

Abstract
Purpose
This non-randomized, patient-access protocol, assessed both safety and efficacy outcomes following liposomal muramyl –tripeptide-phosphatidylethanolamine (L-MTP-PE; mifamurtide) in patients with high-risk, recurrent and/or metastatic osteosarcoma.
Methods
Patients received mifamurtide 2 mg/m2 intravenously twice-weekly ×12 weeks, then weekly ×24 weeks with and without chemotherapy. Serum concentration-time profiles were collected. Adverse events within 24 hours of drug administration were classified as infusion-related adverse events (IRAE); other AEs and overall survival (OS) were assessed.
Results
The study began therapy in January 2008; the last patient completed therapy in October 2012. 205 patients were enrolled; median age was 16.5 years and 143/204 (72%) had active disease. Mifamurtide serum concentrations declined rapidly in the first 30 minutes post-infusion, then in a loglinear manner 2–6 hours post-dose; t1/2 was 2 hours. There were no readily apparent relationships between age and BSA-normalized clearance, half-life, or pharmacodynamic effects, supporting the dose of 2 mg/m2 mifamurtide across the age range. Patients reported 3,415 IRAE after 7,122 mifamurtide infusions. These were very rarely grade 3 or 4 and most commonly included chills+fever or headache+fatigue symptom clusters. One and two year OS was 70.6% and 41.4%. Patients with initial metastatic disease or progression approximated by within 9 months of diagnosis (N=40) had similar 2-year OS (38.8%) as the entire cohort (41.4%)
Conclusions
Mifamurtide had a manageable safety profile; PK/PD of mifamurtide in this patient access study was consistent with prior studies. Two-year OS was 41.4%. A randomized clinical trial would be required to definitively determine impact on patient outcomes.
Keywords: Osteosarcoma, mifamurtide, pharmacokinetics, L-MTP-PE, macrophage activation, biologic therapy
Introduction
Successful treatment of non-metastatic and metastatic and/or recurrent osteosarcoma utilizes both very aggressive and complete local control measures (amputation, limb salvage surgery with negative margins, surgical removal of lung and/or bone metastases) as well as multi-agent chemotherapy [1–7]. Chemotherapy has dramatically improved survival from the historical 10–20% with surgery alone to the current (2013)50–70% in non-metastatic disease [8–10] and 20–35% in those with recurrent and/or metastatic disease [11–13]. For multiple-relapsed patients survival remains extremely poor (5–10%) [13–16]. Chemotherapy agents commonly used to treat osteosarcoma include doxorubicin, cisplatin, high-dose methotrexate, and ifosfamide with mesna +/− etoposide [17, 18]. These commonly used agents are considered the standard of care in osteosarcoma, despite having many serious short-term (hours-days), intermediate (weeks) and long-term toxicities including nausea/vomiting, pancytopenia, liver toxicity, cardiotoxicity [19], renal dysfunction, hearing loss [20, 21] and infertility [22]. Initial treatment of osteosarcoma takes 32–48 weeks and involves many hospitalizations and/or clinic visits for chemotherapy administration, surgery, and close monitoring of chemotherapy-related side effects such as anemia, neutropenia, thrombocytopenia, nausea and vomiting [23]. Outpatient chemotherapy for osteosarcoma is now possible; this can improve quality of life [24].
Although state-of-the art chemotherapy and surgery have resulted in similar patterns of survival after osteosarcoma diagnosis for more than 20 years [4, 25], there are some recent indications that activating the immune system can contribute to better outcome. Infections associated with limb surgery have a positive effect on survival in both dogs and humans with osteosarcoma [26, 27]. Higher absolute lymphocyte count recovery after chemotherapy also appears to be associated with improved survival [28]. Indeed, the best evidence for the immune system being involved in improving survival comes from more than 20 years of studies using a macrophage activator composed of phosphatidyl choline + phosphatidyl serine liposomes containing muramyl tripeptide phosphatidyl ethanolamine (L-MTP-PE; mifamurtide) [29–32]. Previous phase I and II trials demonstrated the biologic effectiveness of mifamurtide using an optimal biologic dose rather than a maximally tolerated dose strategy; best effects were seen if patients were treated for at least 24 weeks [33–39]. In the Intergroup trial INT-0133, 6-year overall survival improved from 70 to 78% in non-metastatic osteosarcoma patients who received mifamurtide with standard chemotherapy [30]. A separate analysis of a smaller cohort in this study who presented with metastatic disease showed the possibility of a larger treatment effect: 13% improved overall survival rate at 5 years; however, because of small numbers (N=91) this did not reach statistical significance [32]. Thus, multiple levels of evidence suggest that mifamurtide, when combined with chemotherapy, can improve osteosarcoma survival [40–42]. The drug is currently available for clinical use in Europe, Mexico, South Korea, Switzerland, and Israel for non-metastatic osteosarcoma.
After initial approval by the European Medical Agency and at the request of the US Food and Drug Administration, we initiated a access study for mifamurtide in osteosarcoma; this study was also designed to better define the pharmacokinetic/pharmacodynamics (PK/PD) and safety profile of mifamurtide in osteosarcoma. Since treatment effect seemed at least as good if not better in those patients with metastases, and we did not want to compete with the on-going multinational EURAMOS-1 study in osteosarcoma, the study population chosen for this patient access and safety trial was patients with metastatic and/or recurrent disease, i.e. a group not eligible for participation in EURAMOS-1, but for whom there was the possibility of benefit and for whom there remains an unmet need for survival improvement. This study (MTP-OS-403; clinical trials.gov NCT00631631; IND 2803) was conducted in the United States. Analysis of outcomes including survival was considered a secondary objective in this heterogeneous cohort of high-risk metastatic and/or recurrent osteosarcoma patients.
Methods
Inclusion criteria
Patients with the diagnosis of high-grade osteosarcoma with relapsed and/or recurrent disease, either locally or metastatic and age 2–50 years were eligible. Resection of recurrence or metastases was not required for study entry, but was encouraged, if possible and also acceptable to the patient. Receipt of an abbreviated chemotherapy regimen due to severe toxicity (e.g. cardiotoxicity from doxorubicin, allergic reaction to methotrexate, or renal dysfunction or severe encephalopathy not allowing treatment with ifosfamide) was also permitted at study entry in a limited number of patients. Other entry criteria included adequate renal and liver function, absence of concurrent active acute infection, and Lansky performance status of 50–100% (≤16 years old), ECOG performance status 0–2, or Karnofsky performance status 50–100% (>16 years old). Exclusion criteria included chronic use of corticosteroids or other immunosuppressive agents and non-metastatic osteosarcoma for whom standard therapy on EURAMOS-1 was possible at the time of the study.
Demographic information and informed consent
At study entry, clinical parameters such as age, gender, date of initial diagnosis, site of initial primary tumor, presence or absence of metastatic or recurrent disease, and disease burden (active or resected) were collected. All patients provided informed consent or assent at a center experienced in the administration of mifamurtide. The first dose was administered at the same center that obtained informed consent; M. D. Anderson Cancer Center was the lead center, N=100. Subsequent doses could be infused under the direction of an experienced pediatric or medical oncologist at outside centers. These centers were, however, required to obtain IRB approval for administration of the investigational agent, have an investigational drug pharmacy, and have sent a memorandum of understanding to provide data concerning infusion toxicities, drug toxicity, and patient survival. Withdrawal from study was allowed for intolerance of side effects, patient or physician discretion, or progression of disease. The study IND (2803) was held by Millennium: the Takeda Oncology Company.
Mifamurtide preparation and administration
Mifamurtide was provided by Millennium: The Takeda Oncology Company (Boston, MA) in glass vials containing 4 mg of the active biologic agent [43, 44]. These were stored in investigational pharmacy at 4°C until use. Preparation involves warming vial to room temperature over 30 minutes, then addition of 0.9% saline and gentle mixing. Dose to be administered (2 mg/m2) was withdrawn using a filter provided with the vial into a 60 cc syringe, then transferred to a minibag and infused using non-filtered tubing intravenously over 60 minutes. Prior to dose #1, all patients were provided with acetaminophen (10–15 mg/kg, max 1000 mg) and ibuprofen (200 or 400 mg) 30 minutes before infusion to reduce common initial infusion related side effects such as chills, fever, and headache. Subsequent doses were given with pre- and post-acetaminophen and/or ibuprofen to control side effects on an as-needed basis. About 75% of patients could be successfully treated with mifamurtide without any pre- and/or post-medication for subsequent doses. Schedule of mifamurtide was twice/week (not on consecutive days) × 12 weeks then weekly for 24 weeks for a total of 48 doses.
Analysis of pharmacokinetics and pharmacodynamics
Blood samples were collected up to 72 hours following the first infusion. Serum MTP-PE concentrations were measured by liquid chromatography with tandem mass spectrometry (LC-MS/MS) using previously described bioanalytical methodology by City of Hope Analytical Pharmacology Core Facility, City of Hope Medical Center, Duarte, CA [45]. The lower limit of quantification was 0.1 nM. TNF-alpha and IL-6 levels were measured by sandwich immunoassay (Viracor-IBT Laboratoriesm Lenexa, KS). The lower limit of quantitation was 0.6 pg/mL for TNF-alpha and 2.4 pg/mL for IL-6. PK and PD data were analyzed by noncompartmental analysis using WinNonlin. PK parameters determined included maximal concentration (Cmax), area under concentration-time curve from time zero to last quantifiable concentration (AUC0-tlast), AUC extrapolated to infinity (AUCinf), clearance (CL), steady state volume of distribution (Vss), terminal phase volume of distribution (Vt), and terminal half-life (t1/2). CL, Vss, and Vt were also normalized by body surface area (BSA) to reflect the BSA–normalized dosing of 2 mg/m2 mifamurtide. PK parameters were descriptively summarized and exploratory graphical assessments of potential relationships between key parameters (e.g. BSA-normalized CL, t1/2) and age were performed. The endpoints used for the PD analysis were the change in concentrations of the cytokines TNF-alpha and IL-6 from time zero to the last quantifiable value of PD effect (AUEC0-tlast), maximum effect (Emax), and time of first occurrence of maximal effect (TEmax). PD parameters were descriptively summarized and exploratory graphical assessments of potential relationships between key parameters (Emax, AUC0-tlast) and age were performed.
Analysis of Infusion Related Adverse Events (IRAE) and Adverse Events (AE)
Very early in the study it became apparent that IRAE, if present, were almost always mild (grade 1 or 2), self-limited and completely gone within 24 hours of infusion. Yet, then quantity and quality of data in the medical record underestimated IRAE. Therefore to more accurately collect data, a 2 page self-report check-list with side effects of fever, chills, tured (fatigue), shortness of breath, bodyaches, pain, nausea, and vomiting and “other” was used to capture not only common IRAE, but also other toxicities with a “fill-in-the blank” option. The form also provided the opportunity to inform the research team about the use of pre- and post-medications such as ibuprofen, acetaminophen and meperidine for treatment of chills. Non-IRAE of grade ≥3 were captured on case report forms as adverse events and verified by monitoring of the medical records.
Other Therapy and Survival Outcomes
Concomitant therapy with chemotherapy agents as well as local control measures (e.g. surgery, radiotherapy) was permitted while on study and after study completion. Agents most commonly used in order of frequency were ifosfamide/mesna +/− etoposide, gemcitabine +/− docetaxel, methotrexate, doxorubicin and cisplatin.
Survival data were collected during treatment and then approximately every three months after treatment or according to institutional norms. Overall survival was calculated from date of enrollment, using Kaplan-Meier estimates. Overall survival was analyzed for patients who were treated ≤9 months or >9 months after diagnosis to provide an approximate means of categorizing patients into primary metastatic or metastatic recurrent disease, respectively.
Results
Demographic and baseline clinical characteristics of this high-risk, metastatic and/or recurrent osteosarcoma cohort are summarized in table I. Most patients were in the typical adolescent+ young adult age range. Importantly, only 29% had recurrent and/or metastatic disease completely resected prior to study entry.
Table I.
Patient Demographics and Baseline Characteristics (Total Population N=205)
| Characteristic | N (% of total) |
|---|---|
| Median age, years (range) | 16.5 (5–51) |
| Male, N (%) | 127 (62%) |
| Caucasian/Hispanic/Black/Asian/other (N) | 149/32/14/6/3 |
| Disease status at study entry | |
| No active disease after surgery, N (%) | 57 (29%) |
| Active disease after surgery, N (%) | 143 (72%) |
| Local Relapse, N (%) | 9 (6%) |
| Metastatic, N (%) | 132 (64%) |
| Pulmonary | 107 (52%) |
| Bone | 9 (4%) |
| Other | 21 (10%) |
| Indicator lesions, N (%) | 79 (39%) |
| Lesion site, N (%) | |
| Primary tumor | 14 (8) |
| Lung metastases | 72 (35%) |
| Other (e.g. bone or soft tissue metastases) | 11 (5%) |
| Median time from initial osteosarcoma diagnosis to first mifamurtide dose, months (range) | 24 (1–136) |
Mifamurtide treatment exposure
The median total amount of mifamurtide received was 123.7 mg and the median number of mifamurtide doses received was 40.5 (range 1–50). 85/204 patients with infusion data completed the study. One hundred nineteen patients did not complete the study for reasons including disease progression (N=74), patient/investigator discretion (N=18), serious illness (N=5) or death (N=3). 83% had concurrent chemotherapy.
Pharmacokinetic and Pharmacodynamic (PK/PD) Analysis
Demographic characteristics of the PK (N=28) and PD (N=27) were very similar to the entire cohort. The PK/PD versus entire cohort had median (range) age 15 (6–39) versus 15 (6–42) years and body surface area (BSA) 1.58 (0.77–2.31) versus 1.55 (0.77–2.24) m2 and 61% versus 56% were male. Table II summarizes PK parameters of mifamurtide; Figure 1 shows the concentration-time profile for mifamurtide after 30 and 60 minute infusions. Data from 28 patients were included in the PK (17/11 had 30-/60-min infusions) and 27 in the PD (13/14 had 30-/60-min infusions) analyses.
Table II.
Summary of Pharmacokinetic Parameters for Mifamurtide*
| Parameter | Mean | % CV |
|---|---|---|
| aAUC(0-inf) (hr.nmol/L) | 21.6 | 34.9 |
| bt ½ (hr) | 2.04 | 22.3 |
| cCL (mL/min) | 1770 | 38.1 |
| cCL (ml/min/m2) | 1250 | 42.5 |
| dVss (L) | 287 | 45.4 |
| dVss (L/m2) | 211 | 54.5 |
| eVz (L) | 336 | 35.0 |
| eVz(L/m2) | 239 | 37.1 |
N=15 except t1/2 N=25.
AUC(0-inf): Area under the concentration-time curve from time zero extrapolated to infinity;
t1/2: terminal half-life;
CL clearance
Vss: volume of distribution at steady state;
Vz: terminal phase volume of distribution
Figure 1.
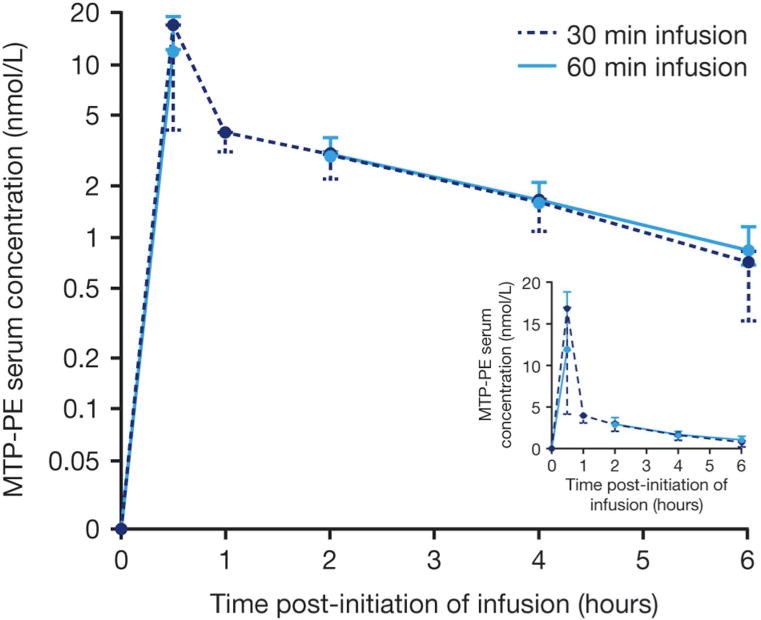
Mean mifamurtide (Standard deviation) serum concentration versus time profiles on a log-linear and linear (inset) scale.
Following an initial rapid decline in mifamurtide serum concentrations during the first 30 min after infusion cessation, mifamurtide serum concentrations declined in a log-linear manner over 2–6 hours post-dose with a mean (%CV) terminal half-life of 2.04 hours (22%). BSA-normalized geometric mean (%CV) BSA-normalized clearance was 1,250 mL/min/m2 (43%) and BSA-normalized steady-state volume of distribution was 262 L/m2 (45%). PK parameters are summarized in Table II. Serum IL-6 levels peaked at a median of 4 hours (regardless of infusion duration) and TNF-alpha peaked at a median of 2 hours in the 30-minute and 4 hours in the 60-minute group, returning to baseline ~24 hours post dose. PD parameters are summarized in table III. No readily apparent relationships were observed between age and BSA-normalized mifamurtide clearance, half-life, or effects on TNF-alpha and IL-6.
Table III.
Pharmacodynamic Parameters for TNF-alpha and IL-6 after Mifamurtide Infusion
| Marker | Mifamurtide Infusion duration, min | aMean (%CV) bEmax (pg/mL) | Mean (%CV) cAUEC0-tlast (hr.pg/ml) | Median (range) TEmax (hr) |
|---|---|---|---|---|
| dTNF-alpha | 60 | 412 (105) | 2460 (82.1) | 4.00 (2.00–4.00) |
| 30 | 582 (96.9) | 2260 (86.0) | 2.05 (1.00–4.00) | |
| eIL-6 | 60 | 895 (94.1) | 4650 (177) | 4.00 (2.00–4.08) |
| 30 | 811 (112.0) | 2200 (174) | 4.00 (2.00–4.08) |
%CV: coefficient of variation;
Emax: maximum pharmacodynamic effect;
AUEC0-tlast: area under the effect the time curve from time 0 to time of last quantifiable effect;
TNF-alpha: tumor necrosis factor alpha;
IL-6: interleukin-6
Infusion related adverse events (IRAE)
Using self-reporting diaries after dosing, patients reported very rare grade 3 or 4 IRAE. In total, 190 of 204 (93%) patients reported any IRAE; 3,415 IRAEs were identified after 7,122 infusions (48%). These were generally grade 1 or 2 and were self-limited and confined to 1 or 2 common symptom clusters (e.g. chills + fever or headache + fatigue). One patient reported grade 1 rash which was related to mifamurtide treatment that resulted in stopping therapy. Figure 2 (top) illustrates the very low frequency and the low intensity of common IRAEs associated with mifamurtide. Although chills + fever and headaches+ fatigue were the most common IRAE symptom clusters, with the use of pre- and/or post-medications these were reported to be tolerable. Figure 2 (bottom) shows pattern of IRAE related to dose 1–12 and 13–24 (2×/week) and 25–36 and 37–50 (weekly). Although more IRAE seemed to occur early in treatment, even with pre-/post-medication, mild IRAE continued in some patients, although not at a level of severity or discomfort requiring active intervention or stopping of mifamurtide. Indeed, some patients with recurring grade 1 symptoms refused to take additional pre-/post-medication.
Figure 2.
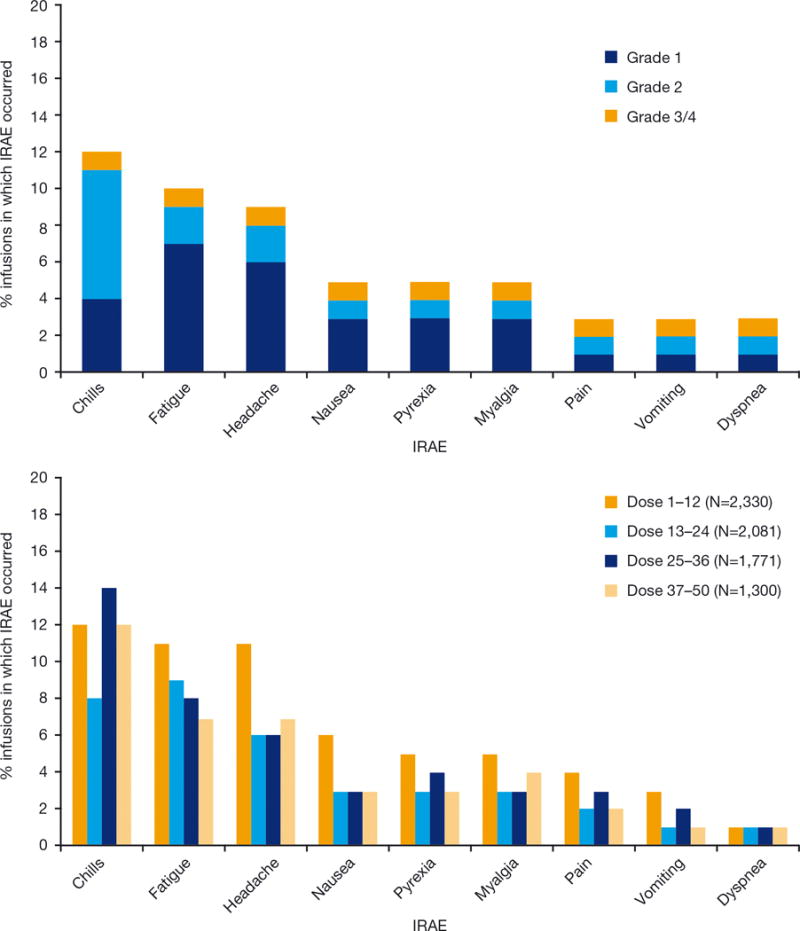
(Top) Very low incidence of grade 3 or 4 infusion related adverse events (IRAE) after mifamurtide. The grade of IRAE according was assessed using NCI-CTCAE v3.0. The graph depicts only IRAE with ≥1% occurrence. Percentages for the number of events was calculated using the total number of infusions (7,122) as a denominator. (Bottom) Except for chills, fewer IRAE were observed with weekly dosing. Number of IRAE observed by dosing period for IRAE with ≥1% total occurrence. Percentages for the number of events were calculated using the total number of infusions in each dosing period as a denominator. Dose 1–12 and 13–24 were twice/week (e.g. M/Th) while doses 25–36 and 37–48 were weekly.
Adverse events
This cohort with high-risk, locally recurrent and/or metastatic disease was also often treated with chemotherapy; 144 of 204 (71%) patients experienced at least one grade 3–5 AE, including 20 (10%) who had mifamurtide-related AEs other than IRAEs. AEs leading to study drug discontinuation, including pyrexia in two patients and respiratory distress in two patients. The SAEs leading to study drug discontinuation, which were (by patient): grade 5 acute respiratory distress (from gemcitabine toxicity, then progression in the lungs), grade 5 hypoxia (same patient), grade 4 malignant pleural effusion (related to renal dysfunction); grade 3 pyrexia; grade 4 pericardial effusion (drug discontinued and resolved in 1 week); and grade 3 respiratory distress. 98/204 (48%) patients reported serious AEs (SAEs) during treatment, including febrile neutropenia (18%), thrombocytopenia (5%), neutropenia (5%), and pyrexia (4%). Only twelve patients (6%) reported a SAE that was considered related to mifamurtide. There were 10 on-study deaths from progressive disease; none were considered related to mifamurtide treatment. Mifamurtide was associated with very few AE compared with those attributed to chemotherapy or concomitant illness.
Survival
Survival was analyzed using the Kaplan-Meier method using all 205 patients entered on the study regardless of how many doses of drug were administered. One patient was enrolled but received no drug because of rapid progression. Median patient follow-up was 595 days (20 months). Clinical characteristics of the cohort impacting overall survival including treatment within 9 months of presentation or not, and active disease at time of study enrollment, and completion of the intended 48 doses or not for patients enrolled more than 9 months from presentation, are depicted in Figure 3.
Figure 3.
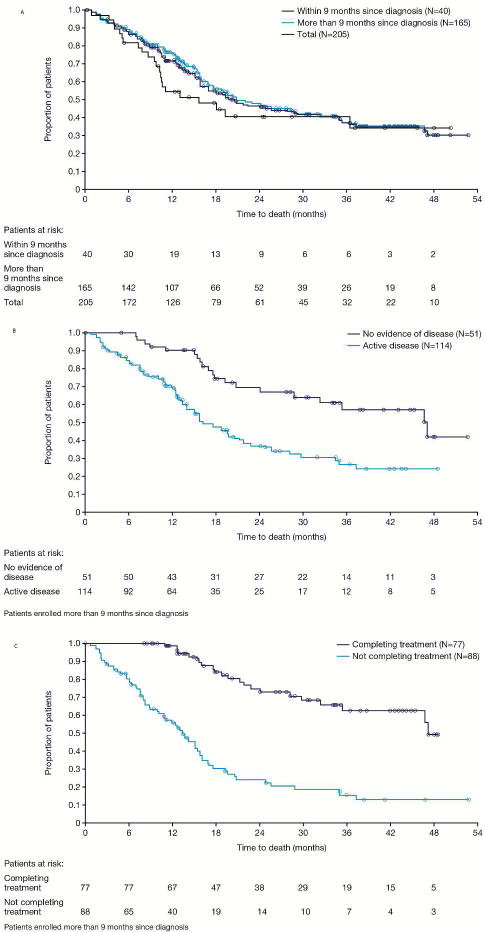
Overall survival (OS) of high-risk, metastatic and/or recurrent osteosarcoma patients after treatment with mifamurtide 2 mg/m2/dose. A: The OS curves for the total population, patients who entered the study within 9 months of diagnosis and those who entered later than 9 months. B: For patients who entered the study later than 9 months from diagnosis, there was superior OS for patients with no evidence of disease at time of study entry versus those with active disease. C: For patients who entered the study later than 9 months from diagnosis, there was superior OS for patients who completed recommended 48 doses of mifamurtide (9 months of therapy) versus those who had <48 doses
Discussion
Surgery remains the cornerstone of successful osteosarcoma therapy in both non-metastatic and metastatic patients [6, 14]. Attempts to provide chemotherapy only when measurable disease is present results in progression and need for surgery in about 90% of cases [7]. In the recurrent, metastatic setting, powering a study with enough patients to conduct a randomized phase II or phase III trial to have a positive effect >10% in this rare bone sarcoma is difficult. This was shown recently when aerosol GM-CSF was tested against resectable pulmonary metastases at first relapse in a Children’s Oncology Group clinical trial – accrual took >5 years for this very defined cohort [46]. Nevertheless, because approximately 30–50% of patients with osteosarcoma will eventually develop metastases and die of disease [4, 25, 29, 47], there remains an unmet need for better therapy to prevent and/or treat lung metastases. Since initial osteosarcoma chemotherapy is so toxic initially, the addition of a safe and effective biologic agent with non-overlapping and no apparent long-term toxicity should be welcomed, not only for initial adjuvant treatment but also in the recurrent situation – if the treatment effect is at least as large as in the adjuvant setting against non-metastatic disease.
The long-term safety of mifamurtide is apparent from 20 years of follow-up in patients treated on phase I and II studies and >10 years of follow-up of the randomized INT-0133 phase III study [29, 30]. Our patient access study treated greater than 200 patients and enabled us to better define incidence and severity of mifamurtide infusion-related toxicities and AEs associated with the drug. Since we obtained self-report data for infusions, the data quality for the common and uncommon grade 1 or grade 2 infusion-related side effects, is more comprehensive than INT0133. We also found that grade ≥3 AEs attributed to mifamurtide were far less common than those attributed to chemotherapy or disease progression, demonstrating a superior toxicity profile versus chemotherapy. Nevertheless in our large cohort, very rare side effects potentially related to mifamurtide such as pericarditis or pleural effusions were seen in 2% of patients. Thus, we recommend echocardiogram and/or chest x-ray investigation(s) if uncommon symptoms (e.g. chest pain, shortness of breath) occur while.
IRAE appeared to be well tolerated; mifamurtide-associated IRAEs were usually controllable with pre-or post-acetaminophen +/−ibuprofen. Occasional patients also received meperidine for chills. Most patients could also be successfully weaned off these pre-/post-medications to try to maximize biologic effectiveness of mifamurtide while having a good quality of life on the drug. Flow diagrams concerning recommended use of pre- and post-medications have been recently published [40, 42] as well as a review of current status of mifamurtide [48].
Compared with chemotherapy, mifamurtide has a unique mode of action in that it modulates the immune system to indirectly effect cytotoxicity rather than having a direct cytotoxic effect on tumor cells [29, 38, 39, 49]. Our study showed t1/2 of the drug was 2 hours. The peak PD effect as measured by IL-6 and TNF-alpha occurred 4 hours after infusion. This would account for the observation that IRAE are not always immediate, but can sometimes occur hours after an infusion. During our study we learned to use a self-report “diary”/questionnaire to obtain data prior to next infusion for effective education and guidance concerning pre-/post-medication. Without the self-report tool, patients, caregivers, doctors, and nurses were sometimes unable to accurately define or recall grade of IRAE that occurred with prior infusions several days or a week earlier and whether tapering off pre-/post-medications or additional therapy was warranted (or not).
Mifamurtide has been extensively tested in phase I, II, and III studies, which highlighted key concepts that underlie the effective use of this drug in osteosarcoma. These include: a) dosing with an optimal biologic dose of 2 mg/m2 rather than the maximally tolerated dose of 6 mg/m2 [40], b) administration of at least 24 weeks of therapy [35], c) use in a minimal residual disease setting [40], and d) the understanding that the drug has similar biologic effectiveness whether given with or without concomitant chemotherapy [34–37, 39, 40, 42]. Updated results for patients with non-metastatic and resectable disease in the INT-0133 study, which led to EMA approval, showed a relative reduction of 28% in the risk of death (p=0.0313, HR = 0.72 [95% CI: 0.53,0.97] with the addition of mifamurtide compared to chemotherapy alone[30]. Similar survival benefit was seen in the metastatic cohort of INT-0133 (HR: 0.72 [95%CI: 0.4–1.3]), although it didn’t reach statistical significance due to the small sample size (N=91; 46 with mifamurtide versus 45 without) [32]. In the present study, therefore, we analyzed patient survival consequent to the use of mifamurtide. Among 50 patients whose disease was completely resected, >50% remained alive >2 years from study entry. Many of these patients were treated at second or subsequent recurrence after treatment with ≥two lines of therapy.
In our study, the PK/PD properties of mifamurtide observed in a largely pediatric and adolescent population are similar to those previously reported in healthy adults [45]. Importantly, there were no readily apparent effects of age on BSA-normalized mifamurtide clearance and the immune modulatory and PD effects. These results support the use of mifamurtide at the current recommended dose of 2 mg/m2 across the age range relevant to its indication in the treatment of osteosarcoma. Mifamurtide is currently approved for use in non-metastatic osteosarcoma in Europe, Mexico, South Korea, Switzerland, and Israel.
Since our study began as a patient access study, which also served to better define toxicities, especially IRAE, the study was not designed to look only at subsets of resectable patients, but also included many non-resectable patients with a substantial burden of disease. We learned that some of these patients could tolerate both radiotherapy and radiosensitizing chemotherapy [50] concomitantly with mifamurtide without any difficulty. In summary our study has provided an increased and accurate body of data from >200 osteosarcoma patients which a) assisted rational understanding of mifamurtide PK/PD parameters, b) better defined incidence, severity, and control of IRAE associated with mifamurtide, and c) has shown “better than expected” overall survival similar to first relapse [16] and possibly better than historical comparison as reported in the literature [14]. Information provided by our study should be useful for pharmacists, regulatory agencies, physicians, nurses, and other caregivers, as well for patient education, as it relates to both current and future mifamurtide indications, but also mifamurtide risks and benefits during osteosarcoma therapy.
Acknowledgments
We wish to acknowledge Dr. Timothy Synld’s laboratory at City of Hope analytical Pharmacology core facility for bioanalysis of mifamurtide concentrations in serum samples as well as Dr. Bruce Green and Dr. Diane R. Mould for their contributions to the PK and PD data analyses. We appreciate the skilled infusion nurses at the M. D. Anderson Cancer Center and Memorial Sloan Kettering Cancer Center who gave the majority of infusions, data analysts who collected IRAE, SAE, and survival data, investigational pharmacies at the major and referral centers for superlative drug accountability, and, of course, the patients who received multiple infusions and provided self-report data to improve our understanding and knowledge of the infusion-related side effects. Dr. Anderson also acknowledges the support of friends and family of Lauren Behr (Sarcoma Research Fund), Haynie Spirit Fund, Sarah’s Garden of Hope, the Curtis family (Distinguished Professor Endowment for Pediatric Cancer Research), and the Wilkes Family for osteosarcoma research funds.
Dr. Meyers and Anderson received no salary but had research support from Millennium: The Takeda Oncology Company for study related data collection and monitoring activities related to IND 2803.
Footnotes
Conflicts of interest statement: Drs. Ventatarkrishnan who performed the PK analysis and B. Wang and Y. Liu who did the statistical analyses and C. Oliva (medical director) are employees of Millennium or Takeda which manufactures and distributes mifamurtide. The clinical oncologists involved in the study (Drs. Anderson, Kleinerman, Hughes, Herzog, Huh, Sutphin, Vyas, Shen, Warwick, Yeager, and Chou) have no conflicts of interest.
References
- 1.Ferguson WS, Goorin AM. Current treatment of osteosarcoma. Cancer Invest. 2001;19(3):292–315. doi: 10.1081/cnv-100102557. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson WS, et al. Presurgical window of carboplatin and surgery and multidrug chemotherapy for the treatment of newly diagnosed metastatic or unresectable osteosarcoma: Pediatric Oncology Group Trial. J Pediatr Hematol Oncol. 2001;23(6):340–8. doi: 10.1097/00043426-200108000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Harris MB, et al. Treatment of metastatic osteosarcoma at diagnosis: a Pediatric Oncology Group Study. J Clin Oncol. 1998;16(11):3641–8. doi: 10.1200/JCO.1998.16.11.3641. [DOI] [PubMed] [Google Scholar]
- 4.Whelan JS, et al. Survival from high-grade localised extremity osteosarcoma: combined results and prognostic factors from three European Osteosarcoma Intergroup randomised controlled trials. Ann Oncol. 2012;23(6):1607–16. doi: 10.1093/annonc/mdr491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bacci G, et al. Preoperative therapy versus immediate surgery in nonmetastatic osteosarcoma. J Clin Oncol. 2003;21(24):4662–3. doi: 10.1200/JCO.2003.99.157. [DOI] [PubMed] [Google Scholar]
- 6.Bielack SS, et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol. 2002;20(3):776–90. doi: 10.1200/JCO.2002.20.3.776. [DOI] [PubMed] [Google Scholar]
- 7.Jaffe N, et al. Can cure in patients with osteosarcoma be achieved exclusively with chemotherapy and abrogation of surgery? Cancer. 2002;95(10):2202–10. doi: 10.1002/cncr.10944. [DOI] [PubMed] [Google Scholar]
- 8.Link MP, et al. Adjuvant chemotherapy of high-grade osteosarcoma of the extremity. Updated results of the Multi-Institutional Osteosarcoma Study. Clin Orthop Relat Res. 1991;(270):8–14. [PubMed] [Google Scholar]
- 9.Link MP, et al. The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med. 1986;314(25):1600–6. doi: 10.1056/NEJM198606193142502. [DOI] [PubMed] [Google Scholar]
- 10.Carrle D, Bielack SS. Current strategies of chemotherapy in osteosarcoma. Int Orthop. 2006;30(6):445–51. doi: 10.1007/s00264-006-0192-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goorin AM, et al. Phase II/III trial of etoposide and high-dose ifosfamide in newly diagnosed metastatic osteosarcoma: a pediatric oncology group trial. J Clin Oncol. 2002;20(2):426–33. doi: 10.1200/JCO.2002.20.2.426. [DOI] [PubMed] [Google Scholar]
- 12.Mialou V, et al. Metastatic osteosarcoma at diagnosis: prognostic factors and long-term outcome–the French pediatric experience. Cancer. 2005;104(5):1100–9. doi: 10.1002/cncr.21263. [DOI] [PubMed] [Google Scholar]
- 13.Crompton BD, et al. Survival after recurrence of osteosarcoma: A 20-year experience at a single institution. Pediatr Blood Cancer. 2005 doi: 10.1002/pbc.20580. [DOI] [PubMed] [Google Scholar]
- 14.Bielack SS, et al. Second and subsequent recurrences of osteosarcoma: presentation, treatment, and outcomes of 249 consecutive cooperative osteosarcoma study group patients. J Clin Oncol. 2009;27(4):557–65. doi: 10.1200/JCO.2008.16.2305. [DOI] [PubMed] [Google Scholar]
- 15.O’Day K, Gorlick R. Novel therapeutic agents for osteosarcoma. Expert Rev Anticancer Ther. 2009;9(4):511–23. doi: 10.1586/era.09.7. [DOI] [PubMed] [Google Scholar]
- 16.Kempf-Bielack B, et al. Osteosarcoma relapse after combined modality therapy: an analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS) J Clin Oncol. 2005;23(3):559–68. doi: 10.1200/JCO.2005.04.063. [DOI] [PubMed] [Google Scholar]
- 17.Ferrari S, et al. Neoadjuvant chemotherapy with methotrexate, cisplatin, and doxorubicin with or without ifosfamide in nonmetastatic osteosarcoma of the extremity: an Italian sarcoma group trial ISG/OS-1. J Clin Oncol. 2012;30(17):2112–8. doi: 10.1200/JCO.2011.38.4420. [DOI] [PubMed] [Google Scholar]
- 18.Meazza C, et al. Prolonged 14-day continuous infusion of high-dose ifosfamide with an external portable pump: feasibility and efficacy in refractory pediatric sarcoma. Pediatr Blood Cancer. 2010;55(4):617–20. doi: 10.1002/pbc.22596. [DOI] [PubMed] [Google Scholar]
- 19.Huh WW, et al. Comparison of doxorubicin cardiotoxicity in pediatric sarcoma patients when given with dexrazoxane versus as continuous infusion. Pediatr Hematol Oncol. 2010;27(7):546–57. doi: 10.3109/08880018.2010.503335. [DOI] [PubMed] [Google Scholar]
- 20.Janeway KA, Grier HE. Sequelae of osteosarcoma medical therapy: a review of rare acute toxicities and late effects. Lancet Oncol. 2010;11(7):670–8. doi: 10.1016/S1470-2045(10)70062-0. [DOI] [PubMed] [Google Scholar]
- 21.Lewis MJ, et al. Ototoxicity in children treated for osteosarcoma. Pediatr Blood Cancer. 2009;52(3):387–91. doi: 10.1002/pbc.21875. [DOI] [PubMed] [Google Scholar]
- 22.Longhi A, et al. Fertility in male patients treated with neoadjuvant chemotherapy for osteosarcoma. J Pediatr Hematol Oncol. 2003;25(4):292–6. doi: 10.1097/00043426-200304000-00005. [DOI] [PubMed] [Google Scholar]
- 23.Anderson P, Salazar-Abshire M. Improving outcomes in difficult bone cancers using multimodality therapy, including radiation: physician and nursing perspectives. Curr Oncol Rep. 2006;8(6):415–22. doi: 10.1007/s11912-006-0069-6. [DOI] [PubMed] [Google Scholar]
- 24.Anderson PM, et al. Outpatient chemotherapy, family-centered care, electronic information, and education in adolescents adn young adults with osteosarcoma. Clinical Oncology in Adolescents adn Young Adults. 2013;3:1–11. [Google Scholar]
- 25.Smith MA, et al. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol. 2010;28(15):2625–34. doi: 10.1200/JCO.2009.27.0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lascelles BD, et al. Improved survival associated with postoperative wound infection in dogs treated with limb-salvage surgery for osteosarcoma. Ann Surg Oncol. 2005;12(12):1073–83. doi: 10.1245/ASO.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 27.Jeys LM, et al. Post operative infection and increased survival in osteosarcoma patients: are they associated? Ann Surg Oncol. 2007;14(10):2887–95. doi: 10.1245/s10434-007-9483-8. [DOI] [PubMed] [Google Scholar]
- 28.Moore C, et al. Prognostic significance of early lymphocyte recovery in pediatric osteosarcoma. Pediatr Blood Cancer. 2010;55(6):1096–102. doi: 10.1002/pbc.22673. [DOI] [PubMed] [Google Scholar]
- 29.Meyers PA. Muramyl tripeptide (mifamurtide) for the treatment of osteosarcoma. Expert Rev Anticancer Ther. 2009;9(8):1035–49. doi: 10.1586/era.09.69. [DOI] [PubMed] [Google Scholar]
- 30.Meyers PA, et al. Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival–a report from the Children’s Oncology Group. J Clin Oncol. 2008;26(4):633–8. doi: 10.1200/JCO.2008.14.0095. [DOI] [PubMed] [Google Scholar]
- 31.Chou AJ, et al. Treatment of osteosarcoma at first recurrence after contemporary therapy: the Memorial Sloan-Kettering Cancer Center experience. Cancer. 2005;104(10):2214–21. doi: 10.1002/cncr.21417. [DOI] [PubMed] [Google Scholar]
- 32.Chou AJ, et al. Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma: a report from the Children’s Oncology Group. Cancer. 2009;115(22):5339–48. doi: 10.1002/cncr.24566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kleinerman ES. Biologic therapy for osteosarcoma using liposome-encapsulated muramyl tripeptide. Hematol Oncol Clin North Am. 1995;9(4):927–38. [PubMed] [Google Scholar]
- 34.Kleinerman ES, et al. Activation of tumoricidal properties in human blood monocytes by liposomes containing lipophilic muramyl tripeptide. Cancer Res. 1983;43(5):2010–4. [PubMed] [Google Scholar]
- 35.Kleinerman ES, et al. Efficacy of liposomal muramyl tripeptide (CGP 19835A) in the treatment of relapsed osteosarcoma. Am J Clin Oncol. 1995;18(2):93–9. doi: 10.1097/00000421-199504000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Kleinerman ES, et al. Phase II study of liposomal muramyl tripeptide in osteosarcoma: the cytokine cascade and monocyte activation following administration. J Clin Oncol. 1992;10(8):1310–6. doi: 10.1200/JCO.1992.10.8.1310. [DOI] [PubMed] [Google Scholar]
- 37.Kleinerman ES, et al. Combination therapy with ifosfamide and liposome-encapsulated muramyl tripeptide: tolerability, toxicity, and immune stimulation. J Immunother Emphasis Tumor Immunol. 1995;17(3):181–93. doi: 10.1097/00002371-199504000-00007. [DOI] [PubMed] [Google Scholar]
- 38.Kleinerman ES, et al. Activation of tumoricidal properties in monocytes from cancer patients following intravenous administration of liposomes containing muramyl tripeptide phosphatidylethanolamine. Cancer Res. 1989;49(16):4665–70. [PubMed] [Google Scholar]
- 39.Kleinerman ES, et al. Unique histological changes in lung metastases of osteosarcoma patients following therapy with liposomal muramyl tripeptide (CGP 19835A lipid) Cancer Immunol Immunother. 1992;34(4):211–20. doi: 10.1007/BF01741788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson PM, Tomaras M, McConnell K. Mifamurtide in osteosarcoma–a practical review. Drugs Today (Barc) 2010;46(5):327–37. doi: 10.1358/dot.2010.46.5.1500076. [DOI] [PubMed] [Google Scholar]
- 41.Frampton JE. Mifamurtide: a review of its use in the treatment of osteosarcoma. Paediatr Drugs. 2010;12(3):141–53. doi: 10.2165/11204910-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 42.Huh WW, Egas-Bejar D, Anderson PM. New Treatment options for non-metastatic osteosarcoma: focus on mifamurtide in adolescents. Clinical Oncology in Adolescents and Young Adults. 2012;2:1–6. [Google Scholar]
- 43.Anderson P. Liposomal muramyl tripeptide phosphatidyl ethanolamine: ifosfamide-containing chemotherapy in osteosarcoma. Future Oncol. 2006;2(3):333–43. doi: 10.2217/14796694.2.3.333. [DOI] [PubMed] [Google Scholar]
- 44.Takeda, MEPACT Summary of Product Characteristics. 2013 http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000802/WC500026565.pdf.
- 45.Venkatakrishnan K, et al. A pharmacokinetic, pharmacodynamic, and electrocardiographic study of liposomal mifamurtide (L-MTP-PE) in healthy adult volunteers. Eur J Clin Pharmacol. 2012;68(10):1347–55. doi: 10.1007/s00228-012-1262-1. [DOI] [PubMed] [Google Scholar]
- 46.Arndt CA, et al. Inhaled granulocyte-macrophage colony stimulating factor for first pulmonary recurrence of osteosarcoma: effects on disease-free survival and immunomodulation. a report from the Children’s Oncology Group. Clin Cancer Res. 2010;16(15):4024–30. doi: 10.1158/1078-0432.CCR-10-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer. 2009;115(7):1531–43. doi: 10.1002/cncr.24121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ando K, et al. Mifamurtide for the treatment of nonmetastatic osteosarcoma. Expert Opin Pharmacother. 2011;12(2):285–92. doi: 10.1517/14656566.2011.543129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asano T, Kleinerman ES. Liposome-encapsulated MTP-PE: a novel biologic agent for cancer therapy. J Immunother. 1993;14(4):286–92. doi: 10.1097/00002371-199311000-00006. [DOI] [PubMed] [Google Scholar]
- 50.Anderson P, et al. Outpatient chemotherapy plus radiotherapy in sarcomas: improving cancer control with radiosensitizing agents. Cancer Control. 2008;15(1):38–46. doi: 10.1177/107327480801500105. [DOI] [PubMed] [Google Scholar]