form of Fas pre-mRNA. RT-PCR analysis of Fas exon 6 alternative splicing was conducted with RNAs extracted from pcDNA3.1 overexpressing, pcDNA-hnRNP A1 overexpressing and non-treated cells. A minigene including exon 5–7 is cloned into pcDNA3.1 plasmid. The primer sets used for RT-PCR are shown as arrows. One primer is basepaired with pcDNA plasmid sequence; the other primer is basepaired with exon 7. The cell lines used are MDA MB 231 cell line (a), HeLa cell line (b) and HCT 116 cell line (c) Quantitation of exon 6 skipping of total RNA is shown in the right panel
hnRNP A1 functions through its highly potential binding site on exon 5
To identify the functional target of hnRNP A1 on Fas pre-mRNA, we started our approach of prediction using Human Splicing Finder. We found that there is a highly potential hnRNP A1 binding site on exon 5 that is located at 1 nt upstream of its 5′ splice site (Fig. 1b). Therefore we hypothesized that hnRNP A1 contacts this site to interfere with the assembly of spliceosome on 5′ splice site of exon 5. Because hnRNP A1 inhibits splicing, we hypothesized that hnRNP A1 makes the 5′ splice site of exon 5 weaker, leading to a relatively stronger 5′ splice site of exon 6. Consequently hnRNP A1 promotes the inclusion of exon 6 because 5′ splice site of exon 6 is selected. To test this hypothesis, we performed mutagenesis analysis on the potential hnRNP A1 binding site on exon 5. As shown in Fig. 4a, the strong hnRNP A1 binding sequence (GAGGAAG) is mutated into CUGGAAG to compromise its binding (hnRNPA1mE5). If our hypothesis is correct, the exon 6 splicing of the mutant minigene would have a similar splicing pattern as that of hnRNP A1 knockdown. In addition, overexpression of hnRNP A1 would not affect exon 6 inclusion of mutant Fas pre-mRNA, as its functional target is disrupted. In consistent with our prediction, exon 6 skipping is significantly increased (~51 %) for the hnRNPA1mE5 minigene (Fig. 4b). Furthermore, overexpression of hnRNP A1 did not affect exon 6 skipping for the mutant minigene. Thus we conclude that hnRNP A1 functions through the potential binding site on exon 5. We next asked if our mutagenesis really disrupted the binding of hnRNP A1 protein. To answer the question, we synthesized biotinylated 10 nt RNA (CAAAGAGGAA), which covers the potential hnRNP A1 binding site on exon 5, as well as the hnRNP A1 binding site mutant 10 nt RNA (CAAACUGGA). We incubated the RNA with HeLa cell extract, and then pulled down the RNA bound proteins with streptavidin beads. We performed immunoblotting analysis with hnRNP A1 antibody for the RNA bound proteins. As shown in Fig. 4c, hnRNP A1 contacted wild type 10 nt RNA sequence. However hnRNP A1 did not contact the mutant 10 nt RNA. Taking the results of Fig. 4b and c together, we conclude that hnRNP A1 promotes exon 6 inclusion by contacting exon 5.
Fig. 4.
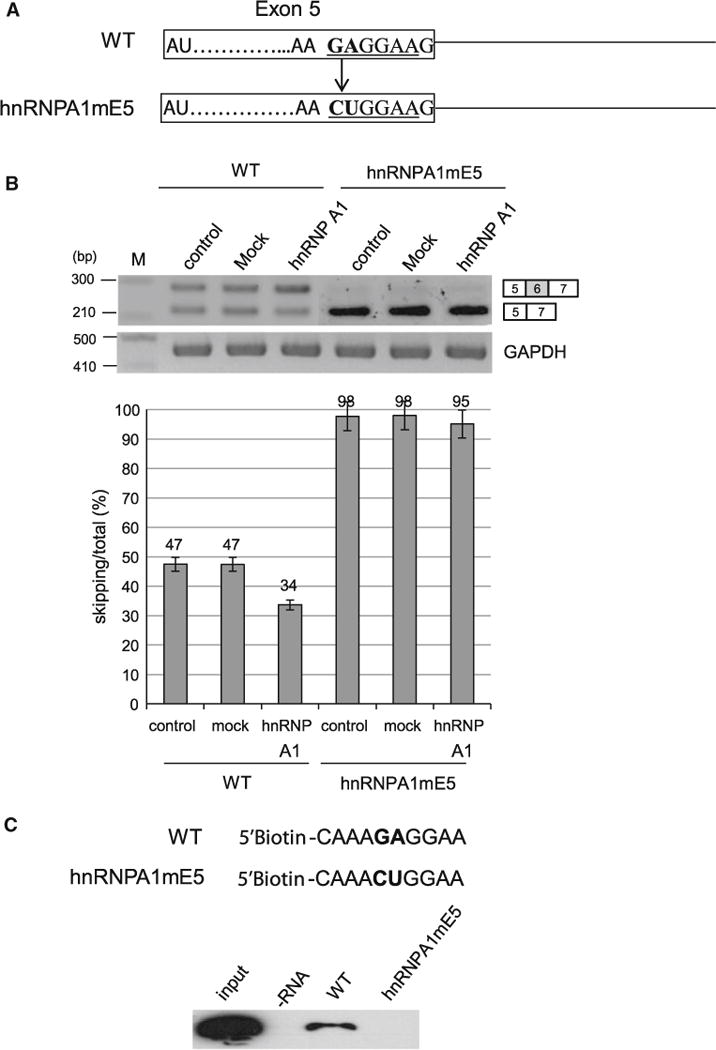
hnRNP A1 functions through its highly potential binding site on exon 5. a The wild type and mutant sequence of potential hnRNP A1 binding site on exon 5. b RT-PCR results of hnRNP A1 mutant minigene with RNAs extracted from overexpressing hnRNP A1, pcDNA3.1 and non-overexpressed cells. c hnRNP A1 analysis with a hnRNP A1 antibody from RNA pulling down assay using 10 nt RNAs of wild type hnRNP A1 binding site on exon 5 and its mutant
hnRNP A1 does not function through the potential binding sites on intron 5, exon 6 and exon 7
We next considered the possibility that hnRNP A1 also functions through other binding sites. We observed that there are also other potential binding sites of hnRNP A1 on Fas pre-mRNA (Fig. 5a). The first one is located at 64 nt upstream from the 3′ splice site of exon 6 (I5). The second one is located at 16 nt downstream from 3′ splice site of exon 6 (E6). The third one is located at 3 nt downstream from the 3′ splice site of exon 7 (E7). We asked if hnRNP A1 also functions through these potential binding sites. To answer the question, we constructed hnRNP A1 binding site mutants on these locations (hnRNPA1mI5, hnRNPA1mE6, hnRNPA1mE7) (Fig. 5a). If hnRNP A1 functions through these potential binding sites, the effects of hnRNP A1 on exon 6 inclusion will be disrupted. We transfected the mutant minigenes alone with hnRNP A1 expressing plasmid into HeLa cells, extracted RNA and then performed RT-PCR analysis. The results in Fig. 5b show that hnRNP A1 promoted exon 6 inclusion significantly for the hnRNPA1mI5 mutant minigene (~12 %) as well as for the hnRNPA1mE6 and hnRNPA1mE7 mutant minigenes (~12 % and ~10 % independently) (Fig. 5b). Therefore we conclude that hnRNP A1 does not function through the potential binding sites on intron 5, exon 6 or exon 7.
Fig. 5.
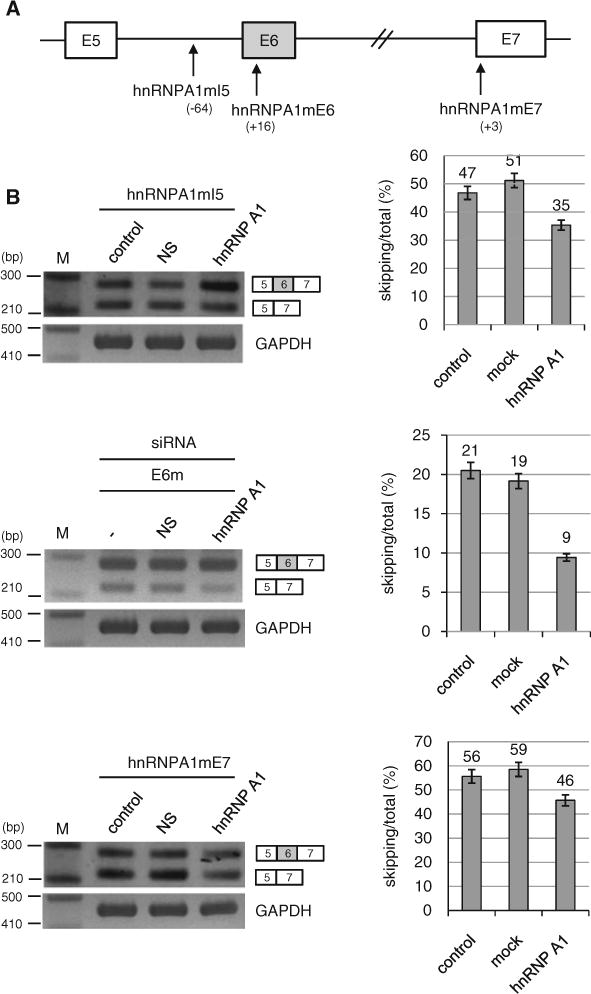
hnRNP A1 does not function through the potential binding sites on intron 5, exon 6 or exon 7. a The locations of potential hnRNP A1 binding sites are shown. b RT-PCR analysis of hnRNP A1 mutant minigenes (hnRNPA1mI5, hnRNPA1mE6 and hnRNPA1mE7) with RNAs extracted from cells overexpressing hnRNP A1 or pcDNA3.1 and cells without treatments. Quantitation results are shown on the right panels
The strength of 5′ splice site on exon 5 is engaged in the function of hnRNP A1 on the splicing of Fas exon 6
Our results showed that hnRNP A1 contacts exon 5 to promote exon 6 inclusion. Because the binding site of hnRNP A1 is close to the 5′ splice site of exon 5, we considered the possibility that hnRNP A1 inhibits splicing at 5′ splice site of nearby exon (exon 5) and then subsequently promotes splicing at 5′ splice site of distant exon (exon 6). If the hypothesis is correct, the strength of 5′ splice site on exon 5 would affect the effects of hnRNP A1 on exon 6 splicing. To test this hypothesis, we mutated the 5′ splice site into a weaker splicing signal (Fig. 6a). As expected, the results in Fig. 6b show that hnRNP A1 did not affect exon 6 splicing of the mutant Fas minigene. Therefore we conclude that the strength of 5′ splice site on exon 5 is engaged in the function of hnRNP A1 on exon 6 inclusion. On the other hand, to investigate whether the exon 6 sequence and the strength of flanking splicing sites of exon 6 also play roles on exon 6 inclusion/skipping, we generated substitution mutations of exon 6 and mutations with stronger flanking splicing sites. However, to this end, our results are inconclusive (Supplementary Figure 1) and more detailed studies are needed in the future to determine the roles of exon 6 sequences and other splicing sites in the skipping/inclusion of exon 6.
Fig. 6.
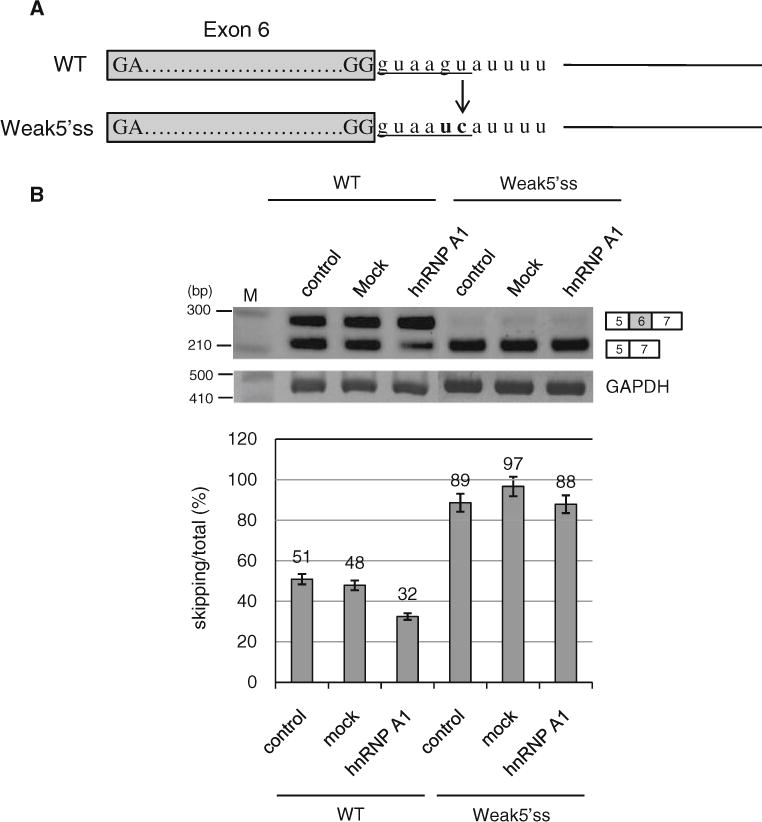
The strength of 5′ splice site on exon 5 is engaged in the hnRNP A1 function on the splicing of Fas exon 6. a The strong wild type and weaker mutant spicing signal sequences of 5′ splice site of exon 5. b RT-PCR results of the mutant minigene with a weaker 5′ splice site of exon 5. RNAs extracted from cells overexpressing hnRNP A1 or pcDNA3.1 and cells without treatments were analyzed
Discussion
In this study, we performed systematic shRNA knockdown and overexpression experiments to determine the role of hnRNP A1 in Fas alternative splicing. We found that the treatment of cells with hnRNP A1 targeting shRNA promotes the exon 6 skipping of Fas pre-mRNA. The results were consistent in all three kinds of cell lines—MDA MB 231, HeLa and HCT116 we tested. By contrast, hnRNP A1 overexpression reduced exon 6 skipping of Fas pre-mRNA. Thus we conclude that hnRNP A1 promotes Fas exon 6 inclusion. To identify the target of hnRNP A1, we destructed the potential hnRNP A1 binding site located at exon 5. As we expected, the destruction of hnRNP A1 binding on exon 6 disrupted the effects of hnRNP A1 on exon 6 inclusion. In addition, we found that other potential binding sites of hnRNP A1 on intron 5, exon 6 and 7 are not functional targets of hnRNP A1. Based on the facts that hnRNP A1 targets exon 5 to promote exon 6 inclusion, and that the functional target of hnRNP A1 is located close to the 5′ splice site, we tested the possibility that the strength of 5′ splice site on exon 5 is engaged in the function of hnRNP A1. As expected the mutation of 5′ splice site on exon 5 into a weaker splicing signal disrupted the effects of hnRNP A1 on exon 6 inclusion. Collectively our results indicate that hnRNP A1 contacts exon 5 to promote Fas exon 6 inclusion. Furthermore the strength of 5′ splice site on exon 5 is engaged in the function of hnRNP A1.
hnRNP A1 promotes the exon 6 inclusion of Fas premRNA
The bioinformatics analysis indicated that hnRNP A1 has the potential to regulate splicing of Fas exon 6. RT-PCR analysis showed that exon 6 skipped isoform is increased significantly after infection of the cells with hnRNP A1 shRNA. The results are confirmed in MDA MB 231, HeLa and HCT 116 cell lines. The effect of hnRNP A1 on Fas alternative splicing was further confirmed by overexpression experiments. In contrast to the effects of hnRNP A1 shRNA treatment, hnRNP A1 overexpression promoted exon 6 inclusion. Through mutagenesis analysis we found that hnRNP A1 functions through exon 5 but does not function through the potential binding sites on intron 5, exon 6 and exon 7. Therefore we conclude that hnRNP A1 promotes Fas exon 6 inclusion. It is well known that hnRNP A1 inhibits exon inclusion [32, 33]. However it was also shown that excess hnRNP A1 promotes distal 5′ splice sites [29]. In addition, hnRNP A1 has been shown to promote splicing [24]. Our results are consistent with previous results that excess hnRNP A1 promotes splicing of distant exon inclusion.
A highly potential binding site of hnRNP A1 is a real functional target
Based on the bioinformatics analysis, we predicted that a strong potential binding site of hnRNP A1 with a high binding score, which is located at exon 5, possibly functions as a target of hnRNP A1. To test this hypothesis, we disrupted the binding potency by mutagenesis. As we expected, the hnRNP A1 binding site mutation destructed the effects of hnRNP A1 on exon 6 splicing. In addition to the strong potential hnRNP A1 binding site on exon 5, there are other potential hnRNP A1 binding sites on intron 5, exon 6 and exon 7 with lower scores. Our results indicate that hnRNP A1 does not target these locations. Thus we conclude that the highly potential binding site of hnRNP A1 at exon 5 was the real functional target of hnRNP A1.
Targeting exon 6 splicing as a treatment of tumors
Alternative splicing has become a target for treatment of numerous diseases. For instance, in the SMN gene, anti-sense oligonucleotides that target a splicing inhibitor increase the inclusion of exon 7, leading to more production of functional SMN protein. Consequently, mice with spinal muscular atrophy (SMA) disease have significantly improved. In this study, we demonstrated that overexpression of hnRNP A1 increases the pro-apoptotic form of Fas that includes exon 6. In addition, we identified an hnRNP A1 binding site in exon 5 that is engaged in the hnRNP A1 effects on exon 6 skipping/inclusion. We deduce that a strategy may be developed to treat tumors that have elevated expression of Fas short form by targeting the exon 5 hnRNP A1 binding site or by manipulating hnRNP A1 expression.
Methods
Cell culture
293T cells and HeLa cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10 % fetal bovine serum (FBS), 2 mM glutamine, 100 IU/ml penicillin and 100 μg/ml streptomycin. HCT116 and MDA MB 231 cells were maintained in RPMI medium supplemented with the same components as for 293T or HeLa cells. All these cells were incubated at 37 °C in a humidified 5 % CO2 condition.
shRNA knockdown
293T cells were transfected with psPAX2 (the packaging vector), pMD2.G (the envelope vector) and shRNA plasmid (Open Biosystems) DNA with 0.67 μg/ml polyethyleneimide (PEI) (Sigma) reagent. After 12 h, media was changed. After 24 h, the viral particle containing supernatant was filtered through a 0.45 μm filter. MDA MB 231, HeLa and HCT116 cells were infected with either non-silencing shRNA or hnRNP A1 shRNA containing viral particles with the addition of 5 mg/ml polybrene (Sigma). After 72 h, total RNAs were extracted.
Transfection
Transfection of Fas minigene or Fas mutant minigenes was carried out in MDA MB 231, HeLa and HCT116 cells using PEI. 2 μg of plasmid was mixed with two times of PEI in 100 μl media. The mixtures were moved to cells in 900 μl of media supplemented with FBS. 4 h later, media was replaced. Following 48 h, total RNAs were extracted from the cells.
Reverse transcription polymerase chain reaction (RT-PCR)
Total RNAs were extracted from MDA MB 231, HeLa and HCT116 cells using RiboEX reagent (GeneAll) according to the manufacturer’s protocol. 1 μg of total RNA was reverse transcribed using oligo-dT18 primer and ImProm-II™ reverse transcriptase (Promega) following the manufacturer’s instruction. 1 μl of reverse transcribed cDNA was used for PCR amplification. Primer sequences for endogenous Fas exon 5–7 are as follows: forward primer 5′-ccc aag ctt ggg atg tga aca tgg aat cat caa gg-3′ and reverse primer 5′-ccg gaa ttc cgg agg att taa ggt tgg aga ttc atg-3′. Primers for detection of minigene of Fas exon 5–7 are as follows: forward primer 5′-taa tac gac tca cta tag gg-3′ and reverse primer is the same as above. Primers for detection of hnRNP A1 gene are as follows: forward primer 5′- ACA TAT GCC ACT GTG GAG GAG-3′ and reverse primer 5′-CTT GCT TTG ACA GGG CTT TTC-3′. Primers for detection of GAPDH gene are as follows: forward primer 5′-ACC ACA GTC CAT GCC ATC AC-3′ and reverse primer 5′-TCC ACC ACC CTG TTG CTG TA-3′.
Immunoblotting
Cells were lysed in lysis buffer (0.1 % triton X-100, 50 mM Tris–Cl pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 mM beta-mercaptoethanol) and incubated for 1 h at 4 °C. The lysed cell mixture was spun down at 12,000 rpm for 30 min at 4 °C. The supernatants were gathered and stored at −80 °C. The proteins were separated on 12 % SDS–PAGE gels and transferred to a nitrocellulose membrane. The membranes were blocked using 5 % skim milk in TBST and incubated with the following antibodies: anti-hnRNP A1 (Santa Cruz biotechnology) and anti-U2AF65 (MC3), and then HRP conjugated anti-mouse secondary antibody.
RNA-hnRNP A1 binding assay
Wild type (CAAAGAGGAA) and mutant (CAAACUGG AA) of 5′ biotin tagged 10 nt RNA was chemically synthesized (Bioneer). Biotinylated RNA was covalently linked with Streptavidin agarose conjugate (Millipore) by mixing together for 1 h in buffer D (20 mM Tris–Cl, 100 mM KCl, 2 mM EDTA, 20 % glycerol, 0.5 mM DTT, 0.5 mM PMSF, pH 7.5). Streptavidin agarose linked biotin-RNA was incubated with HeLa nuclear extract for 3.5 h at 4 °C in buffer D. After washing with buffer for five times, we added SDS–PAGE loading dye and then boiled the sample. Supernatants were analyzed by immunoblotting with hnRNP A1 antibody (Santa Cruz biotechnology).
In vitro mutagenesis
The Fas sequence was amplified using human genomic DNA as a template to construct a wild type minigene. A minigene containing sequence from exon 5–7 of Fas was produced using a primer set FasforP (E5-Hind) and FasrevP (E7-EcoR) that are described in Table. 1. PCR products were cloned into pcDNA3.1(+) vector. To generate hnRNP A1 binding site mutant and weak 5′ splice site mutant of exon 5, we performed overlapping PCR. Each construct was produced by PCR with the following primers: hnRNP A1 binding site mutant primers (forward: A1-E5mut F and reverse: A1-E5mut R; forward: A1-I5mut F and reverse: A1-I5mut R; forward: A1-E6mut F and reverse: A1-E6mut R; forward: A1-E7mut F and reverse: A1-E7mut R), weaker 5′ splice site mutant primers (forward: E6mut5′ss F and reverse: E6mut5′ss R), splice site mutant primers (forward: I53′-I6 F and reverse: I53′-I6 R; forward: I63′-I5 F and reverse: I63′-I5 R). The products were cloned into pcDNA3.1(+) vector using a Fas primer set (forward: FasforP (E5-Hind) and reverse: FasrevP (E7-EcoR)).
Table 1.
Primer list
| Name | Sequence |
|---|---|
| FasforP (E5-Hind) | 5′-cccaagcttgggatgtgaacatggaatcatcaagg-3′ |
| FasrevP (E7-EcoR) | 5′-ccggaattccggaggatttaaggttggagattcatg-3′ |
| A1-E5mut F | 5′-GTGCAAAGAGGAAGgtaatt-3′ |
| A1-E5mut R | 5′-aattacCTTCCTCTTTGCAC-3′ |
| A1-I5mut F | 5′-cttgattacctacctgtcctttat-3′ |
| A1-I5mut R | 5′- ataaaggacaggtaggtaatcaag-3′ |
| A1-E6mut F | 5′- GATCTAACTCTACCTGGCTTTGTC-3′ |
| A1-E6mut R | 5′ GACAAAGCCAGGTAGAGTTAGATC-3′ |
| A1-E7mut F | 5′-ctttcagTGctacccAAGGAAGTAC-3′ |
| A1-E7mut R | 5′-GTACTTCCTTgggtagCActgaaag-3′ |
| E6mut5′ss F | 5′-TGTTTGGGgtaatctcttgc-3′ |
| E6mut5′ss R | 5′-gcaagagattacCCCAAACA-3′ |
| I53′-I6 F | 5′-cctttatttaatcttaaagcaaggctgagacctgagttgataaaatttctttgttctttcagGATCCAGATCTAAC-3′ |
| I53′-I6 R | 5′-GTTAGATCTGGATCCTGAAAGAACAAAGAAATTTTATCAACTCAGGTCTCAGCCTTGCTTTAAGATTAAATAAAGG-3′ |
| I63′-I5 F | 5′-ctcacatgcattctaattgcttattttcatataaaatgtccaatgttccaacctacagTGAAGAGAAAGG-3′ |
| I63′-I5 R | 5′-CCTTTCTCTTCACTGTAGGTTGGAACATTGGACATTTTATATGAAAATAAGCAATTAGAATGCATGTGAG-3′ |
Supplementary Material
Acknowledgments
We thank Juan Valcarcel for providing the mutant fas minigene plasmids (e1, E23, 6-6, 5-5). This work was supported by Mid-career Researcher Program through a National Research Foundation (NRF) grant (2011-0000188 and 20120005340) funded by the Ministry of Education, Science, and Technology (MEST), Korea; and a Systems Biology Infrastructure Establishment grant provided by Gwangju Institute of Science and Technology (GIST) in 2012.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10495-013-0824-8) contains supplementary material, which is available to authorized users.
Contributor Information
Hyun kyung Oh, School of Life Sciences, Gwangju Institute of Science and Technology, Gwangju 500-712, South Korea.
Eunkyung Lee, School of Life Sciences, Gwangju Institute of Science and Technology, Gwangju 500-712, South Korea.
Ha Na Jang, School of Life Sciences, Gwangju Institute of Science and Technology, Gwangju 500-712, South Korea.
Jaehoon Lee, School of Life Sciences, Gwangju Institute of Science and Technology, Gwangju 500-712, South Korea.
Heegyum Moon, School of Life Sciences, Gwangju Institute of Science and Technology, Gwangju 500-712, South Korea.
Zhi Sheng, Virginia Tech Carilion Research Institute and the Department of Biomedical Sciences and Pathobiology, College of Veterinary Medicine, Virginia Tech, Blacksburg, VA, USA.
Youngsoo Jun, School of Life Sciences, Gwangju Institute of Science and Technology, Gwangju 500-712, South Korea.
Tiing Jen Loh, School of Life Sciences, Gwangju Institute of Science and Technology, Gwangju 500-712, South Korea.
Sunghee Cho, School of Life Sciences, Gwangju Institute of Science and Technology, Gwangju 500-712, South Korea.
Jianhua Zhou, Nantong University, Nantong, China; iBioinfo Group, Lexington, MA, USA.
Michael R. Green, Howard Hughes Medical Institute and Programs in Gene Function and Expression and Molecular Medicine, University of Massachusetts Medical School, Worcester, MA 01605, USA
Xuexiu Zheng, School of Life Sciences, Gwangju Institute of Science and Technology, Gwangju 500-712, South Korea.
Haihong Shen, Email: haihongshen@gist.ac.kr, School of Life Sciences, Gwangju Institute of Science and Technology, Gwangju 500-712, South Korea.
References
- 1.Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, et al. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991;66(2):233–243. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- 2.Trauth BC, Klas C, Peters AM, Matzku S, Moller P, Falk W, et al. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science. 1989;245(4915):301–305. doi: 10.1126/science.2787530. [DOI] [PubMed] [Google Scholar]
- 3.Oehm A, Behrmann I, Falk W, Pawlita M, Maier G, Klas C, et al. Purification and molecular cloning of the APO-1 cell surface antigen, a member of the tumor necrosis factor/nerve growth factor receptor superfamily. Sequence identity with the Fas antigen. J Biol Chem. 1992;267(15):10709–10715. [PubMed] [Google Scholar]
- 4.Lee SH, Kim SY, Lee JY, Shin MS, Dong SM, Na EY, et al. Detection of soluble Fas mRNA using in situ reverse transcription-polymerase chain reaction. Lab Invest. 1998;78(4):453–459. [PubMed] [Google Scholar]
- 5.Wahl MC, Will CL, Luhrmann R. The spliceosome: Design principles of a dynamic RNP machine. Cell. 2009;136(4):701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 6.Black DL. Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell. 2000;103(3):367–370. doi: 10.1016/s0092-8674(00)00128-8. [DOI] [PubMed] [Google Scholar]
- 7.Graveley BR. Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001;17(2):100–107. doi: 10.1016/s0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- 8.Modrek B, Resch A, Grasso C, Lee C. Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res. 2001;29(13):2850–2859. doi: 10.1093/nar/29.13.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 10.Matlin AJ, Clark F, Smith CW. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005;6(5):386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- 11.Jurica MS, Moore MJ. Pre-mRNA splicing: awash in a sea of proteins. Mol Cell. 2003;12(1):5–14. doi: 10.1016/s1097-2765(03)00270-3. [DOI] [PubMed] [Google Scholar]
- 12.Reed R. Initial splice-site recognition and pairing during pre-mRNA splicing. Curr Opin Genet Dev. 1996;6(2):215–220. doi: 10.1016/s0959-437x(96)80053-0. [DOI] [PubMed] [Google Scholar]
- 13.Dreyfuss G, Matunis MJ, Pinol-Roma S, Burd CG. hnRNP proteins and the biogenesis of mRNA. Annu Rev Biochem. 1993;62:289–321. doi: 10.1146/annurev.bi.62.070193.001445. [DOI] [PubMed] [Google Scholar]
- 14.Hertel KJ. Combinatorial control of exon recognition. J Biol Chem. 2008;283(3):1211–1215. doi: 10.1074/jbc.R700035200. [DOI] [PubMed] [Google Scholar]
- 15.Senapathy P, Shapiro MB, Harris NL. Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods Enzymol. 1990;183:252–278. doi: 10.1016/0076-6879(90)83018-5. [DOI] [PubMed] [Google Scholar]
- 16.Zhang XH, Chasin LA. Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev. 2004;18(11):1241–1250. doi: 10.1101/gad.1195304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graveley BR. Sorting out the complexity of SR protein functions. RNA. 2000;6(9):1197–1211. doi: 10.1017/s1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manley JL, Krainer AR. A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins) Genes Dev. 2010;24(11):1073–1074. doi: 10.1101/gad.1934910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blencowe BJ, Bowman JA, McCracken S, Rosonina E. SR-related proteins and the processing of messenger RNA precursors. Biochem Cell Biol. 1999;77(4):277–291. [PubMed] [Google Scholar]
- 20.Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J. 2009;417(1):15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 21.Fu XD. The superfamily of arginine/serine-rich splicing factors. RNA. 1995;1(7):663–680. [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu J, Mayeda A, Krainer AR. Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol Cell. 2001;8(6):1351–1361. doi: 10.1016/s1097-2765(01)00409-9. [DOI] [PubMed] [Google Scholar]
- 23.Tange TO, Damgaard CK, Guth S, Valcarcel J, Kjems J. The hnRNP A1 protein regulates HIV-1 tat splicing via a novel intron silencer element. EMBO J. 2001;20(20):5748–5758. doi: 10.1093/emboj/20.20.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Contreras R, Fisette JF, Nasim FU, Madden R, Cordeau M, Chabot B. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol. 2006;4(2):e21. doi: 10.1371/journal.pbio.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Busch A, Hertel KJ. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip Rev RNA. 2012;3(1):1–12. doi: 10.1002/wrna.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoffman DW, Query CC, Golden BL, White SW, Keene JD. RNA-binding domain of the A protein component of the U1 small nuclear ribonucleoprotein analyzed by NMR spectroscopy is structurally similar to ribosomal proteins. Proc Natl Acad Sci USA. 1991;88(6):2495–2499. doi: 10.1073/pnas.88.6.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beyer AL, Christensen ME, Walker BW, LeStourgeon WM. Identification and characterization of the packaging proteins of core 40S hnRNP particles. Cell. 1977;11(1):127–138. doi: 10.1016/0092-8674(77)90323-3. [DOI] [PubMed] [Google Scholar]
- 28.Han SP, Tang YH, Smith R. Functional diversity of the hnRNPs: past, present and perspectives. Biochem J. 2010;430(3):379–392. doi: 10.1042/BJ20100396. [DOI] [PubMed] [Google Scholar]
- 29.Mayeda A, Krainer AR. Regulation of alternative pre-mRNA splicing by hnRNP A1 and splicing factor SF2. Cell. 1992;68(2):365–375. doi: 10.1016/0092-8674(92)90477-t. [DOI] [PubMed] [Google Scholar]
- 30.Mayeda A, Munroe SH, Caceres JF, Krainer AR. Function of conserved domains of hnRNP A1 and other hnRNP A/B proteins. EMBO J. 1994;13(22):5483–5495. doi: 10.1002/j.1460-2075.1994.tb06883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez-Contreras R, Cloutier P, Shkreta L, Fisette JF, Revil T, Chabot B. hnRNP proteins and splicing control. Adv Exp Med Biol. 2007;623:123–147. doi: 10.1007/978-0-387-77374-2_8. [DOI] [PubMed] [Google Scholar]
- 32.Cho S, Moon H, Yang X, Zhou J, Kim HR, Shin MG, et al. Validation of trans-acting elements that promote exon 7 skipping of SMN2 in SMN2-GFP stable cell line. Biochem Biophys Res Commun. 2012;423(3):531–535. doi: 10.1016/j.bbrc.2012.05.161. [DOI] [PubMed] [Google Scholar]
- 33.Goina E, Skoko N, Pagani F. Binding of DAZAP1 and hnRNPA1/A2 to an exonic splicing silencer in a natural BRCA1 exon 18 mutant. Mol Cell Biol. 2008;28(11):3850–3860. doi: 10.1128/MCB.02253-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tavanez JP, Madl T, Kooshapur H, Sattler M, Valcarcel J. hnRNP A1 proofreads 3′ splice site recognition by U2AF. Mol Cell. 2012;45(3):314–329. doi: 10.1016/j.molcel.2011.11.033. [DOI] [PubMed] [Google Scholar]
- 35.Bonnal S, Martinez C, Forch P, Bachi A, Wilm M, Valcarcel J. RBM5/Luca-15/H37 regulates Fas alternative splice site pairing after exon definition. Mol Cell. 2008;32(1):81–95. doi: 10.1016/j.molcel.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 36.Izquierdo JM, Majos N, Bonnal S, Martinez C, Castelo R, Guigo R, et al. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell. 2005;19(4):475–484. doi: 10.1016/j.molcel.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 37.Izquierdo JM. Cell-specific regulation of Fas exon 6 splicing mediated by Hu antigen R. Biochem Biophys Res Commun. 2010;402(2):324–328. doi: 10.1016/j.bbrc.2010.10.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.