

Abstract
Attention-deficit hyperactive disorder (ADHD) is the most commonly studied and diagnosed psychiatric disorder in children. Methylphenidate (MPH, e.g., Ritalin) has been used to treat ADHD for over 50 years. It is the most commonly prescribed treatment for ADHD, and in the past decade it was the drug most commonly prescribed to teenagers. In addition, MPH has become one of the most widely abused drugs on college campuses. In this study, we examined the effects of MPH on hippocampal synaptic plasticity, which serves as a measurable quantification of memory mechanisms. Field potentials were recorded with permanently implanted electrodes in freely-moving mice to quantify MPH modulation of perforant path synaptic transmission onto granule cells of the dentate gyrus. Our hypothesis was that MPH affects hippocampal synaptic plasticity underlying learning because MPH boosts catecholamine signaling by blocking the dopamine and norepinephrine transporters (DAT and NET respectively). In vitro hippocampal slice experiments indicated MPH enhances perforant path plasticity, and this MPH enhancement arose from action via D1-type dopamine receptors and β-type adrenergic receptors. Similarly, MPH boosted in vivo initiation of long-term potentiation (LTP). While there was an effect via both dopamine and adrenergic receptors in vivo, LTP induction was more dependent on the MPH-induced action via D1-type dopamine receptors. Under biologically reasonable experimental conditions, MPH enhances hippocampal synaptic plasticity via catecholamine receptors.
Keywords: dentate gyrus, perforant path, memory, reward, long-term potentiation, LTP
INTRODUCTION
Attention deficit/hyperactivity disorder (ADHD) is a behavioral disorder characterized by inattention, hyperactivity, and impulsivity. Although newer drugs have been developed, methylphenidate (MPH) is one of the most highly prescribed drugs for the treatment of ADHD (Chai et al., 2012). MPH dose-dependently increases extracellular dopamine (DA) and norepinephrine (NE) indirectly by blocking the transporters, DAT and NET (Biederman and Spencer, 1999; Kuczenski and Segal, 1997; Kuczenski and Segal, 2002). Functionally, MPH is remarkably similar to cocaine in both DAT blockade and subjective “high” when administered by the same route (Stoops et al., 2003; Volkow et al., 1995; Volkow and Swanson, 2003). Human studies using PET imaging show that intravenous MPH increases DA neurotransmission in the striatum (Volkow et al., 2004), which is an earmark of addictive drugs (Di Chiara, 2002).
MPH increases hippocampal NE and DA in vivo (Kuczenski and Segal, 2002; Weikop et al., 2007), both of which are known to affect synaptic plasticity such as long-term depression (LTD) and long-term potentiation (LTP) (Hopkins and Johnston, 1984; Hyman et al., 2006; Izumi et al., 1992; Jones and Bonci, 2005; Kauer, 2004; Lisman and Grace, 2005; Thomas et al., 1996). As well as influencing hippocampal plasticity (Kulla and Manahan-Vaughan, 2000; Sajikumar and Frey, 2004; Tang and Dani, 2009), DA neurotransmission also influences hippocampal-related function (Rossato et al., 2009). These results are consistent with evidence indicating that addictive drugs act upon synaptic plasticity mechanisms that normally underlie learning and memory (Dani and Harris, 2005; Hyman et al., 2006; Jay, 2003; Jones and Bonci, 2005; Kauer, 2004; Kelley, 2004; Ungless et al., 2004; Winder et al., 2002).
One of the most important pathways for the formation of associative memory is the perforant path, which originates in the entorhinal cortex and transmits convergent information from the neocortex to the hippocampus (Deadwyler et al., 1979; Lavenex and Amaral, 2000). The accumulated evidence supports that synaptic plasticity along the perforant path is a substrate for memory (Lynch, 2004; Martinez and Derrick, 1996; McHugh et al., 2008; Rumpel et al., 2005), and the medial perforant path in particular carries place and spatial information (Hargreaves et al., 2005) that is important for drug associated memory. Both DA and NE release in the hippocampus enhance LTP and learning, which establishes a functional link between memory systems and the catecholamine-releasing “reward” centers (Lisman and Grace, 2005; Moore and Bloom, 1979). Recent work suggests that these two catecholamine systems may be more interactive in the hippocampus than previously anticipated (Agnati et al., 1995; Borgkvist et al., 2012; Smith and Greene, 2012). In this study, we examined MPH’s influence over perforant path synaptic plasticity owing to signaling via DA or NE receptors.
MATERIALS AND METHODS
Experimental Procedures
All animal experiments were been carried out in accordance with the NIH guide for the care and use of laboratory animals and according to protocols submitted to the Institutional Animal Care and Use Committee at Baylor College of Medicine. All efforts were made to minimize animal suffering, to reduce the number of animals used, and to use alternatives to in vivo techniques when available.
Slice Preparation and Perforated Patch Clamp Recording
Hippocampal slices containing the dentate gyrus were prepared as previously described (Zhang et al., 2010). Briefly, C57BL/6 mice of either sex (24–30 day old) were anesthetized via injection of a mixture of ketamine (42.8 mg/ml), xylazine (8.6 mg/ml), and acepromazine (1.4 mg/ml). When the animal was deeply anesthetized, it was decapitated. Then the brain was rapidly removed and sliced with a vibratome (Leica VT 1000S). Four horizontal brain slices (270 μm thick) were first recovered in a homemade chamber filled with low-calcium, high-magnesium artificial cerebrospinal fluid (ACSF) containing (in mM): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 7 MgSO4, 0.5 CaCl2, and 25 D-(+)-glucose continuously saturated with 95% O2 and 5% CO2 at 32 °C for 30 min and then maintained at room temperature (22 ± 1 °C) for at least 1 hour until the recordings were begun.
The recordings were made with a 200B amplifier (Axon Inst.) applied to a hippocampal slice that was placed in a homemade recording chamber continuously perfused with well oxygenated ACSF (1–2 ml/min) containing (in mM): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 1 MgSO4, 2 CaCl2, 25 D-(+)-glucose, maintained at 32–34 °C by an automatic temperature controller (TC-324B, Warner Instrument Corp, Hamden, CT). Picrotoxin (100 μM, Sigma-Aldrich), a GABAA receptor antagonist, was routinely included in the ACSF. Patch-clamp recording electrodes (3–4 MΩ) were filled with the following intracellular solution (in mM): 140 potassium gluconate, 10 KCl, 10 HEPES, 5 MgCl2, (pH 7.2 with KOH) freshly supplemented with 200 μg/ml amphotericin B (Sigma-Aldrich) (Yang et al., 2009). The rest of the pipette solution containing amphotericin B was kept in a 4°C refrigerator and was discarded after 6 h.
After achieving a gigaohm seal (> 2 GΩ), a perforated whole-cell recording conformation was obtained within 5–20 min as judged by gradual reduction of the access resistance to < 20 MΩ. Under current clamp, excitatory postsynaptic potentials (EPSPs) were evoked by stimulation pulses generated by a stimulus isolator (A365, WPI, FL) every 30 s via a stereotrode tungsten electrode (WPI, FL) placed at the medial perforant pathway. A stable baseline was required for 10 min before beginning the experiment by applying drugs or beginning the spike timing dependent plasticity (STDP) protocol. In order to monitor the perforated patch recording conditions, a small hyperpolarizing current (0.01 nA) was injected (200 ms) through the recording pipette to measure input resistance. If the input resistance changed by 30% or more, then the experiment was stopped and the data were discarded. All drugs were bath applied for at least 15 min, and the STDP protocol (presynaptic stimulation followed by postsynaptic depolarization separated by a 10-ms time interval) was delivered 60 times at 0.33 Hz in 3 min. The magnitude of the synaptic plasticity change was defined as the mean peak amplitude of 40 consecutive EPSPs taken 30–60 min after the end of STDP induction protocol normalized by the average of the initial 20 consecutive EPSPs (i.e., baseline).
Fast-Scan Cyclic Voltammetry in Striatal Slices
Wild-type C57BL/6J male mice between the ages of 3 and 5 months were used for these experiments. Animals were anesthetized via injection of a ketamine, xylazine and acepromazine combination anesthetic. When the animal was deeply anesthetized, it was decapitated, and the brain quickly dissected out. Horizontal slices (300 μm) containing the dorsal striatum were incubated at 32 ± 0.5 °C for 10 minutes and held at room temperature for ≥ 1 hour. The slices were studied at 34 ± 1 °C in ACSF: 125 mM NaCl, 2.5 mM KCl, 1.3 mM MgCl2, 2.5 mM CaCl2, 1.25 mM Na2HPO3, 26 mM NaHCO3 and 10 mM glucose saturated with 95% O2 and 5% CO2.
Fast-Scan cyclic voltammetry was performed using homemade carbon-fiber microelectrodes. The potential applied to the electrode was scanned linearly from 0 to −400 to 1000 to −400 to 0 mV at a rate of 300 mV/ms against a silver/silver chloride reference. The tip of the carbon fiber recording electrode was centered approximately 200 μm away from the tips of the bipolar stimulating electrode. Voltammograms were sampled at 50 kHz and the background current was digitally subtracted. An oxidation peak between 400 and 600 mV and a reduction peak between −200 and −250 mV were used to identify the signal as a dopamine signal. The peak oxidation currents for DA in each voltammogram were converted to a DA concentration during a post experimental calibration of the carbon-fiber electrode against solutions of 0.1 to 10 μM DA (Zhang et al., 2012; Zhang et al., 2009; Zhang et al., 2010).
The dorsal striatum was usually stimulated (1-ms duration, 0.6-mA constant current) with a bipolar tungsten electrode with 120 s between stimulations to ensure recovery of the DA signal. After recording stable DA release for 10 minutes in ACSF, MPH was bath administered and allowed to equilibrate for 45 minutes before data were collected for MPH induced changes in DA release. For some of the experiments, stimulus trains were applied. When stimulation trains were applied, they combined a tonic and phasic component. The tonic stimulation was given at 1.8 Hz, and the phasic stimulation was given as 5 pulses at 20 Hz. The phasic stimulation matches the average intraburst firing frequency of approximately 20 Hz that has been measured in vivo from rodents (Benoit-Marand et al., 2001; Hyland et al., 2002; Zhang et al., 2009). The average frequency of the total stimulation train was 4 Hz, which is consistent with the average baseline firing rate of DA neurons in vivo. The stimulation train was applied before and after bath administration of MPH (50 nM). This MPH concentration is close to the peak serum concentrations observed following moderate therapeutic oral dosages (Swanson and Volkow, 2003).
In Vivo Electrophysiology
Animal Care and Surgery
Adult male C57BL/6J mice (Jackson Laboratory), 3–4 months old, were used in all of these experiments. For in vivo electrophysiology, the mice were housed individually with food and water ad libitum in a temperature-controlled room (23 ± 1 °C) with a reversed 12 hr artificial light-dark cycle (light on at 9 p.m.).
On day 0 mice were anesthetized with sodium pentobarbital (80 mg/kg, intraperitoneal injection, i.p., with additional 15 mg/kg supplements as necessary) and secured into the stereotaxic apparatus. A concentric bipolar stimulating electrode consisting of a tungsten wire placed inside a 30 gauge stainless steel tube was positioned in the medial part of the perforant path (0.2 mm posterior and between 2.8–3.0 mm lateral of lambda, 1.0–1.3 mm below the dura). The recording electrode (Teflon-coated tungsten wire, bare diameter 50 μm) attached to a stainless steel guide tube (outer diameter 310 μm) was targeted ipsilaterally to the hilus of the dentate gyrus (DG) (1.8–2.0 mm posterior, 1.4–1.6 mm lateral of bregma, 2.2–2.3 mm below the skull) (Franklin and Paxinos, 1997). The final depth and position of the electrodes were determined by judging the real-time electrophysiological signal. A silver electrode placed contralaterally in the cortex served as the recording reference and ground. Dental cement was used to anchor the electrodes and the connecting device for chronic recordings. Mice were given at least 2 weeks to recover before beginning the experimental recordings.
Handling, Habituation, Stimulation, and Recording Procedures
Beginning on day 10 post-surgery and continuing for 5 days, each animal received 5 min of handling followed by administration of 0.3 ml saline (i.p.) in the home cage in the experimental room both in the morning and afternoon. Then the mice were individually habituated to the recording chamber: a Plexiglas cylinder with bedding within a sound-reducing box (Med Associates, VT). At the same time the mice were habituated to the recording headstage system. Mice received 1 hr of habituation daily for 4 consecutive days starting from day 11. Monophasic square pulses (100 μs) were delivered to the perforant path (A360R, WPI, FL). Recorded signals were amplified (x 100), filtered (bandpass 0.1 Hz to 5 kHz), digitized at 10 kHz, and stored on disk for off-line analysis. On day 15, an input-output curve (I/O) was generated. Single test stimuli were then delivered at 30 s intervals at an intensity (35–100 mA) that evoked 40% of the maximal amplitude of the population spike.
In a typical experiment examining evoked synaptic responses in the perforant path-dentate pathway, there were two or three counterbalanced sessions for each mouse: e.g., saline, 5 mg/kg MPH, and 20 mg/kg MPH. Each data acquisition session continued for 4 days. On day minus one (−1d), the baseline amplitude of the population spike was examined for stability for 30 min. On 0d, another 30 min stable baseline recording was required before administering saline, 5 mg/kg, or 20 mg/kg MPH (i.p., 0.1 ml per 10 g body weight). The responses were usually monitored for another 3 hr following the injection, but in some cases the responses were monitored for 5 hr. In some cases, the baselines also were recorded between injection of drug or saline and theta burst stimulation (TBS) to ensure stability (e.g., Figure 4). In the experiments for Figure 4, after the baseline was recorded, mice were disconnected from the recording amplifier, given an injection of either saline or MPH, reconnected to the recording amplifier, and baseline recording continued for the 15 minutes before the TBS was administered. However, during the multiple injections necessary for the experiments of Figures 5 and 6, we did not record during the manipulations to avoid electrical noise. The mice were left in the recording chamber, but unconnected to the recording system in between the injections and TBS. The mice were reconnected 1–2 min prior to TBS administration. Baseline recordings were taken on 1d and 2d after treatment to follow the overall stability of the recordings. I/O curves were recorded systematically between sessions to verify the stability and the recovery after the treatment of each session. Throughout the electrophysiological recording, the mouse’s behavior was video monitored, and the EEG derived from the dentate gyrus recording also was displayed on an oscilloscope to ensure the absence of electrical after discharges and to ensure the mouse was in a “still-alert” state (Doyere et al., 2003).
Figure 4.

Moderate dose of MPH enhanced TBS induced LTP. (A) Example traces compare before (black trace) and after treatment (grey) trace for MPH (5 mg/kg) alone without TBS, for saline with TBS, and for MPH (5 mg/kg) + TBS. (B) Saline (grey, n = 5) or 5 mg/kg MPH (black, n = 5) was injected 15 minutes before TBS stimulation was given. This lower dose of MPH will not induce LTP alone (open circles, n = 5). Each mouse received 2 series of TBS with saline or MPH in a counterbalanced manner over 2 weeks. An injection of 5 mg/kg MPH followed by TBS results in significant LTP of the population spike amplitude (F(1,551) = 176.17, p < 0.0001).
Figure 5.
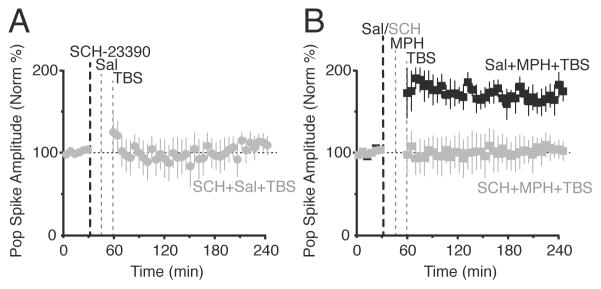
Inhibiting DA D1-type receptors blocked MPH’s effect. (A) Systemic injection of a D1/D5 receptor antagonist, SCH-23390, alone did not affect synaptic transmission following the weak TBS (n = 5). (B) Saline administered before MPH and TBS (black, n = 5) gave robust LTP, whereas SCH-23390 administered before MPH and TBS (grey, n = 5) did not show any synaptic transmission change (F(1,319) = 248.19, p < 0.0001). Treatments were counterbalanced and given at least 7 days apart for each mouse.
Figure 6.
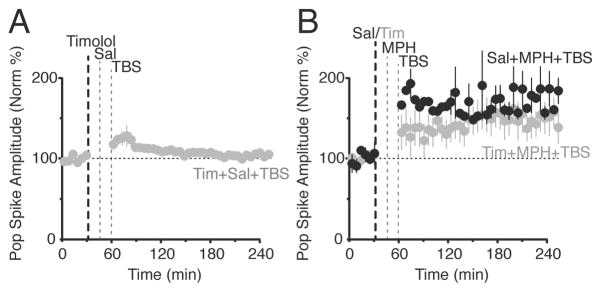
Inhibiting NE β-receptors decreased but did not prevent MPH’s effect. (A) Systemic injection of a non-specific NE β-receptor antagonist, timolol, alone did not affect synaptic transmission following the weak TBS (n = 5). (B) Saline administered before MPH and TBS (black, n = 5) gave robust LTP, while timolol administered before MPH and TBS (grey, n = 7) decreased MPH’s effect on the resulting synaptic transmission, but LTP was not blocked. Treatments were counterbalanced and given at least 7 days apart for each mouse. Injection of timolol before the MPH/TBS LTP induction protocol significantly decreased the resulting synaptic transmission in the dentate gyrus when compared to the saline control, (F(1,339) = 252.4, p < 0.05).
Theta Burst Stimulation
Theta burst stimulation (TBS) was designed to model the firing patterns of hippocampal neurons recorded during exploratory behavior in intact animals. A single TBS train consisted of 6 individual 0.1 ms pulses in a 400 Hz burst with 6 bursts at 5 Hz per train. This train was repeated at an interval of 20 seconds to induce different amounts of synaptic potentiation. A single train produced no change in synaptic transmission, while 6 trains (a strong TBS protocol) consistently induced LTP of the perforant path connection to dentate granule cells. We titrated the strength of the TBS and found that 2 trains on average produced slight long-term depression and did not induce significant LTP on its own. This version of TBS was used in all of the TBS experiments.
Drug Administration and Histology
All the drugs were from Sigma (St. Louis, MO). For i.p. injection, sodium pentobarbitol, dl-threo-methylphenidate hydrochloride, R(+)-SCH-23390 hydrochloride, and timolol maleate were dissolved in nonpyrogenic saline and adjusted for injection volume of 0.1 ml per 10 g body weight. After all experimentation, the mice were killed with an overdose of isoflurane. An anodal current (30 mA, 10 s) was passed through the tungsten wire for identification of the electrode placements. Frozen 30 μm coronal sections were cut and stained with hematoxylin.
Data Analysis
During the in vivo experiments, the amplitude of the population (pop) spike was measured for each stimulus evoked postsynaptic response by calculating the depth of the local field potential. The pop spike amplitude was measured from the point directly above the minimum of the local field potential on an imaginary line extending between the local maxima before and after the pop spike itself. The calculated values from every ten consecutive pop spikes were averaged. The averaged values before any drug treatment were normalized to 100%, and those averaged values defined the baseline. The mean baseline values were obtained during the 30 min of recording immediately before the first drug treatment.
The in vivo data of Figures 3–6 were collected in 3 continuous sections on day 0 (i.e., the treatment day). The baseline (30 min) collected before the treatments began, the treatment section, and the post-treatment section (3 hours). The baseline was always normalized to 100%, and the value for synaptic transmission (i.e., the LTP or LTD value) was calculated by averaging the data collected in the last hour of the post-treatment section. To determine whether a treatment data set had an effect, we ultimately compared the treatment group and its paired control group. The data groups were analyzed with multi-way repeated measures analysis of variance (ANOVA). In the cases where 3 or more groups were compared (i.e., Figures 4–6), the multi-way ANOVA indicated whether the groups were different. Then, a Tukey HSD post-hoc test was performed to determine the relationship of significant differences between groups, and this process yielded the reported p value. That is, the process indicated whether the treatment was different from its paired control, which also indicated the induction of LTP or LTD.
Figure 3.

High dose of MPH induced LTP. (A) Anatomical track verification of a recording electrode location in the dentate gyrus (dotted circle, left). The black dots show the range of recording locations in the dentate gyrus (right). (B) Anatomical lesion of a stimulating electrode in the medial perforant path (arrow, left). The black dots show the range of stimulation-site locations in the medial perforant path (right). (C) MPH-induced potentiation of the perforant path recorded in the DG (grey squares). The time course of each procedure is across 4 days, from the day before (−1d) until 2 days after (2d) the MPH administration. A 30 min baseline was obtained on −1d and confirmed to be stable for 30 min on day 0 (0d). Saline (black, n = 5) or 20 mg/kg MPH (grey, n = 5) was injected (i.p.) at 30 min on day 0 (vertical dashed line). The amplitudes of the population spike of the fEPSC are plotted over time for 4 days (−1d to 2d). Significant LTP was measured for 20 mg/kg MPH on 0d (F(2,389) = 7.8, p < 0.0005). The stability of the recordings and the return to baseline are shown on 1d and 2d.
RESULTS
MPH Enhanced STDP in Hippocampal Slices
EPSPs were evoked by stimulation pulses applied every 30 s via an electrode placed in the medial perforant pathway and recorded under current clamp through a perforated patch on dentate granule cells (Figure 1A). Figure 1B indicates the timing of the STDP protocol that was applied. The presynaptic stimulation occurred 10 ms before the postsynaptic depolarization. Postsynaptic current was injected to cause the postsynaptic cell to fire two action potentials at 100 Hz. This pairing was repeated 60 times at 0.33 Hz over 3 min after recording the baseline for 15 min to ensure stability of the recording. EPSPs evoked by stimulation were recorded for 60 min following the STDP pairing. The STDP stimulation protocol alone did not produce consistent synaptic plasticity, with the average being not significantly different from baseline (94% ± 19%, n = 7, p = 0.77; grey circle, Figure 1G).
Figure 1.

MPH enhanced STDP in hippocampal slices. (A) A schematic diagram of a patch-clamp recording (R) from a granule cell in a mouse dentate gyrus (DG) slice. A stimulating electrode (S) was placed in the medial perforant path (mPP) originating in the medial entorhinal cortex (EC). (B) An illustration of the STDP induction protocol with Δt = 10 ms, meaning the presynaptic stimulation preceded by 10 ms the postsynaptic action potential (AC) firing induced by current injection via the recording pipette. STDP induction by the protocol in the presence of 500 nM MPH (C), 20 μM SCH-23390 (SCH) + 500 nM MPH (D), 20 μM timolol (TIM) + 500 nM MPH (E), and SCH+TIM+MPH (F), respectively. The open horizontal bar in each left panel indicates the period of drug application, and the arrows indicate application of the 10 ms STPD protocol. The right panel of the corresponding figures shows tabulated individual plasticity values and averaged values of the magnitude of synaptic plasticity. (G) A summary of the mean magnitude of STDP induced by the 10 ms protocol shown in C–F and for control experiments. * p < 0.05 compared to the baseline, # p < 0.05 comparison between the two groups indicated.
The literature indicates MPH doses for hippocampal slice experiments ranging from about 50 nM to 300 μM (Dommett et al., 2008; Ishimatsu et al., 2002). MPH has a higher affinity for the DAT (Ki = 34 nM) than for the NET (Ki = 339 nM) (Bymaster et al., 2002; Pristupa et al., 1994), however, the median effective doses for DAT and NET blockade are close (Hannestad et al., 2010; Volkow et al., 1998), and therapeutic doses injected i.p. in rats show similar increases in DA and NE concentrations in the prefrontal cortex (Bymaster et al., 2002). Based on the literature and our voltammetry experiments below, we used a moderate concentration of 0.5 μM MPH, and found that MPH (0.5 μM) significantly enhanced LTP induced by the STDP protocol (154% ± 21% of baseline, n = 7, p = 0.02, Figure 1C; black circle, Figure 1G).
SCH-23390, a D1/D5 receptor antagonist, reduced the STDP enhancement so that it was no longer significantly different from baseline (SCH, Figure 1D: 128% ± 48% of baseline, n = 8, p = 0.29; black square, Figure 1G). Timolol, a nonspecific NE β receptor antagonist also reduced the STDP enhancement so that it was no longer significant (TIM, Figure 1E: 148% ± 46% of baseline, n = 8, p = 0.17; black upward triangle, Figure 1G). With a single antagonist, the stimulation protocol for a given recording could produce LTP, LTD, or no change, with the average being not significantly different from baseline (Figure 1D and 1E, right panels). However, when both antagonists (SCH-23390 and timolol) were applied along with MPH before the STDP protocol, no change in synaptic strength was observed, and individual recordings were much less likely to vary greatly (Figure 1F: 81% ± 15% of baseline, n = 6, p = 0.13; black downward triangle, Figure 1G).
The compiled data (Figure 1G) show that MPH with the STDP protocol (black circle) significantly enhanced STDP-induced LTP in hippocampal slices compared to the baseline, compared to a control in which the STDP protocol was applied without MPH (light grey circle), and compared to a control in which MPH was perfused in the bath but no STDP stimulation was applied (light grey square). Adding either a DA D1/D5 receptor antagonist or a NE β receptor antagonist reduced MPH-enhancement below the level of significance. Combining the two antagonists (black downward triangle) potently blocked the increase in synaptic strength induced by MPH during the STPD protocol.
MPH Increased Dopamine Release in Striatal Slices
To better understand MPH’s influence over DA release and MPH dosing for our experiments, we used fast-scan cyclic voltammetry (FSCV) to measure dynamic DA release from in vitro striatal slices, which provide robust, measureable DA release. Our intent was to find the MPH dose that increases DA signaling without producing an overly severe alteration in the DA signaling process. In that way, we hoped to better model the more conservative dosing expected in therapeutic situations. We used FSCV to obtain high fidelity information on the time course of the evoked DA response. First we completed a dose-response curve for concentrations of MPH ranging from 1 nM to 300 μM (Figure 2A–C), which covers the dose range commonly used for in vitro hippocampal studies (Dommett et al., 2008; Ishimatsu et al., 2002). The dose-response curve showed an inverted U-shape curve. Up to 1 μM MPH, the DA release is well behaved, but above that concentration the DA peak falls with increasing MPH concentrations. Therefore, we used MPH concentrations well below 1 μM for the slice experiments, and we considered doses in this range to be biologically reasonable.
Figure 2.

The MPH dose dependence for influencing DA release measured by fast-scan cyclic voltammetry. (A) Example DA traces before (0 MPH) and after bath administration of the indicated concentration of MPH. The average DA responses peak magnitude (B) and dopamine signal area (C) are plotted against the MPH concentrations. (D) MPH (50 nM) increases DA release. An example DA trace is shown before (black trace) and after MPH bath administration (grey trace). The bar graphs show that the average DA peak and area under the DA curve increase after MPH administration. (E) MPH (50 nM) increases the phasic/tonic ratio of DA release. The phasic-to-tonic ratio increases following treatment with 50 nM MPH. The individual paired traces are averaged and plotted over each other. When treated with 50 nM MPH (larger amplitude grey trace), the tonic and phasic DA release both increased. However, the phasic DA release increased more, and this result is reflected in the increased phasic-to-tonic ratios of the peak amplitudes and areas following MPH application (bar graphs, right).
DA neurons discharge tonically at low frequencies that consist mainly of individual action potentials (Grace and Bunney, 1984). Periodically, DA neurons fire phasic bursts that average near 20 Hz in rodents (Hyland et al., 2002; Robinson et al., 2004; Zhang et al., 2009). After bath application of MPH (50 nM), the DA release in the dorsolateral striatum increased (Figure 2D). For a single electrical stimulus (1p), the peak DA concentration increased to 1.40 ± 0.15 μM from 0.99 ± 0.11 μM in the control (n = 13, p < 0.0002), and the total DA signal, measured as area under the curve, increased to 0.89 ± 0.13 μM*s from 0.47 ± 0.07 μM*s in the control (n = 13, p < 0.005).
To examine the tonic and phasic components of DA release, a tonic 1.8 Hz stimulation was given before a phasic burst (5 pulses at 20 Hz). In the presence of MPH (50 nM), each DA response was larger (Figure 2E). Also the phasic-to-tonic ratios of the peak amplitudes and areas were significantly increased following MPH application. The phasic/tonic ratio of the DA concentration peaks increased to 3.07 ± 0.36 in MPH from 2.05 ± 0.26 in the control solutions (n = 11, p = 0.009) and the phasic/tonic ratio of the area under the DA curves increased to 5.91 ± 1.02 in MPH from 3.40 ± 0.59 in the control (n = 11, p = 0.014). Thus, MPH increased phasic DA release more than it increased tonic DA release under these conditions.
Behavioral Observations Defined In Vivo MPH Dosing
We set out to find an appropriate in vivo MPH dose that recapitulates some of the behavioral effects of cocaine, but not so high as to induce stereotypy or impair performance on a behavioral memory task. Increased locomotion and stereotypies are side effects of moderate to high therapeutic doses of MPH in humans. The therapeutic range of MPH doses in humans has been suggested to be 0.5–2.5 mg/kg (Kuczenski and Segal, 2001). However, the route of administration and species differences in drug metabolism significantly affect plasma and brain MPH concentrations. Although MPH has higher affinity for the human DAT over the NET, 3 mg/kg MPH injected i.p. in rats showed similar increases in DA and NE concentrations in the prefrontal cortex, striatum, and nucleus accumbens as a percentage of baseline (Bymaster et al., 2002). Results also suggest that mice are less sensitive to MPH than rats, and chronic injections of 3 mg/kg preferentially increased DA and NE in the prefrontal cortex without affecting levels in the striatum and without causing sensitization over 21 days of daily injections (Koda et al., 2010). Guided by these data we performed open field experiments on several doses of MPH looking for increased locomotor behavior without stereotypies, defined as repetitive turning in tight circles or excessive self-grooming.
As a second test of a moderate dose of MPH, we ran mice through a hippocampal dependent, delayed non-match to sample T-maze task designed to test spatial working memory, a type of memory that MPH is specifically known to enhance (Repantis et al., 2010; Wright and White, 2003). A boost in T-maze performance should mean we have a moderate therapeutic dose, as indicated in the literature by behavioral experiments in mice (Heredia et al., 2013; Huang and Huang, 2012; Lloyd et al., 2013). A dose of 1 mg/kg did not significantly increase locomotor behavior, while 5 mg/kg increased locomotor behavior without inducing stereotypy. We found, however, that a dose of 20 mg/kg did induce stereotypy, and we therefore labeled 20 mg/kg a high dose of MPH. For the remaining experiments we used 5 mg/kg as our medium dose of MPH. After being thoroughly trained on the task, administration of 5 mg/kg MPH (i.p.) significantly increased correct arm choice in the T-maze compared to mice that received saline (88% ± 7% after MPH versus 69% ± 11% after saline as a control, p < 0.05, n = 16 for both groups, data not shown). This gave us our basis for dosing to begin the in vivo electrophysiological experiments.
MPH Enhanced LTP In Vivo
Field potentials evoked by stimulation of the medial perforant path were recorded in the hilar region of the hippocampal dentate gyrus from freely moving C57BL/6 mice (Figure 3A,B). An example of the lesion from a recording site is shown in Figure 3A (left), and all the recording sites in the dentate gyrus are indicated in Figure 3A (right). An example of the lesion from a stimulation site is shown in Figure 3B (arrow, left), and all the stimulation sites in the medial perforant path are indicated in Figure 3B (right). We focused on the medial perforant path because it relays convergent information from the neocortex that is rich in contextual, place, and spatial content (Hargreaves et al., 2005), and evidence indicates that such information is associated with the drug experience (Biala et al., 2005; Kilts et al., 2001; Tang and Dani, 2009). Field recordings were made from the hilus of the dentate gyrus to follow the synaptically generated field excitatory postsynaptic potential (fEPSP) and the population (pop) spike, which corresponds to the synchronous discharge of a large number of granule cells (Buzsaki and Czeh, 1981). The magnitudes of these components are directly related to the number of neurons activated by the stimulus (Andersen et al., 1971; Lomo, 1971). Synaptic transmission was quantified by measuring the pop spike amplitude because the slope of the fEPSP is often obscured in vivo by an increase in the pop spike that occurs after synaptic potentiation induction in awake animals.
Two weeks after surgical implantation of the electrodes and habituation to the recording situation, mice were treated with three 4-day counterbalanced sessions of systemic i.p. injection of saline, 5, or 20 mg/kg MPH. We tested whether MPH alone would induce any directional plasticity that could be measured by the pop spike of the fEPSPs. Neither saline nor 5 mg/kg MPH affected synaptic transmission: for saline, 104% ± 1.1%, n = 3, p > 0.05 (Figure 3C, saline, black squares); for 5 mg/kg MPH (see Figure 4B, open circles, to simplify Figure 3C). However, 20 mg/kg MPH induced long-term synaptic potentiation (Figure 3C, grey squares) as measured in the pop spike amplitude 2 hr after administration: 109% ± 1.1%, n = 5, p < 0.001 for 20 mg/kg.
To simulate a signal along the perforant path to the dentate gyrus, we applied relatively weak theta burst stimulation (TBS) and combined that TBS with MPH administration. Based on the in vitro experiments (Figure 1), we hypothesized that MPH should boost this sub-threshold TBS paradigm to produce long-term potentiation (LTP) owing to blockade of the NET and/or DAT (Kulla and Manahan-Vaughan, 2000; Moore and Bloom, 1979; Rossato et al., 2009; Sajikumar and Frey, 2004). A saline injection 15 min before the standard theta burst stimulation resulted in slight long-term depression (Figure 4B, light grey circles, 93.1% ± 2.0% of baseline, n = 5, p = 0.008). A moderate dose of MPH (5 mg/kg) alone did not produce significant synaptic change (Figure 4B, open circles): 104% ± 1.1%, n = 3, p > 0.05. However, when that dose was given 15 min before the standard TBS, it produced long lasting potentiation of the perforant path to dentate gyrus pathway (Figure 4B, black circles): 131% ± 2.3%, n = 5, p < 0.0001.
To determine whether primarily DA or NE receptors were responsible for the MPH induced synaptic potentiation, we systemically inhibited DA D1/D5 receptors (0.2 mg/kg, i.p., SCH-23390) or NE β receptors (10 or 20 mg/kg, timolol). Injections of SCH-23390 (0.2 mg/kg, i.p) alone did not significantly alter synaptic transmission (Figure 5A): 102.5% ± 2.8% of baseline, n = 5, p > 0.05. In the paired control experiments where saline (i.p.) preceded the MPH administration, MPH induced a significant potentiation over the baseline (Figure 5B, black squares): 164.2% ± 2.9% of baseline, n = 5, p < 0.0001. Systemic injection of SCH-23390 preceding MPH (5 mg/kg) administration completely prevented synaptic potentiation induced by MPH combined with TBS (Figure 5B, grey squares): 100.1% ± 18.6% of baseline, n = 5, p > 0.05. Injections of timolol (10 or 20 mg/kg, i.p.) alone did not significantly alter synaptic transmission (Figure 6A): 103.2% ± 5.9% of baseline, n = 5, p > 0.05. In the paired control experiments where saline (i.p.) preceded MPH administration, MPH followed by TBS induced a significant potentiation over baseline (Figure 6B, black circles): 174% ± 13%, n = 7, p < 0.0001. Systemic injection of timolol decreased the change in synaptic transmission induced by MPH preceding TBS, but significant LTP was still induced: 151% ± 3.4% of baseline, n = 5, p = 0.0037. The 3-way ANOVA and Tukey HSD post-hoc tests report that all 3 groups are statistically different, indicating that when timolol inhibits NE β receptors there is a difference in MPH’s effect. As always, the reported LTP value above was calculated for the last hour of the 3 hour time course after TBS administration (Figure 6B). The greatest difference caused by timolol occurs at earlier times during the first hour after TBS when inhibition of the NE β receptors by timolol resulted in a stronger suppression of the early synaptic potentiation: 136% ± 3.1%, n = 5, p = 0.0004.
DISCUSSION
Methylphenidate (MPH) is widely used for the treatment of ADHD to improve attention, learning, and memory. The main mechanistic action of MPH is to inhibit DA and NE reuptake transporters. Based on its action and on previous findings (Dommett et al., 2008; Urban et al., 2013), we hypothesized that MPH directly alters hippocampal long-term synaptic plasticity. Therefore, the influence of MPH over plasticity of the perforant path to the dentate gyrus was examined. We focused on the medial perforant path because it specifically carries place and spatial information (Hargreaves et al., 2005) that is important for associative memory. The accumulated evidence also supports that synaptic plasticity along this pathway is a contributing substrate of memory (Lynch, 2004; Martinez and Derrick, 1996; McHugh et al., 2008; Rumpel et al., 2005). Using in vitro slices we found that MPH enhances the induction of STDP. A STDP protocol that induced no net synaptic change by itself would induce LTP when the stimulus protocol was applied in the presence of MPH (0.5 μM). When either the D1-type DA receptors were inhibited with SCH-23390 or the NE β receptors were inhibited by timolol, the average LTP amplitude induced by the MPH protocol was reduced (Figure 1). In slices, it is noteworthy that with either D1-type receptors or NE β receptors inhibited there is still a trend toward potentiation, and there is significant scatter among the individual plasticity recordings (Figure 1D,E, right bar plots). When both sets of catecholamine receptors were inhibited, however, LTP induction by the STDP protocol was eliminated on average and for each individual experiment (Figure 1F, right bar plot). Chronic recordings from freely moving mice showed that similar catecholamine mechanisms are at play in vivo. Field potentials evoked by stimulation of the medial perforant path were recorded in the hilar region of the dentate gyrus (Tang and Dani, 2009). Weak theta burst stimulation (TBS) that induced no synaptic change or slight LTD on its own, induced LTP when applied in the presence of MPH (5 mg/kg). The LTP induced by TBS + MPH was reduced when NE β receptors were inhibited by timolol (Figure 6), but the LTP induction was absent when D1-type DA receptors were inhibited with SCH-23390 (Figure 5).
While both the in vitro and in vivo experiments suggest influence by both catecholamines, the results are quantitatively different. The in vitro experiments show influence by inhibiting either the D1-type receptors or NE β receptors, but the strongest inhibition of STDP occurred when both receptor types were inhibited. The in vivo experiments show greater and complete inhibition of the MPH-influenced plasticity when the D1-type receptors were inhibited. These differences could arise from the difference in the preparations. The in vitro slices were from 24–30 day old mice while the in vivo experiments were conducted on young adult mice (3–4 months old). Possibly more importantly, the scatter among the individual experiments in vitro (Figure 1D,E, right) suggest that the individual placements of stimulation and recording electrodes, coupled with the reduced slice preparation, could produce more extremes in the local catecholamine signaling. This variability appears less likely in the intact, in vivo situation where mature, more consistent catecholamine signaling is present. In that case, the actions of the receptors and the receptor inhibitors may reflect the final more refined and consistent catecholamine signaling into the dentate gyrus seen in the adult mice.
Evidence indicates that NET and DAT blockade influences individual areas of the hippocampus differently (Hopkins and Johnston, 1988; Huang and Kandel, 1996; Ishihara et al., 1991; Lacaille and Harley, 1985; Munro et al., 2001). In the hippocampal CA1 region of rat slices, MPH boosted tetanic LTP based mainly on its ability to block NETs because the NE β receptor inhibitor, timolol, blocked MPH enhancement of tetanic LTP (Dommett et al., 2008). In our experiments applied to the dentate gyrus, however, the DA D1-type receptors and NE β receptors both seemed important during our in vitro (relatively weak) STDP protocol applied to slices from young mice (24–30d). Furthermore, the D1-type receptors seemed even more important than the NE receptors during our in vivo TBS protocol in adult mice.
Previous reports showed mixed results for the ability of MPH to increase DA and NE in slice preparations (Heal et al., 2008). We found effects of both DA and NE in our hippocampal slice LTP paradigm, suggesting that the STDP stimulation protocol can cause variable DA and NE release from terminals in the dentate gyrus slice. Furthermore, MPH significantly increased these catecholamine signals to enhance synaptic plasticity. In vivo, TBS enhancement by MPH in the dentate gyrus was more dependent on the activity of DA receptors. Whether the DA driven enhancement was due to endogenous DA release or preferential release of DA by the TBS protocol itself is unknown. These differences in the results that arise from different plasticity induction protocols applied to different hippocampal locations are consistent with region-specific forms of neuromodulation (Hopkins and Johnston, 1988; Huang and Kandel, 1996; Ishihara et al., 1991; Lacaille and Harley, 1985; Munro et al., 2001). Therefore, the cumulative results suggest that MPH influences hippocampal plasticity owing to effects on both the DA and NE systems.
Throughout these experiments we attempted to use stimulus protocols and MPH doses that are reasonable. A TBS protocol was used in vivo to mimic the theta mode of hippocampal firing that is observed during states of active, alert behavior in freely-moving rodents (Hyman et al., 2003; Orr et al., 2001). That TBS protocol only produced LTP after administration of MPH. Similarly, the STDP protocol used for the in vitro experiments was mild. In addition, the perforated-patch methodology, rather than the whole-cell configuration, was used for the in vitro recordings to preserve the intracellular milieu. Unlike tetanic stimulation to induce synaptic plasticity, STDP pairs a presynaptic stimulation followed by a postsynaptic depolarization to mimic the biological case in which the afferent activity drives a postsynaptic response in the target area. The mild STDP protocol used for the in vitro experiments induced synaptic plasticity (LTP) only in the presence of MPH (0.5 μM). The doses of MPH arose from the literature, our voltammetry data, and our behavioral data. Based on our voltammetry experiments, the DA release was well behaved up to 1 μM MPH, which resulted in the largest peak evoked DA release. Therefore, we used 0.5 μM MPH for the hippocampal slice experiments. This concentration is higher than blood plasma levels in patients treated for ADHD, which is usually in the range of 50–200 nM, but MPH reaches higher concentrations in the brain than in the blood (Hoffman and Lefkowitz, 1996). The MPH concentration (5 mg/kg) used for the in vivo experiments with freely-moving mice had no effect over in vivo synaptic transmission before the TBS was applied (Figure 4). This MPH dose (5 mg/kg) increased locomotor behavior without inducing stereotypy, and this dose increased correct arm choices in the delayed non-match to sample T-maze task. That task is designed to test spatial working memory, which is a type of memory that MPH is specifically known to enhance (Repantis et al., 2010; Wright and White, 2003). Therefore, the 5 mg/kg dose of MPH seems like a biologically appropriate concentration for the behavioral tasks and in vivo measurements of synaptic plasticity in mice. Under these biologically reasonable experimental conditions, MPH enhanced synaptic plasticity involving catecholamine receptors.
Unpredicted rewards are known to produce brief burst firing by DA neurons (Schultz et al., 1997), and phasic bursts induce greater extracellular DA release compared with tonic, single-spike firing activity (Floresco et al., 2003; Garris et al., 1994; Gonon, 1988; Grace, 1991; Rice and Cragg, 2004; Zhang et al., 2009). Dopamine neurons operate in these distinct tonic and phasic timescales to differentiate behaviorally relevant information (Schultz, 2007). By inhibiting DATs and NETs, MPH increases and lengthens the catecholamine signal after release, which may share similarities to the DA/NE signal after a long phasic burst. Our voltammetry results showed that MPH enhanced the DA signaling from phasic bursts even more than it enhanced the tonic signals. Likewise, MPH is expected to enhance the size and length of adrenergic signaling. Dopamine and adrenergic receptors are expressed in the hippocampus (Huang et al., 1992; Jurgens et al., 2005), and the catecholamines have been shown to enhance synaptic plasticity via D1-type DA receptors and β-type NE receptors (Lisman and Grace, 2005; Tang and Dani, 2009; Thomas et al., 1996). Our results support the importance of catecholamines in synaptic plasticity, and the results indicate that MPH influences these synaptic processes. Because the in vivo experiments were conducted on freely-moving mice under biologically reasonable conditions, those results provide strong support for the importance of MPH acting via catecholamines in the hippocampus.
Highlights.
Weak perforant path stimulation induced LTP in the presence of methylphenidate
Inhibition of D1/D5 DA and β-type NE receptors prevented this LTP induced in vitro
During in vivo protocols, D1/D5 DA receptors more potently regulated this LTP
Methylphenidate doses that regulated LTP preferentially increased phasic DA signals
Methylphenidate doses that regulated LTP enhanced a working memory task in mice
Acknowledgments
The work was supported by grants from the National Institutes of Health, NIDA DA09411 and NINDS NS21229.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agnati LF, Zoli M, Stromberg I, Fuxe K. Intercellular communication in the brain: wiring versus volume transmission. Neuroscience. 1995;69:711–726. doi: 10.1016/0306-4522(95)00308-6. [DOI] [PubMed] [Google Scholar]
- Andersen P, Bliss TV, Skrede KK. Unit analysis of hippocampal polulation spikes. Exp Brain Res. 1971;13:208–221. doi: 10.1007/BF00234086. [DOI] [PubMed] [Google Scholar]
- Benoit-Marand M, Borrelli E, Gonon F. Inhibition of dopamine release via presynaptic D2 receptors: time course and functional characteristics in vivo. J Neurosci. 2001;21:9134–9141. doi: 10.1523/JNEUROSCI.21-23-09134.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biala G, Betancur C, Mansuy IM, Giros B. The reinforcing effects of chronic D-amphetamine and morphine are impaired in a line of memory-deficient mice overexpressing calcineurin. Eur J Neurosci. 2005;21:3089–3096. doi: 10.1111/j.1460-9568.2005.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederman J, Spencer T. Attention-deficit/hyperactivity disorder (ADHD) as a noradrenergic disorder. Biol Psychiatry. 1999;46:1234–1242. doi: 10.1016/s0006-3223(99)00192-4. [DOI] [PubMed] [Google Scholar]
- Borgkvist A, Malmlof T, Feltmann K, Lindskog M, Schilstrom B. Dopamine in the hippocampus is cleared by the norepinephrine transporter. Int J Neuropsychopharmacol. 2012;15:531–540. doi: 10.1017/S1461145711000812. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Czeh G. Commissural and perforant path interactions in the rat hippocampus. Field potentials and unitary activity. Exp Brain Res. 1981;43:429–438. doi: 10.1007/BF00238387. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Katner JS, Nelson DL, Hemrick-Luecke SK, Threlkeld PG, Heiligenstein JH, Morin SM, Gehlert DR, Perry KW. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2002;27:699–711. doi: 10.1016/S0893-133X(02)00346-9. [DOI] [PubMed] [Google Scholar]
- Chai G, Governale L, McMahon AW, Trinidad JP, Staffa J, Murphy D. Trends of outpatient prescription drug utilization in US children, 2002–2010. Pediatrics. 2012;130:23–31. doi: 10.1542/peds.2011-2879. [DOI] [PubMed] [Google Scholar]
- Dani JA, Harris RA. Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat Neurosci. 2005;8:1465–1470. doi: 10.1038/nn1580. [DOI] [PubMed] [Google Scholar]
- Deadwyler SA, West M, Lynch G. Activity of dentate granule cells during learning: differentiation of perforant path input. Brain Res. 1979;169:29–43. doi: 10.1016/0006-8993(79)90371-8. [DOI] [PubMed] [Google Scholar]
- Di Chiara G. Nucleus accumbens shell and core dopamine: differential role in behavior and addiction. Behav Brain Res. 2002;137:75–114. doi: 10.1016/s0166-4328(02)00286-3. [DOI] [PubMed] [Google Scholar]
- Dommett EJ, Henderson EL, Westwell MS, Greenfield SA. Methylphenidate amplifies long-term plasticity in the hippocampus via noradrenergic mechanisms. Learn Mem. 2008;15:580–586. doi: 10.1101/lm.1092608. [DOI] [PubMed] [Google Scholar]
- Doyere V, Schafe GE, Sigurdsson T, LeDoux JE. Long-term potentiation in freely moving rats reveals asymmetries in thalamic and cortical inputs to the lateral amygdala. Eur J Neurosci. 2003;17:2703–2715. doi: 10.1046/j.1460-9568.2003.02707.x. [DOI] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat Neurosci. 2003;6:968–973. doi: 10.1038/nn1103. [DOI] [PubMed] [Google Scholar]
- Franklin KB, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Academic Press; London: 1997. [Google Scholar]
- Garris PA, Ciolkowski EL, Pastore P, Wightman RM. Efflux of dopamine from the synaptic cleft in the nucleus accumbens of the rat brain. J Neurosci. 1994;14:6084–6093. doi: 10.1523/JNEUROSCI.14-10-06084.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonon FG. Nonlinear relationship between impulse flow and dopamine released by rat midbrain dopaminergic neurons as studied by in vivo electrochemistry. Neuroscience. 1988;24:19–28. doi: 10.1016/0306-4522(88)90307-7. [DOI] [PubMed] [Google Scholar]
- Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–24. doi: 10.1016/0306-4522(91)90196-u. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984;4:2866–2876. doi: 10.1523/JNEUROSCI.04-11-02866.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannestad J, Gallezot JD, Planeta-Wilson B, Lin SF, Williams WA, van Dyck CH, Malison RT, Carson RE, Ding YS. Clinically relevant doses of methylphenidate significantly occupy norepinephrine transporters in humans in vivo. Biol Psychiatry. 2010;68:854–860. doi: 10.1016/j.biopsych.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves EL, Rao G, Lee I, Knierim JJ. Major dissociation between medial and lateral entorhinal input to dorsal hippocampus. Science. 2005;308:1792–1794. doi: 10.1126/science.1110449. [DOI] [PubMed] [Google Scholar]
- Heal DJ, Smith SL, Kulkarni RS, Rowley HL. New perspectives from microdialysis studies in freely-moving, spontaneously hypertensive rats on the pharmacology of drugs for the treatment of ADHD. Pharmacol Biochem Behav. 2008;90:184–197. doi: 10.1016/j.pbb.2008.03.016. [DOI] [PubMed] [Google Scholar]
- Heredia L, Torrente M, Colomina MT, Domingo JL. Assessing anxiety in C57BL/6J mice: A pharmacological characterization of the zero maze test. J Pharmacol Toxicol Methods. 2013 doi: 10.1016/j.vascn.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Hoffman BB, Lefkowitz RJ. Goodman and Gilman’s The pharmacological basis of therapeutics, Catecholamines, sympathomimetic drugs, and adrenergic receptor antagonists. McGraw-Hill; New York: 1996. [Google Scholar]
- Hopkins WF, Johnston D. Frequency-dependent noradrenergic modulation of long-term potentiation in the hippocampus. Science. 1984;226:350–352. doi: 10.1126/science.6091272. [DOI] [PubMed] [Google Scholar]
- Hopkins WF, Johnston D. Noradrenergic enhancement of long-term potentiation at mossy fiber synapses in the hippocampus. J Neurophysiol. 1988;59:667–687. doi: 10.1152/jn.1988.59.2.667. [DOI] [PubMed] [Google Scholar]
- Huang FL, Huang KP. Methylphenidate improves the behavioral and cognitive deficits of neurogranin knockout mice. Genes Brain Behav. 2012;11:794–805. doi: 10.1111/j.1601-183X.2012.00825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Zhou D, Chase K, Gusella JF, Aronin N, DiFiglia M. Immunohistochemical localization of the D1 dopamine receptor in rat brain reveals its axonal transport, pre- and postsynaptic localization, and prevalence in the basal ganglia, limbic system, and thalamic reticular nucleus. Proc Natl Acad Sci USA. 1992;89:11988–11992. doi: 10.1073/pnas.89.24.11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER. Modulation of both the early and the late phase of mossy fiber LTP by the activation of beta-adrenergic receptors. Neuron. 1996;16:611–617. doi: 10.1016/s0896-6273(00)80080-x. [DOI] [PubMed] [Google Scholar]
- Hyland BI, Reynolds JN, Hay J, Perk CG, Miller R. Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience. 2002;114:475–492. doi: 10.1016/s0306-4522(02)00267-1. [DOI] [PubMed] [Google Scholar]
- Hyman JM, Wyble BP, Goyal V, Rossi CA, Hasselmo ME. Stimulation in hippocampal region CA1 in behaving rats yields long-term potentiation when delivered to the peak of theta and long-term depression when delivered to the trough. J Neurosci. 2003;23:11725–11731. doi: 10.1523/JNEUROSCI.23-37-11725.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Ishihara K, Katsuki H, Kawabata A, Sasa M, Satoh M, Takaori S. Effects of thyrotropin-releasing hormone and a related analog, CNK-602A, on long-term potentiation in the mossy fiber-CA3 pathway of guinea pig hippocampal slices. Brain Res. 1991;554:203–208. doi: 10.1016/0006-8993(91)90190-7. [DOI] [PubMed] [Google Scholar]
- Ishimatsu M, Kidani Y, Tsuda A, Akasu T. Effects of methylphenidate on the membrane potential and current in neurons of the rat locus coeruleus. J Neurophysiol. 2002;87:1206–1212. doi: 10.1152/jn.00463.2001. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Clifford DB, Zorumski CF. Norepinephrine reverses N-methyl-D-aspartate-mediated inhibition of long-term potentiation in rat hippocampal slices. Neurosci Lett. 1992;142:163–166. doi: 10.1016/0304-3940(92)90364-d. [DOI] [PubMed] [Google Scholar]
- Jay TM. Dopamine: a potential substrate for synaptic plasticity and memory mechanisms. Prog Neurobiol. 2003;69:375–390. doi: 10.1016/s0301-0082(03)00085-6. [DOI] [PubMed] [Google Scholar]
- Jones S, Bonci A. Synaptic plasticity and drug addiction. Curr Opin Pharmacol. 2005;5:20–25. doi: 10.1016/j.coph.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Jurgens CW, Rau KE, Knudson CA, King JD, Carr PA, Porter JE, Doze VA. Beta1 adrenergic receptor-mediated enhancement of hippocampal CA3 network activity. J Pharmacol Exp Ther. 2005;314:552–560. doi: 10.1124/jpet.105.085332. [DOI] [PubMed] [Google Scholar]
- Kauer JA. Learning mechanisms in addiction: synaptic plasticity in the ventral tegmental area as a result of exposure to drugs of abuse. Annu Rev Physiol. 2004;66:447–475. doi: 10.1146/annurev.physiol.66.032102.112534. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Ventral striatal control of appetitive motivation: role in ingestive behavior and reward-related learning. Neurosci Biobehav Rev. 2004;27:765–776. doi: 10.1016/j.neubiorev.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Kilts CD, Schweitzer JB, Quinn CK, Gross RE, Faber TL, Muhammad F, Ely TD, Hoffman JM, Drexler KP. Neural activity related to drug craving in cocaine addiction. Arch Gen Psychiatry. 2001;58:334–341. doi: 10.1001/archpsyc.58.4.334. [DOI] [PubMed] [Google Scholar]
- Koda K, Ago Y, Cong Y, Kita Y, Takuma K, Matsuda T. Effects of acute and chronic administration of atomoxetine and methylphenidate on extracellular levels of noradrenaline, dopamine and serotonin in the prefrontal cortex and striatum of mice. J Neurochem. 2010;114:259–270. doi: 10.1111/j.1471-4159.2010.06750.x. [DOI] [PubMed] [Google Scholar]
- Kuczenski R, Segal DS. Effects of methylphenidate on extracellular dopamine, serotonin, and norepinephrine: comparison with amphetamine. J Neurochem. 1997;68:2032–2037. doi: 10.1046/j.1471-4159.1997.68052032.x. [DOI] [PubMed] [Google Scholar]
- Kuczenski R, Segal DS. Locomotor effects of acute and repeated threshold doses of amphetamine and methylphenidate: relative roles of dopamine and norepinephrine. J Pharmacol Exp Ther. 2001;296:876–883. [PubMed] [Google Scholar]
- Kuczenski R, Segal DS. Exposure of adolescent rats to oral methylphenidate: preferential effects on extracellular norepinephrine and absence of sensitization and cross-sensitization to methamphetamine. J Neurosci. 2002;22:7264–7271. doi: 10.1523/JNEUROSCI.22-16-07264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulla A, Manahan-Vaughan D. Depotentiation in the dentate gyrus of freely moving rats is modulated by D1/D5 dopamine receptors. Cereb Cortex. 2000;10:614–620. doi: 10.1093/cercor/10.6.614. [DOI] [PubMed] [Google Scholar]
- Lacaille JC, Harley CW. The action of norepinephrine in the dentate gyrus: beta-mediated facilitation of evoked potentials in vitro. Brain Res. 1985;358:210–220. doi: 10.1016/0006-8993(85)90965-5. [DOI] [PubMed] [Google Scholar]
- Lavenex P, Amaral DG. Hippocampal-neocortical interaction: a hierarchy of associativity. Hippocampus. 2000;10:420–430. doi: 10.1002/1098-1063(2000)10:4<420::AID-HIPO8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46:703–713. doi: 10.1016/j.neuron.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Lloyd SA, Oltean C, Pass H, Phillips B, Staton K, Robertson CL, Shanks RA. Prenatal exposure to psychostimulants increases impulsivity, compulsivity, and motivation for rewards in adult mice. Physiol Behav. 2013;119C:43–51. doi: 10.1016/j.physbeh.2013.05.038. [DOI] [PubMed] [Google Scholar]
- Lomo T. Patterns of activation in a monosynaptic cortical pathway: the perforant path input to the dentate area of the hippocampal formation. Exp Brain Res. 1971;12:18–45. [PubMed] [Google Scholar]
- Lynch MA. Long-Term Potentiation and Memory. Physiol Rev. 2004;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- Martinez JL, Derrick BE. Long-term potentiation and learning. Annu Rev Psychol. 1996;47:173–203. doi: 10.1146/annurev.psych.47.1.173. [DOI] [PubMed] [Google Scholar]
- McHugh SB, Niewoehner B, Rawlins JN, Bannerman DM. Dorsal hippocampal N-methyl-D-aspartate receptors underlie spatial working memory performance during non-matching to place testing on the T-maze. Behav Brain Res. 2008;186:41–47. doi: 10.1016/j.bbr.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RY, Bloom FE. Central catecholamine neuron systems: anatomy and physiology of the norepinephrine and epinephrine systems. Annu Rev Neurosci. 1979;2:113–168. doi: 10.1146/annurev.ne.02.030179.000553. [DOI] [PubMed] [Google Scholar]
- Munro CA, Walling SG, Evans JH, Harley CW. Beta-adrenergic blockade in the dentate gyrus in vivo prevents high frequency-induced long-term potentiation of EPSP slope, but not long-term potentiation of population spike amplitude. Hippocampus. 2001;11:322–328. doi: 10.1002/hipo.1046. [DOI] [PubMed] [Google Scholar]
- Orr G, Rao G, Houston FP, McNaughton BL, Barnes CA. Hippocampal synaptic plasticity is modulated by theta rhythm in the fascia dentata of adult and aged freely behaving rats. Hippocampus. 2001;11:647–654. doi: 10.1002/hipo.1079. [DOI] [PubMed] [Google Scholar]
- Pristupa ZB, Wilson JM, Hoffman BJ, Kish SJ, Niznik HB. Pharmacological heterogeneity of the cloned and native human dopamine transporter: disassociation of [3H]WIN 35,428 and [3H]GBR 12,935 binding. Mol Pharmacol. 1994;45:125–135. [PubMed] [Google Scholar]
- Repantis D, Schlattmann P, Laisney O, Heuser I. Modafinil and methylphenidate for neuroenhancement in healthy individuals: A systematic review. Pharmacol Res. 2010;62:187–206. doi: 10.1016/j.phrs.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ. Nicotine amplifies reward-related dopamine signals in striatum. Nature Neuroscience. 2004;7:583–584. doi: 10.1038/nn1244. [DOI] [PubMed] [Google Scholar]
- Robinson S, Smith DM, Mizumori SJ, Palmiter RD. Firing properties of dopamine neurons in freely moving dopamine-deficient mice: effects of dopamine receptor activation and anesthesia. Proc Natl Acad Sci USA. 2004;101:13329–13334. doi: 10.1073/pnas.0405084101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossato JI, Bevilaqua LR, Izquierdo I, Medina JH, Cammarota M. Dopamine controls persistence of long-term memory storage. Science. 2009;325:1017–1020. doi: 10.1126/science.1172545. [DOI] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Sajikumar S, Frey JU. Late-associativity, synaptic tagging, and the role of dopamine during LTP and LTD. Neurobiol Learn Mem. 2004;82:12–25. doi: 10.1016/j.nlm.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Schultz W. Multiple dopamine functions at different time courses. Annu Rev Neurosci. 2007;30:259–288. doi: 10.1146/annurev.neuro.28.061604.135722. [DOI] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Smith CC, Greene RW. CNS dopamine transmission mediated by noradrenergic innervation. J Neurosci. 2012;32:6072–6080. doi: 10.1523/JNEUROSCI.6486-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoops WW, Glaser PE, Rush CR. Reinforcing, subject-rated, and physiological effects of intranasal methylphenidate in humans: a dose-response analysis. Drug Alcohol Depend. 2003;71:179–186. doi: 10.1016/s0376-8716(03)00131-5. [DOI] [PubMed] [Google Scholar]
- Swanson JM, Volkow ND. Serum and brain concentrations of methylphenidate: implications for use and abuse. Neurosci Biobehav Rev. 2003;27:615–621. doi: 10.1016/j.neubiorev.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Tang J, Dani JA. Dopamine enables in vivo synaptic plasticity associated with the addictive drug nicotine. Neuron. 2009;63:673–682. doi: 10.1016/j.neuron.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Moody TD, Makhinson M, O’Dell TJ. Activity-dependent beta-adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Magill PJ, Bolam JP. Uniform inhibition of dopamine neurons in the ventral tegmental area by aversive stimuli. Science. 2004;303:2040–2042. doi: 10.1126/science.1093360. [DOI] [PubMed] [Google Scholar]
- Urban KR, Li YC, Gao WJ. Treatment with a clinically-relevant dose of methylphenidate alters NMDA receptor composition and synaptic plasticity in the juvenile rat prefrontal cortex. Neurobiol Learn Mem. 2013;101:65–74. doi: 10.1016/j.nlm.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Ding YS, Fowler JS, Wang GJ, Logan J, Gatley JS, Dewey S, Ashby C, Liebermann J, Hitzemann R. Is methylphenidate like cocaine? Studies on their pharmacokinetics and distribution in the human brain. Arch Gen Psychiatry. 1995;52:456–463. doi: 10.1001/archpsyc.1995.03950180042006. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Swanson JM. Dopamine in drug abuse and addiction: results from imaging studies and treatment implications. Mol Psychiatry. 2004;9:557–569. doi: 10.1038/sj.mp.4001507. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Swanson JM. Variables that affect the clinical use and abuse of methylphenidate in the treatment of ADHD. Am J Psychiatry. 2003;160:1909–1918. doi: 10.1176/appi.ajp.160.11.1909. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Gatley SJ, Logan J, Ding YS, Hitzemann R, Pappas N. Dopamine transporter occupancies in the human brain induced by therapeutic doses of oral methylphenidate. Am J Psychiatry. 1998;155:1325–1331. doi: 10.1176/ajp.155.10.1325. [DOI] [PubMed] [Google Scholar]
- Weikop P, Yoshitake T, Kehr J. Differential effects of adjunctive methylphenidate and citalopram on extracellular levels of serotonin, noradrenaline and dopamine in the rat brain. European Neuropsychopharmacology. 2007;17:658–671. doi: 10.1016/j.euroneuro.2007.02.014. [DOI] [PubMed] [Google Scholar]
- Winder DG, Egli RE, Schramm NL, Matthews RT. Synaptic plasticity in drug reward circuitry. Curr Mol Med. 2002;2:667–676. doi: 10.2174/1566524023361961. [DOI] [PubMed] [Google Scholar]
- Wright FK, White KG. Effects of methylphenidate on working memory in pigeons. Cogn Affect Behav Neurosci. 2003;3:300–308. doi: 10.3758/cabn.3.4.300. [DOI] [PubMed] [Google Scholar]
- Yang K, Hu J, Lucero L, Liu Q, Zheng C, Zhen X, Jin G, Lukas RJ, Wu J. Distinctive nicotinic acetylcholine receptor functional phenotypes of rat ventral tegmental area dopaminergic neurons. J Physiol. 2009;587:345–361. doi: 10.1113/jphysiol.2008.162743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Dong Y, Doyon WM, Dani JA. Withdrawal from chronic nicotine exposure alters dopamine signaling dynamics in the nucleus accumbens. Biol Psychiatry. 2012;71:184–191. doi: 10.1016/j.biopsych.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Zhang L, Liang Y, Siapas AG, Zhou FM, Dani JA. Dopamine signaling differences in the nucleus accumbens and dorsal striatum exploited by nicotine. J Neurosci. 2009;29:4035–4043. doi: 10.1523/JNEUROSCI.0261-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang TA, Tang J, Pidoplichko VI, Dani JA. Addictive nicotine alters local circuit inhibition during the induction of in vivo hippocampal synaptic potentiation. J Neurosci. 2010;30:6443–6453. doi: 10.1523/JNEUROSCI.0458-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]