

Abstract
Sphingosine kinase (SphK) 1 and 2 phosphorylate sphingosine to generate sphingosine-1-phosphate (S1P), a pluripotent lipophilic mediator implicated in a variety of cellular events. Here we show that the activity of β-site APP cleaving enzyme-1 (BACE1), the rate-limiting enzyme for amyloid-β peptide (Aβ) production, is modulated by S1P in mouse neurons. Treatment by SphK inhibitor, RNA interference knockdown of SphK, or overexpression of S1P degrading enzymes decreased BACE1 activity, which reduced Aβ production. S1P specifically bound to full-length BACE1 and increased its proteolytic activity, suggesting that cellular S1P directly modulates BACE1 activity. Notably, the relative activity of SphK2 was upregulated in the brains of patients with Alzheimer's disease. The unique modulatory effect of cellular S1P on BACE1 activity is a novel potential therapeutic target for Alzheimer's disease.
Introduction
Amyloid-β peptide (Aβ) is the major component of senile plaques deposited in the brains of patients with Alzheimer's disease (AD). Several lines of evidence suggest that the accumulation of Aβ is linked to the pathogenesis of AD (Tomita, 2009; De Strooper et al., 2010). Aβ is derived from amyloid-β precursor protein (APP) that is sequentially cleaved by two aspartate proteases, β- and γ-secretases. The major β-secretase is a type-1 transmembrane protein termed BACE1 (β-site APP cleaving enzyme 1) (Vassar et al., 2009). BACE1-deficient mice do not generate Aβ (Cai et al., 2001; Luo et al., 2001), but they exhibited hypomyelination (Hu et al., 2006; Willem et al., 2006) and altered neurological phenotype (Laird et al., 2005; Savonenko et al., 2008; Hu et al., 2010). However, modest reduction of BACE1 activity is sufficient for a significant reduction in brain Aβ deposition in AD model mice (McConlogue et al., 2007; Chow et al., 2010). Moreover, several reports indicate that the protein levels and/or the activity of BACE1 were increased in the brains of patients with sporadic AD (Fukumoto et al., 2002; Yang et al., 2003; Li et al., 2004; Ahmed et a., 2010), suggesting that subtle changes in BACE1 activity significantly impact on the pathomechanism of AD. BACE1 resides in the lipid raft, a membrane microdomain enriched in cholesterol and sphingolipids, and a significant role of lipids and microdomain is implicated in the regulation of the β-cleavage (Kalvodova et al., 2005; Rajendran et al., 2008; Vetrivel and Thinakaran, 2010). In this study, we focused on a biologically active lipid metabolite, sphingosine-1-phosphate (S1P). S1P functions as a ligand for G-protein-coupled receptor (GPCR)-type receptors from the extracellular side; alternatively, S1P has been shown to directly act on intracellular targets (Alvarez et al., 2007; Takabe et al., 2008; Pyne and Pyne, 2010). S1P is produced by phosphorylation of sphingosine by two related rate-limiting kinases, sphingosine kinase 1 (SphK1) and SphK2 (see Fig. 1A). Although SphK1 and SphK2 show different kinetic properties and tissue expression patterns (Blondeau et al., 2007; Spiegel and Milstien, 2007), both kinases are functionally redundant in the production of S1P in vivo (Mizugishi et al., 2005). Here, we show that modulation of SphK and S1P degrading enzymes alters the Aβ generation by regulating the β-cleavage via direct action of S1P on BACE1 protein. Furthermore, we found that SphK2 activity is increased in the brains of patients with sporadic AD. These data unveil a novel regulatory mechanism of BACE1 linked to S1P levels in neurons, supporting the view that SphK2/S1P is a novel potential therapeutic target for AD.
Figure 1.

Sphingosine kinase inhibitors decreased the Aβ secretion from neuronal cells. A, Schematic depiction of synthetic pathway for S1P. Five conserved regions of SphK are indicated by rectangles. Location of catalytic region is also indicated in this diagram. B–E, Effects of SKI on secretion of Aβ40 and Aβ42 from neuronal cells. The levels of secreted Aβ in conditioned media were quantified by ELISAs. Mean ± SEM percentages of the relative ratio of secreted Aβ to levels in untreated control are indicated. *p < 0.05, ***p < 0.001 by Student's t test. B, Levels of secreted Aβ from mouse primary cortical neurons (7 d in vitro) after treatment with SKI II for 24 h (n = 4). C, Levels of secreted Aβ from N2a cells after treatment with SKI II for 24 h (n = 4). D, Levels of secreted Aβs from N2a cells after treatment with DMS or SKI V for 24 h (n = 4–7). E, Levels of secreted Aβ measured by human Aβ-specific ELISA from N2a cells stably expressing Swedish mutant of APP after treatment with SKI II for 24 h (n = 4).
Materials and Methods
Compounds.
N-[N-(3,5-Difluorophenacetyl)-l-alanyl]-(S)-phenylglycine t-butyl ester (DAPT) was synthesized as described previously (Kan et al., 2003). ABC294640 [3-(4-chlorophenyl)-adamantane-1-carboxylic acid (pyridin-4-ylmethyl)amide] was synthesized according to the reported procedure (U.S. Patent 2006287317). Sphingosine kinase inhibitor (SKI) II (Sigma-Aldrich), SKI V (Sigma-Aldrich), N, N-dimethylsphingosine (DMS) (Cayman Chemical), β-secretase inhibitor IV (Calbiochem), S1P (Calbiochem), and 2-acetyl-4-tetrahydroxybutylimidazole (THI) (Matreya) were purchased from the indicated vendors. DAPT, β-secretase inhibitor IV, SKI II, and SKI V were dissolved in DMSO, DMS were dissolved in ethanol, and S1P was dissolved in 3 mm NaOH. Lipid immobilized agarose beads were purchased from Echelon. Synthetic Aβ1–42 peptides (Peptide Institute) were solubilized at a concentration of 0.6 mg/ml in PBS and incubated at 37°C for 24 h to form Aβ fibrils (Hori et al., 2007).
Antibodies and immunological methods.
PS1 C-terminal fragment (CTF) (G1L3) was raised as described previously (Tomita et al., 1999). The following antibodies were purchased from the indicated vendors: human Aβ 82E1 (catalog #10323; Immuno-Biological Laboratories), APP (18) (catalog #28053; Immuno-Biological Laboratories), BACE1 (c) (catalog #18711; Immuno-Biological Laboratories), APP (c) (catalog #18961; Immuno-Biological Laboratories), anti-mouse/rat APP (597) (catalog #28055; Immuno-Biological Laboratories), anti-BACE1 (42) (catalog #28051; Immuno-Biological Laboratories), anti-sAPPβwt (catalog #18957; Immuno-Biological Laboratories), SphK2 (P-19) (SC-22704; Santa Cruz Biotechnology), TRAF2 (C-20) (SC-876; Santa Cruz Biotechnology), SphK1 (catalog #10006822; Cayman Chemical), α-Tubulin DM1A (T9026; Sigma-Aldrich), α-actin AC-40 (A4700; Sigma-Aldrich), anti-βIII-tubulin Tuj1 (MAB1195; R & D Systems), APP N-terminal 22C11 (MAB348; Millipore), and anti-Myc 9B11 (catalog #2276; Cell Signaling Technology). The samples were analyzed by immunoblotting or two-site ELISAs for the detection of Aβ as described previously (Iwatsubo et al., 1994; Tomita et al., 1997). For immunoblot detection for sAPPα and sAPPβ in cultured media, anti-human/mouse APP (597) and anti-sAPPβwt antibodies were used. Specificities of APP antibodies were shown previously (Fukumoto et al. 2010). For lipid binding assay, naive Neuro-2a (N2a) cell lysates or recombinant BACE1 [corresponding to its extracellular domain with a 10×His tag (catalog #931AS; R & D Systems)] were solubilized with 40 mm HEPES buffer, pH 7.4, containing 150 mm NaCl, 0.5% NP-40, and Complete protease inhibitor cocktail (Roche Applied Science) and spun down. Supernatants were coincubated with the lipid coated beads (Echelon Biosciences) or Nickel-NTA beads (Qiagen) that were preincubated with 3% BSA in HEPES buffer. After 3 h, bound proteins were eluted by sample buffer and subjected to immunoblotting.
Cell culture and transfection.
Expression constructs for human APP carrying Swedish mutation (APPNL), SC100, and NΔE were described previously (Kopan et al., 1996; Tomita et al., 1997). cDNAs encoding SphK2, S1P phosphatase (SGPP1), and S1P lyase (SGPL1) were inserted into pcDNA3.1DV5–His/TOPO (Invitrogen) and mutant cDNAs were generated by long PCR-based Quikchange strategy (Stratagene). All constructs were sequenced using Thermosequenase (GE Healthcare) on an automated Sequencer (Li-Cor). N2a cells are maintained as described (Tomita et al., 1997). Primary cortical neurons were prepared from BALB/c mice at embryonic day 16 and grown in Neurobasal medium supplemented with B27 (Invitrogen) for 7 d (Fukumoto et al., 1999). Plasmid transfection was performed using Lipofectamine2000 (Invitrogen) or Fugene 6 (Roche Applied Science) following the instructions of the manufacturer. Small interfering RNA (siRNA) duplexes targeting to control, mouse Sphk1 and Sphk2 (target sequences: Sphk1, 5′-CTG GAC CAG TTG CAT ATA GAA-3′; Sphk2, 5′-TAG GCC TGG CCT CGT TGC ATA-3′) were purchased from Qiagen. Each siRNA was reversely transfected in N2a cells using LipofectAMINE RNAiMax (Invitrogen) following the instructions of the manufacturer.
In vitro secretase activity assay.
For in vitro β-secretase assay, recombinant human BACE1 (catalog #931AS; R & D Systems) or cell membranes of N2a, primary neuronal cells, or mouse brain (Hashimoto et al., 2002; Takasugi et al., 2003) were used as enzyme sources. After homogenization in 10 mm Tris, pH 7.0, the enzyme fractions were acidified by 25 mm CH3COONa, pH 4.5, and incubated with the β-secretase-specific substrate JMV2236 (Bachem) at 37°C at the indicated times. Fluorescence of the fractions was measured at 320 and 420/430 nm as excitation and emission wavelengths, respectively. In vitro α-secretase assay was performed using SensoLyte 520 TACE (α-Secretase) Activity Assay kit (Anaspec) following the instructions of the manufacturer. N2a cells were treated with the indicated reagent for 24 h and collected cell membrane. Ten micrograms of protein were used as enzymatic source, and reaction were performed for 30 min.
In vitro SphK2 activity assay.
Specific SphK2 activity assay was performed according to a previous report (Zemann et al., 2006; Don et al., 2007). After 48 h incubation, cells were washed with iced PBS and lysed by freeze–thaw cycle in 50 mm HEPES, pH 7.4, 10 mm KCl, 15 mm MgCl2, 0.1% Triton X-100, 20% glycerol, 2 mm orthovanadate, 2 mm dithiothreitol, 10 mm NaF, 1 mm deoxypyridoxine, and EDTA-free complete protease inhibitor (Roche Applied Science). Lysates were cleared by centrifugation at 15,000 rpm for 5 min. The lysates and NBD-Sphingosine (10 μm final; Avanti Polar Lipids) were mixed in the reaction buffer (50 mm HEPES, pH 7.4, 15 mm MgCl2, 0.5 mm KCl, 10% glycerol, and 2 mm ATP) and incubated for 30 min at 30°C. The reactions were stopped by the addition of equal amount of 1 m potassium phosphate, pH 8.5, followed by addition of 2.5-fold chloroform/methanol (2:1), and then centrifuged at 15,000 rpm for 1 min. Only the reactant NBD-S1P, but not the substrate NBD-Sphingosine, was collected in alkaline aqueous phase. After aqueous phase was combined with an equal amount of dimethylformamide, the fluorescence value was read. For the analysis of human brains, Tris-soluble fractions were used as an enzyme source. Specificity of this method has been described previously (Zemann et al., 2006).
SKI II treatment in wild-type and AD model mice.
All experiments using animals in this study were performed according to the guidelines provided by the Institutional Animal Care Committee of the Graduate School of Pharmaceutical Sciences, The University of Tokyo. All animals were maintained on food and water with a 12 h light/dark cycle. Wild-type female mice (C57BL; SLC Japan) at 8 weeks of age were used. SKI II was dissolved at 2 μm in 40% DMSO/PBS. Each 2 μl solution was administered by stereotaxic injection into the hippocampus (bregma −2.6 mm, 3.1 mm lateral, 2.4 mm depth). After 8 h, the hippocampus of injected and uninjected site were isolated. Hippocampus samples were solubilized with 10 mm Tris buffer containing 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate and subjected to the sandwich ELISA for Aβ (Wako Chemical). A7 transgenic mice overexpress human APP695 harboring K670N, M671L, and T714I FAD mutations in neurons under the control of Thy1.2 promoter (Yamada et al., 2009). Female A7 mice at 6 months of age were used for subchronic treatment of SKI II. SKI II was dissolved in corn oil and injected orally for 7 d (50 mg · kg−1 · d−1).
Human brain samples.
Human brain samples from AD and aged control patients were derived from tissue bank at the University of Pennsylvania Alzheimer's Disease Core Center (ADCC) and the Center for Neurodegenerative Disease Research (CNDR). Control and AD brains were diagnosed symptomatically and pathologically at ADCC–CNDR as described (Arnold et al., 2010). All samples used for experimental measures were derived from frontal cortex under approval by the institutional review board, ADCC–CNDR, and institutional ethical committee of Graduate School of Pharmaceutical Sciences, The University of Tokyo. Brain samples were homogenized in TSI buffer (50 mm Tris HCl, pH 7.6, 150 mm NaCl, 0.5 mm diisopropyl fluorophosphate, 0.5 mm phenylmethylsulfonyl fluoride, 1 mm EGTA, 1 mg/ml antipain, 1 mg/ml leupeptin, 1 mg/ml pepstatin, 1 mg/ml Na-p-tosyl-l-lysine chloromethyl ketone) and centrifuged at 260,000 × g for 20 min. Supernatant was collected as Tris-soluble fraction and used for SphK assay.
Results
SphK inhibitors decreased Aβ secretion by reducing the β-cleavage of APP
To investigate the relationship between S1P and Aβ production, we focused on the activity of SphKs (Fig. 1A). Recently, several small compounds that specifically inhibit SphKs have been developed as anti-cancer drugs (Pyne and Pyne, 2010). Treatment with a SphK-selective inhibitor, SKI II (French et al., 2003, 2006), decreased the secretion of endogenous Aβ from mouse primary cortical neurons (Fig. 1B), as well as in mouse neuroblastoma N2a cells, in a dose-dependent manner (Fig. 1C). Two additional selective SphK inhibitors with different chemical structure (i.e., N,N-dimethylsphingosine and SKI V) also decreased the Aβ secretion from N2a cells (Fig. 1D). SKI II treatment decreased the Aβ secretion from N2a cells overexpressing the Swedish mutant form of human APP (APPNL) (Fig. 1E). These data suggest an inhibitory effect of SphK inhibitors on Aβ secretion. Notably, both Aβ40 and Aβ42 levels were affected in a similar manner in all following experiments. Next we analyzed the APP metabolism in SKI II-treated cells. SKI II treatment did not affect the expression levels of either BACE1 or presenilin-1, which is the γ-secretase catalytic component, in primary cortical neuron (Fig. 2A) or N2a cells (Fig. 2B), respectively. Because the treatment of authentic BACE1 inhibitor IV abolished the secretion of β-secretase-mediated cleavage product of APP, i.e., sAPPβ, in conditioned media, SKI II treatment also caused a moderate but significant decrease in sAPPβ in primary cortical neurons (Fig. 2C) or N2a cells (Fig. 2D). In contrast, SKI II showed no effect on the cleavage of the C-terminal stub of human APP (SC100) that serves as a direct substrate of γ-secretase (Fig. 2E). Moreover, SKI II treatment caused neither an increase in in vitro α-secretase activity in cell membranes (78.9 ± 2.3% compared with DMSO treatment, n = 4) nor change in the level of APP on the plasma membrane (Fig. 2F). Collectively, these data strongly suggest that SKI II directly affected the β-cleavage of APP.
Figure 2.

SKI II decreased the β-secretase cleavage products. A–D, Immunoblot analyses of the protein levels of APP derivatives and BACE1 in neuronal cells. Immunoblot analysis of cell lysates (A) and cultured media (B) of mouse primary cortical neurons (7 d in vitro) treated with BACE inhibitor IV (BSI IV; 1 μm) or SKI II (1 μm) for 24 h in duplicate (n = 3; representative results are shown). Immunoblot analysis of cell lysates (C) and cultured media (D) of naive N2a cells treated with BACE inhibitor IV (BSI IV; 1 μm) or SKI II (3 μm) for 24 h. E, Levels of secreted human Aβ from N2a cells overexpressing SC100 after treatment with SKI II for 24 h. Secreted human Aβ40 and Aβ42 were detected by human Aβ-specific ELISA (n = 4; mean ± SEM). F, Effect of SKI II (1 μm) on the cell-surface levels of APP in naive N2a cells. After treatment with vehicle or SKI II for 24 h, N2a cells were biotinylated by sulfo-NHS-biotin and pulled down by streptavidin beads.
S1P metabolism coordinately modulates the β-cleavage of APP
To test whether SKI II directly inhibited the enzymatic activity of BACE1, a major β-secretase in neurons, we coincubated SKI II in in vitro assay using recombinant soluble BACE1 corresponding to its extracellular domain. However, SKI II itself did not affect the catalytic activity of recombinant BACE1 (Fig. 3). This result suggests that SKI II modulates the β-cleavage through the inhibition of SphK. Consistently, RNA interference (RNAi) against either SphK1 or 2 significantly decreased the Aβ production in N2a cells (Fig. 4A). Notably, knockdown of SphK2 showed a potent inhibitory effect. Supporting this result, SphK2-selective inhibitor ABC294640 (French et al., 2010) inhibited the Aβ generation in N2a cells similarly to that by SKI II (Fig. 4B), indicating that SphK2 plays a major role in the β-cleavage modulation. Thus, we focused on SphK2 in the following part of the study. RNAi against SphK2 resulted in a significant decrease in the levels of SphK2 expression (Fig. 4C), kinase activity (Fig. 4D), βCTF (Fig. 4E), as well as the secretion of sAPPβ (Fig. 4F), whereas the levels of secreted sAPPα was increased (Fig. 4G). In contrast, overexpression of SphK2, but not of an inactive mutant (G243D), significantly increased the levels of βCTF and secreted Aβ (Fig. 4H,I), along with an augmentation in SphK2 activity in vitro (Fig. 4J). Notably, coexpression of inactive SphK2 mutant decreased the levels of secreted Aβ (Fig. 4I), indicating that this mutant functions in a dominant-negative manner (Yoshimoto et al., 2003). These data indicate that cellular SphK2 activity is tightly correlated with the β-cleavage of APP.
Figure 3.

Effect of SKI II on the catalytic activity of BACE1. In vitro BACE1 activity assay using a fluorogenic BACE1-specific substrate. BACE inhibitor IV (BSI IV; A) or SKI II (B) was coincubated with recombinant soluble BACE1 protein at indicated duration and concentrations. Relative fluorescence units were shown (n = 3).
Figure 4.

SphK2 activity modulated the β-secretase cleavage products. A, N2a cells were transiently transfected with siRNAs against endogenous SphKs. After 48 h transfection, media were replaced and further incubated for 24 h. Levels of secreted Aβ were quantified by ELISA (n = 3; mean ± SEM; *p < 0.05, **p < 0.01). B, Levels of secreted Aβ from N2a cells treated with BACE inhibitor IV (BACEi IV), SKI II, or SphK2-selective inhibitor ABC294640 (ABC) for 24 h (n = 3; mean ± SEM; ***p < 0.001). C–G, Effect of transient SphK2 knockdown on APP derivatives in N2a cells. Representative immunoblot analysis was shown in C. In vitro SphK2 activity (D) as well as βCTF (E) in cell lysates and the amount of sAPPβ in conditioned media (F) were significantly decreased by knockdown of SphK2. In contrast, the level of sAPPα in conditioned media (G) was significantly increased (quantitated by densitometric analysis; n = 4; mean ± SEM; *p < 0.05). H–J, Effect of SphK2 on N2a cells coexpressing Swedish mutant of APP. wt, Wild type. Representative immunoblot analysis was shown in H. Overexpression of SphK2, but not inactive mutant (G243D), increased the levels of Aβ production (I) as well as SphK2 activity in vitro (J) (n = 4; mean ± SEM; *p < 0.05, **p < 0.01).
To further test whether S1P, which is produced by SphK activity, is the regulator of β-cleavage, we examined the effects of S1P degrading enzymes on APP processing. SGPP1 dephosphorylates S1P to sphingosine, and SGPL1 irreversibly cleaves S1P to generate phosphoethanolamine and a long-chain aldehyde (Fig. 5A) (Alvarez et al., 2007; Takabe et al., 2008). Thus, these enzymes decrease the cellular S1P levels with different end products. Overexpression of either SGPP1 or SGPL1 in N2a cells strongly reduced the levels of βCTF and secreted Aβ (Fig. 5B–D). In contrast, the expression of catalytically inactive SGPL1 harboring K353L mutation (Reiss et al., 2004) showed no effect. Moreover, the inhibition of SGPL1 by THI (Schwab et al., 2005) caused a significant increase in Aβ secretion from mouse primary neurons (Fig. 5E), suggesting that the enzymatic activity of S1P degrading enzymes is important for the modulation of β-cleavage. Together, these data indicate that the S1P metabolism coordinately modulates the β-cleavage of APP.
Figure 5.
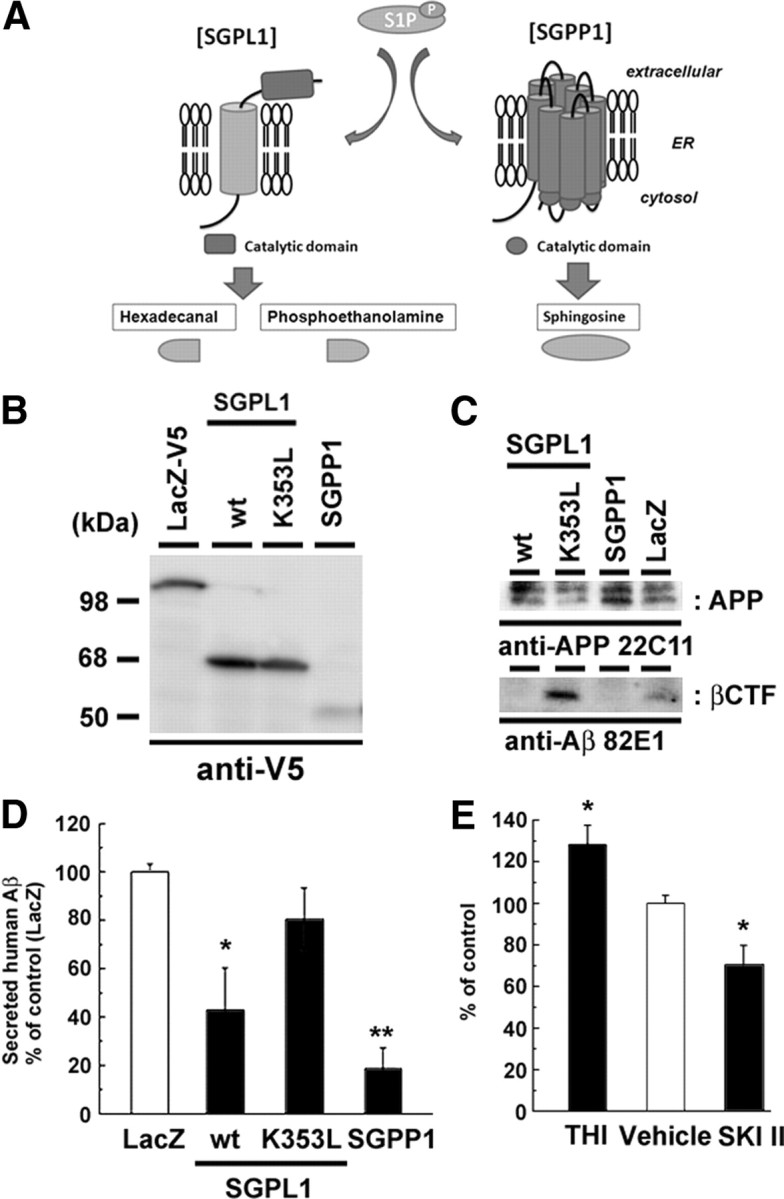
Effect of S1P degrading enzymes on Aβ production. A, Schematic view of S1P degradation pathway. Note that SGPL1 and SGPP1 generate different degradation products of S1P. ER, Endoplasmic reticulum. B–D, Effects of the overexpression of V5-tagged S1P degrading enzymes on APP metabolism in N2a cells. N2a cells were cotransfected with S1P degrading enzymes and Swedish mutant of APP. After 24 h transfection, media were replaced and further incubated for 24 h. Immunoblot analysis of S1P degrading enzymes (B) and APP derivatives (C) are shown. Human APP-derived βCTF was specifically detected by an anti-human Aβ N-terminus antibody (82E1). wt, Wild type. D, The levels of secreted human Aβ was detected by human Aβ-specific ELISA (n = 4; mean ± SEM; *p < 0.05, **p < 0.01). Note that overexpression of SGPL1 or SGPP1, but not SGPL1 carrying catalytically inactive mutation (K353L), decreased the generation of βCTF and the Aβ secretion from N2a cells. E, The levels of secreted Aβ from mouse primary cortical neurons (7 d in vitro) treated with SGPL1 inhibitor THI (50 μg/ml) or SKI II (1 μm) for 24 h (n = 4; mean ± SEM; *p < 0.05).
Cell-associated S1P directly modulates BACE1 activity
A proportion of newly synthesized S1P is secreted, whereas others remain associated with cells. In general, extracellular S1P poorly permeates into the cells (Kihara et al., 2003) and functions as a ligand for cell-surface GPCR-type receptors (Alvarez et al., 2007; Takabe et al., 2008). However, extracellular application of S1P failed to restore the reduced Aβ secretion by SKI II treatment or SphK2 knockdown (Fig. 6A,B), suggesting that cell-associated S1P is involved in the regulation of β-cleavage. Next we tested the effect of S1P on the intrinsic activity of membrane-bound BACE1 in a cell-free assay. Both pretreatment of SKI II (Fig. 6C) and SphK2 knockdown (Fig. 6D) on N2a cells significantly decreased the BACE1 activity in the membrane fractions in vitro, implicating that the levels of S1P within cells correlate with BACE1 activity. Supporting this notion, addition of S1P into the microsome fraction significantly increased the intrinsic BACE1 activity (Fig. 6E). These data implicate the direct action of S1P on BACE1 activity rather than the cell-surface receptor-mediated modulation. To provide additional evidence that S1P directly modulates BACE1, we examined binding of endogenous BACE1 in N2a cell lysates to S1P immobilized on agarose beads (Fig. 6F). We confirmed the specific binding of TRAF2 to S1P beads as described recently (Alvarez et al., 2010). Furthermore, endogenous BACE1, but not APP, was specifically pulled down by matrices carrying S1P. In contrast, recombinant BACE1 protein that lacks the transmembrane and cytoplasmic domains was never bound to S1P beads (Fig. 6G). Collectively, these results strongly suggest that the cell-associated S1P modulates the proteolytic activity of membrane-bound form of BACE1 via direct interaction (Fig. 6H).
Figure 6.
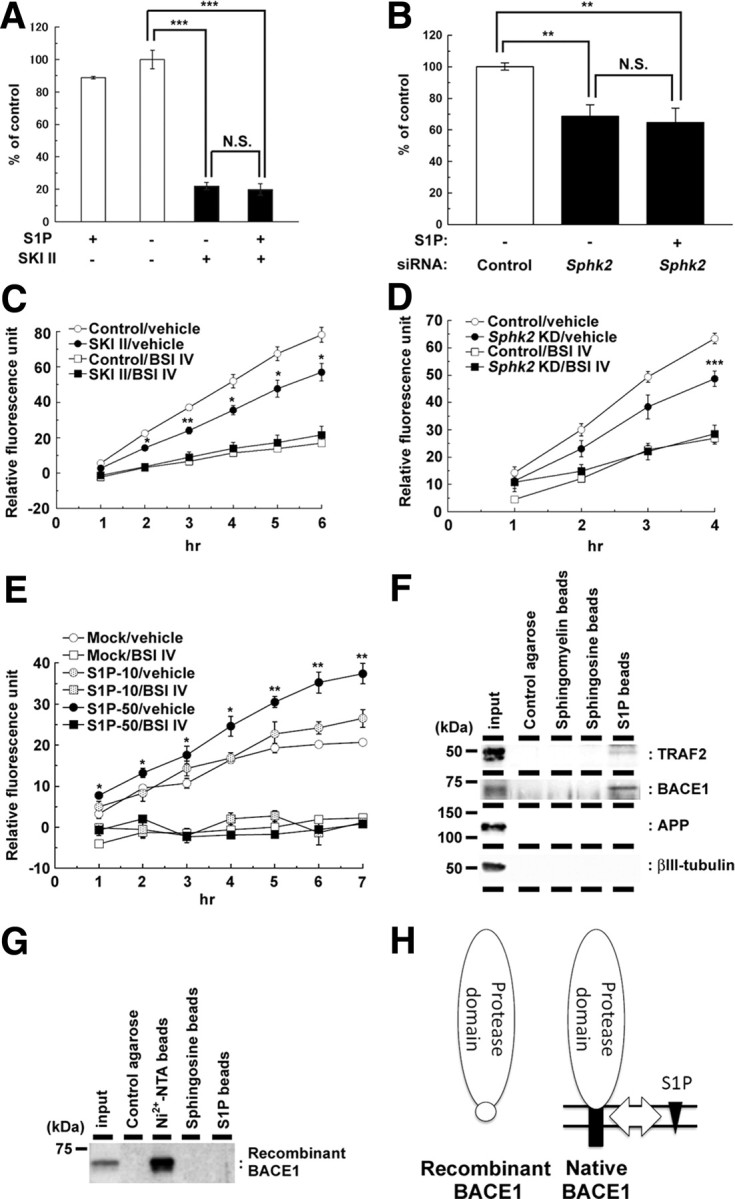
SKI II treatment decreased the β-secretase activity in cellular membrane. A, B, Effect of extracellularly added S1P (10 μm) on levels of secreted Aβ from mouse primary cortical neurons (7 d in vitro) after treatment with SKI II (1 μm) for 24 h (A) (n = 4; mean ± SEM) or from N2a cells after 48 h SphK2 knockdown (B) (n = 3; mean ± SEM; **p < 0.01, ***p < 0.001; N.S., no significant difference). Note that S1P failed to rescue the decrease in Aβ production either by SKI II or SphK2 knockdown. C, β-Secretase activity in the membrane fractions of N2a cells treated with vehicle or SKI II (1 μm) for 24 h. BACE inhibitor IV (BSI IV; 1 μm) was added to the in vitro assay (n = 3; mean ± SEM; *p < 0.05, **p < 0.01 vs control/vehicle). D, β-Secretase activity in the membrane fractions of SphK2 knockdown N2a cells. BACE inhibitor IV (BSI IV; 1 μm) was added to the in vitro assay (n = 3; mean ± SEM; ***p < 0.001 vs control/vehicle). E, Effect of S1P on β-secretase activity in the membrane fractions of mouse brain. S1P (10 or 50 μm) and BACE inhibitor IV (BSI IV; 1 μm) were added to the in vitro assay (n = 3; mean ± SEM; *p < 0.05, **p < 0.01 vs mock/vehicle). F, G, Association of BACE1 holoprotein with immobilized S1P. N2a cell lysates (F) or recombinant BACE1 with 10×His tag that lacks the transmembrane and cytoplasmic domains (G) were incubated with control agarose (no lipid), Nickel-NTA agarose, sphingomyelin, sphingosine, or S1P-coated affinity matrices (as indicated), and bound proteins were analyzed by immunoblotting. H, Schematic model of the binding of BACE1 and S1P. S1P (black triangles) interacts with the C-terminal region of BACE1 (black squares), including the transmembrane domain, but not with the extracellular protease domain (white ovals). Location of 10×His tag is indicated by a white circle.
Roles of SphK2 activity in Alzheimer's disease
Data shown above indicate that the intracellular S1P is one of the important determinants for BACE1 activity. We further examined the impact of reduced S1P levels on Aβ levels in vivo. Stereotaxic injection of SKI II into wild-type mouse brain significantly decreased the amount of endogenous Aβ at hippocampus (Fig. 7A). Because SKI II exhibited a favorable bioavailability (French et al., 2006), we next orally administered SKI II to APP transgenic mice A7 overexpressing human APP carrying Swedish and Austrian mutations (Yamada et al., 2009). After 6 d treatment with SKI II in 6-month-old female mice, in which pathologically detectable amyloid plaques have not been developed, the total Aβ levels in brains were significantly decreased (Fig. 7B). These data indicate that the inhibition of SphK activity in APP transgenic mouse brains has beneficial effects against Aβ production.
Figure 7.

Role of SphK2 activity in AD brains. A, Effect of the direct injection of SKI II into the hippocampus of nontransgenic wild-type female mice (C57BL) at 8 weeks of age. The levels of Tris-soluble Aβ in the injected side of hippocampus were divided by those in the uninjected side. Data represent relative ratio of each group (n = 4; mean ± SEM; **p < 0.01). B, Levels of Tris-soluble Aβ in the cerebral cortices of female A7 mice at 6 months of age after a 7 d treatment with SKI II (50 mg · kg−1 · d−1, p.o.). Total brain Aβ levels were measured by human-specific sandwich ELISA (n = 5; mean ± SEM; **p < 0.01). C, Effect of Aβ fibril on SphK2 activity in N2a cells. N2a cells were treated with Aβ42 fibril (30 μm) overnight, and cell lysates were subjected to an in vitro SphK2 activity assay (n = 3; mean ± SEM; ***p < 0.001). D–F, Immunoblot analysis of Tris-soluble fractions (15 μg of protein in each lane) from cortices of AD (denoted as A) or non-demented (denoted as N) individuals. Average protein levels of SphK2 (E) and βIII-tubulin (F) in each individual were analyzed by densitometric analyses (*p < 0.05). G, Average of in vitro SphK2 enzymatic activity of Tris-soluble fractions from brains of AD and non-demented individuals. The enzymatic activities of SphK2 were normalized by the protein levels of SphK2 quantified in D.
SphK activity is regulated by various stimuli and stress (Spiegel and Milstien, 2007). Intriguingly, treatment of N2a cells with fibrillized Aβ42 caused a significant increase in the SphK2 activity (Fig. 7C), raising the possibility that the Aβ deposits in AD brains in turn augment Aβ production through SphK-mediated BACE1 activation in neurons. To further clarify the significance of SphK2 activity in the pathogenesis of sporadic AD, we compared the protein levels and the activities of SphK2 in cerebral cortices between sporadic AD patients and non-demented individuals (Fig. 7D). In contrast to the levels of α-actin, the levels of neuronal βIII-tubulin were significantly decreased in AD brains because of neuronal loss (Hempen and Brion, 1996). The protein levels of SphK2 also showed a decreased trend in AD brains (Fig. 7E,F), in accord with the previous description that the major SphK2-expressing cells are neurons (Blondeau et al., 2007). However, the relative in vitro activities of SphK2 were significantly upregulated in AD brains (Fig. 7G). These results provide compelling evidence that changes in the levels of cell-associated S1P in neurons, which is increased by a variety of stimuli including Aβ fibrils, modulate the proteolytic activity of BACE1, thereby forming a vicious cycle in the etiology of AD (Fig. 8).
Figure 8.

Schematic representation of the role of S1P metabolism in AD.
Discussion
Here we show that lowering the activity of SphK or increasing that of S1P degrading enzymes decreased the Aβ production by the inhibition of BACE1 activity in vitro and in vivo. Notably, SphK2 activity was increased by exposing cells to Aβ fibrils, and it also was increased in the postmortem cerebral cortices of AD patients. These results suggest that SphK2 and S1P are involved in the etiology of AD and novel potential therapeutic targets for AD.
Metabolites of sphingolipids are functionally interrelated with each other. Inhibition of SphK activity diminishes the generation of S1P and simultaneously increases the cellular levels of sphingosine and ceramide (Spiegel and Milstien, 2007; Pyne and Pyne, 2010). However, overexpression of SGPP1 or SGPL1, which decreases S1P levels by dephosphorylation or irreversible degradation, also decreased the levels of Aβ in N2a cells (Fig. 5). Moreover, direct addition of S1P to the membrane, but not on living cells, increased the β-secretase activity, suggesting that the cell-associated form of S1P per se plays a critical role for the modulation of BACE1 activity in neurons. Importantly, we observed a specific interaction of S1P with BACE1 holoprotein (Fig. 6F), in which transmembrane and intracellular domains of BACE1 are required (Fig. 6G). Thus, we hypothesize that S1P binds to BACE1 transmembrane/intracellular domain (Fig. 6H) and affects the proteolytic activity by altering the conformation or substrate accessibility. In good accordance with this, recent findings implicate intracellular S1P as a novel modulator for enzymes; S1P specifically binds to the histone deacetylases HDAC1 and HDAC2 and inhibited their enzymatic activity (Hait et al., 2009). Moreover, S1P targets to TRAF2 at the RING domain to stimulate E3 ligase activity (Alvarez et al., 2010). It has been shown that functions of membrane-embedded as well as membrane-associated proteins are modulated by direct interaction with sphingolipids [e.g., activation of TrkA receptor by GM1 (Mutoh et al., 1995), inhibition of epidermal growth factor receptor by GM3 (Kawashima et al., 2009), activation of synaptobrevin (Darios et al., 2009), and functional modulation of stargazin by sphingosine (Sumioka et al., 2010)]. In these cases, lipid interactions are predicted to affect the conformation of functionally active domains located at the luminal or cytoplasmic sides. Moreover, it was shown previously that RTN3 inhibits the BACE1 activity via interaction with the transmembrane domain of BACE1 (He et al., 2004; Murayama et al., 2006). In addition, we have recently identified that a lipophilic, noncompetitive BACE1 inhibitor, TAK-070, directly targets the transmembrane domain of BACE1 (Fukumoto et al., 2010). These results collectively support the notion that targeting the transmembrane domain of BACE1, which harbors an allosteric modulatory function on the catalytic domain, might be a novel approach for the inhibition of the β-cleavage. Additional detailed analysis of molecular effects of S1P on BACE1 should be performed.
Several reports indicate that intrinsic activity of BACE1 is increased in AD brains (Fukumoto et al., 2002; Yang et al., 2003; Li et al., 2004; Ahmed et al., 2010), although the underlying molecular mechanism is essentially unknown. In this study, we found that treatment of cultured cells with Aβ fibrils augmented SphK2 activity, which was increased in AD brains, as well. Notably, intrinsic SphK2 activity is modulated by extracellular signal-regulated kinase (ERK) and fyn kinase (Olivera et al., 2006; Hait et al., 2007), which have been implicated in Aβ-mediated neurotoxicity (Crews and Masliah, 2010), suggesting the possibility that upregulation of SphK2 activity was mediated by aberrant phosphorylation by ERK and/or fyn kinase activity. Additional analysis would be required to understand the molecular connection between Aβ and SphK2 activity. Moreover, SphK2 activity was upregulated by neuronal stress, such as ischemia (Blondeau et al., 2007), which is also correlated with modulation of BACE1 activity (Wen et al., 2004; Tesco et al., 2007). Nevertheless, the increased SphK2 activity by Aβ fibril in neurons thereby may form a vicious cycle in the pathophysiology of AD (Fig. 8). Finally, SKI II treatment decreased the brain Aβ levels in APP transgenic mice, supporting the feasibility of SphK inhibition as a potential AD therapy. Especially, SphK2 single knock-out mice did not show significant developmental defects (Mizugishi et al., 2005). Moreover, SphK2 has been implicated in proapoptotic function, whereas SphK1 harbors anti-apoptotic effects (Liu et al., 2003; Maceyka et al., 2005). Thus, SphK2 selective inhibitors, e.g., ABC294640 (French et al., 2010), may be tolerable and suitable therapeutic agents for AD therapeutics. In conclusion, SphK2/S1P in brain might be a novel molecular target for AD therapeutics, and additional analysis for the regulatory mechanisms of β-secretase activity by SphK/S1P will facilitate the understanding of the pathogenesis of sporadic AD.
Footnotes
The authors declare no competing financial interests.
This work is supported in part by Grants-in-Aid for Young Scientists (S) (T.T.) and (B) (N.T.) from Japan Society for the Promotion of Science (JSPS), by the Ministry of Health, Labor, and Welfare of Japan (Comprehensive Research on Aging and Health) (T.T.), by the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (N.T., T.T., and T.I.), by Scientific Research on Priority Areas “Research on Pathomechanisms of Brain Disorders” from the Ministry of Education, Culture, Sports, Science, and Technology (T.T., T.I.), by Core Research for Evolutional Science and Technology of Japan Science and Technology Corporation (T.T., T.I.), and by National Institutes of Health Grant AG10124 (J.Q.T.). I.H. and Y.H. are research fellows of JSPS. We are grateful to Dr. G. Thinakaran (The University of Chicago, Chicago, IL) for valuable reagents, Takeda Pharmaceutical Company for Aβ ELISA, and our current and previous laboratory members for helpful discussions and technical assistance.
References
- Ahmed RR, Holler CJ, Webb RL, Li F, Beckett TL, Murphy MP. BACE1 and BACE2 enzymatic activities in Alzheimer's disease. J Neurochem. 2010;112:1045–1053. doi: 10.1111/j.1471-4159.2009.06528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez SE, Milstien S, Spiegel S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol Metab. 2007;18:300–307. doi: 10.1016/j.tem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T, Milstien S, Spiegel S. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–1088. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SE, Lee EB, Moberg PJ, Stutzbach L, Kazi H, Han LY, Lee VM, Trojanowski JQ. Olfactory epithelium amyloid-β and paired helical filament-tau pathology in Alzheimer disease. Ann Neurol. 2010;67:462–469. doi: 10.1002/ana.21910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondeau N, Lai Y, Tyndall S, Popolo M, Topalkara K, Pru JK, Zhang L, Kim H, Liao JK, Ding K, Waeber C. Distribution of sphingosine kinase activity and mRNA in rodent brain. J Neurochem. 2007;103:509–517. doi: 10.1111/j.1471-4159.2007.04755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Aβ peptides by neurons. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Chow VW, Savonenko AV, Melnikova T, Kim H, Price DL, Li T, Wong PC. Modeling an anti-amyloid combination therapy for Alzheimer's disease. Sci Transl Med. 2010;2:13ra1. doi: 10.1126/scitranslmed.3000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews L, Masliah E. Molecular mechanisms of neurodegeneration in Alzheimer's disease. Hum Mol Genet. 2010;19:R12–R20. doi: 10.1093/hmg/ddq160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darios F, Wasser C, Shakirzyanova A, Giniatullin A, Goodman K, Munoz-Bravo JL, Raingo J, Jorgacevski J, Kreft M, Zorec R, Rosa JM, Gandia L, Gutiérrez LM, Binz T, Giniatullin R, Kavalali ET, Davletov B. Sphingosine facilitates SNARE complex assembly and activates synaptic vesicle exocytosis. Neuron. 2009;62:683–694. doi: 10.1016/j.neuron.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Vassar R, Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol. 2010;6:99–107. doi: 10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Don AS, Martinez-Lamenca C, Webb WR, Proia RL, Roberts E, Rosen H. Essential requirement for sphingosine kinase 2 in a sphingolipid apoptosis pathway activated by FTY720 analogues. J Biol Chem. 2007;282:15833–15842. doi: 10.1074/jbc.M609124200. [DOI] [PubMed] [Google Scholar]
- French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- French KJ, Upson JJ, Keller SN, Zhuang Y, Yun JK, Smith CD. Antitumor activity of sphingosine kinase inhibitors. J Pharmacol Exp Ther. 2006;318:596–603. doi: 10.1124/jpet.106.101345. [DOI] [PubMed] [Google Scholar]
- French KJ, Zhuang Y, Maines LW, Gao P, Wang W, Beljanski V, Upson JJ, Green CL, Keller SN, Smith CD. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J Pharmacol Exp Ther. 2010;333:129–139. doi: 10.1124/jpet.109.163444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto H, Tomita T, Matsunaga H, Ishibashi Y, Saido TC, Iwatsubo T. Primary cultures of neuronal and non-neuronal rat brain cells secrete similar proportions of amyloid β peptides ending at Aβ40 and Aβ42. Neuroreport. 1999;10:2965–2969. doi: 10.1097/00001756-199909290-00017. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. β-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Takahashi H, Tarui N, Matsui J, Tomita T, Hirode M, Sagayama M, Maeda R, Kawamoto M, Hirai K, Terauchi J, Sakura Y, Kakihana M, Kato K, Iwatsubo T, Miyamoto M. A noncompetitive BACE1 inhibitor TAK-070 ameliorates Aβ pathology and behavioral deficits in a mouse model of Alzheimer's disease. J Neurosci. 2010;30:11157–11166. doi: 10.1523/JNEUROSCI.2884-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hait NC, Bellamy A, Milstien S, Kordula T, Spiegel S. Sphingosine kinase type 2 activation by ERK-mediated phosphorylation. J Biol Chem. 2007;282:12058–12065. doi: 10.1074/jbc.M609559200. [DOI] [PubMed] [Google Scholar]
- Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, Spiegel S. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–1257. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Wakabayashi T, Watanabe A, Kowa H, Hosoda R, Nakamura A, Kanazawa I, Arai T, Takio K, Mann DM, Iwatsubo T. CLAC: a novel Alzheimer amyloid plaque component derived from a transmembrane precursor, CLAC-P/collagen type XXV. EMBO J. 2002;21:1524–1534. doi: 10.1093/emboj/21.7.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Lu Y, Qahwash I, Hu XY, Chang A, Yan R. Reticulon family members modulate BACE1 activity and amyloid-β peptide generation. Nat Med. 2004;10:959–965. doi: 10.1038/nm1088. [DOI] [PubMed] [Google Scholar]
- Hempen B, Brion JP. Reduction of acetylated α-tubulin immunoreactivity in neurofibrillary tangle-bearing neurons in Alzheimer's disease. J Neuropathol Exp Neurol. 1996;55:964–972. doi: 10.1097/00005072-199609000-00003. [DOI] [PubMed] [Google Scholar]
- Hori Y, Hashimoto T, Wakutani Y, Urakami K, Nakashima K, Condron MM, Tsubuki S, Saido TC, Teplow DB, Iwatsubo T. The Tottori (D7N) and English (H6R) familial Alzheimer disease mutations accelerate Aβ fibril formation without increasing protofibril formation. J Biol Chem. 2007;282:4916–4923. doi: 10.1074/jbc.M608220200. [DOI] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Hu X, Zhou X, He W, Yang J, Xiong W, Wong P, Wilson CG, Yan R. BACE1 deficiency causes altered neuronal activity and neurodegeneration. J Neurosci. 2010;30:8819–8829. doi: 10.1523/JNEUROSCI.1334-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- Kalvodova L, Kahya N, Schwille P, Ehehalt R, Verkade P, Drechsel D, Simons K. Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J Biol Chem. 2005;280:36815–36823. doi: 10.1074/jbc.M504484200. [DOI] [PubMed] [Google Scholar]
- Kan T, Tominari Y, Morohashi Y, Natsugari H, Tomita T, Iwatsubo T, Fukuyama T. Solid-phase synthesis of photoaffinity probes: highly efficient incorporation of biotin-tag and cross-linking groups. Chem Commun (Camb) 2003:2244–2245. doi: 10.1039/b306970b. [DOI] [PubMed] [Google Scholar]
- Kawashima N, Yoon SJ, Itoh K, Nakayama K. Tyrosine kinase activity of epidermal growth factor receptor is regulated by GM3 binding through carbohydrate to carbohydrate interactions. J Biol Chem. 2009;284:6147–6155. doi: 10.1074/jbc.M808171200. [DOI] [PubMed] [Google Scholar]
- Kihara A, Ikeda M, Kariya Y, Lee EY, Lee YM, Igarashi Y. Sphingosine-1-phosphate lyase is involved in the differentiation of F9 embryonal carcinoma cells to primitive endoderm. J Biol Chem. 2003;278:14578–14585. doi: 10.1074/jbc.M211416200. [DOI] [PubMed] [Google Scholar]
- Kopan R, Schroeter EH, Weintraub H, Nye JS. Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci U S A. 1996;93:1683–1688. doi: 10.1073/pnas.93.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Lindholm K, Yang LB, Yue X, Citron M, Yan R, Beach T, Sue L, Sabbagh M, Cai H, Wong P, Price D, Shen Y. Amyloid β peptide load is correlated with increased β-secretase activity in sporadic Alzheimer's disease patients. Proc Natl Acad Sci U S A. 2004;101:3632–3637. doi: 10.1073/pnas.0205689101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Toman RE, Goparaju SK, Maceyka M, Nava VE, Sankala H, Payne SG, Bektas M, Ishii I, Chun J, Milstien S, Spiegel S. Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J Biol Chem. 2003;278:40330–40336. doi: 10.1074/jbc.M304455200. [DOI] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R. Mice deficient in BACE1, the Alzheimer's β-secretase, have normal phenotype and abolished β-amyloid generation. Nat Neurosci. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- Maceyka M, Sankala H, Hait NC, Le Stunff H, Liu H, Toman R, Collier C, Zhang M, Satin LS, Merrill AH, Jr, Milstien S, Spiegel S. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem. 2005;280:37118–37129. doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- McConlogue L, Buttini M, Anderson JP, Brigham EF, Chen KS, Freedman SB, Games D, Johnson-Wood K, Lee M, Zeller M, Liu W, Motter R, Sinha S. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J Biol Chem. 2007;282:26326–26334. doi: 10.1074/jbc.M611687200. [DOI] [PubMed] [Google Scholar]
- Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL. Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol. 2005;25:11113–11121. doi: 10.1128/MCB.25.24.11113-11121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama KS, Kametani F, Saito S, Kume H, Akiyama H, Araki W. Reticulons RTN3 and RTN4-B/C interact with BACE1 and inhibit its ability to produce amyloid β-protein. Eur J Neurosci. 2006;24:1237–1244. doi: 10.1111/j.1460-9568.2006.05005.x. [DOI] [PubMed] [Google Scholar]
- Mutoh T, Tokuda A, Miyadai T, Hamaguchi M, Fujiki N. Ganglioside GM1 binds to the Trk protein and regulates receptor function. Proc Natl Acad Sci U S A. 1995;92:5087–5091. doi: 10.1073/pnas.92.11.5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera A, Urtz N, Mizugishi K, Yamashita Y, Gilfillan AM, Furumoto Y, Gu H, Proia RL, Baumruker T, Rivera J. IgE-dependent activation of sphingosine kinases 1 and 2 and secretion of sphingosine 1-phosphate requires Fyn kinase and contributes to mast cell responses. J Biol Chem. 2006;281:2515–2525. doi: 10.1074/jbc.M508931200. [DOI] [PubMed] [Google Scholar]
- Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T, Schwille P, Schulz JB, Schroeder C, Simons M, Jennings G, Knölker HJ, Simons K. Efficient inhibition of the Alzheimer's disease β-secretase by membrane targeting. Science. 2008;320:520–523. doi: 10.1126/science.1156609. [DOI] [PubMed] [Google Scholar]
- Reiss U, Oskouian B, Zhou J, Gupta V, Sooriyakumaran P, Kelly S, Wang E, Merrill AH, Jr, Saba JD. Sphingosine-phosphate lyase enhances stress-induced ceramide generation and apoptosis. J Biol Chem. 2004;279:1281–1290. doi: 10.1074/jbc.M309646200. [DOI] [PubMed] [Google Scholar]
- Savonenko AV, Melnikova T, Laird FM, Stewart KA, Price DL, Wong PC. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Natl Acad Sci U S A. 2008;105:5585–5590. doi: 10.1073/pnas.0710373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, Cyster JG. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309:1735–1739. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. Functions of the multifaceted family of sphingosine kinases and some close relatives. J Biol Chem. 2007;282:2125–2129. doi: 10.1074/jbc.R600028200. [DOI] [PubMed] [Google Scholar]
- Sumioka A, Yan D, Tomita S. TARP phosphorylation regulates synaptic AMPA receptors through lipid bilayers. Neuron. 2010;66:755–767. doi: 10.1016/j.neuron.2010.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60:181–195. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T. The role of presenilin cofactors in the γ-secretase complex. Nature. 2003;422:438–441. doi: 10.1038/nature01506. [DOI] [PubMed] [Google Scholar]
- Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, Hiltunen M, Yang SH, Zhong Z, Shen Y, Simpkins JW, Tanzi RE. Depletion of GGA3 stabilizes BACE and enhances β-secretase activity. Neuron. 2007;54:721–737. doi: 10.1016/j.neuron.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita T. Secretase inhibitors and modulators for Alzheimer's disease treatment. Expert Rev Neurother. 2009;9:661–679. doi: 10.1586/ern.09.24. [DOI] [PubMed] [Google Scholar]
- Tomita T, Maruyama K, Saido TC, Kume H, Shinozaki K, Tokuhiro S, Capell A, Walter J, Grünberg J, Haass C, Iwatsubo T, Obata K. The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid β protein ending at the 42nd (or 43rd) residue. Proc Natl Acad Sci U S A. 1997;94:2025–2030. doi: 10.1073/pnas.94.5.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita T, Takikawa R, Koyama A, Morohashi Y, Takasugi N, Saido TC, Maruyama K, Iwatsubo T. C terminus of presenilin is required for overproduction of amyloidogenic Aβ42 through stabilization and endoproteolysis of presenilin. J Neurosci. 1999;19:10627–10634. doi: 10.1523/JNEUROSCI.19-24-10627.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Kovacs DM, Yan R, Wong PC. The beta-secretase enzyme BACE in health and Alzheimer's disease: regulation, cell biology, function, and therapeutic potential. J Neurosci. 2009;29:12787–12794. doi: 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivel KS, Thinakaran G. Membrane rafts in Alzheimer's disease β-amyloid production. Biochim Biophys Acta. 2010;1801:860–867. doi: 10.1016/j.bbalip.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Onyewuchi O, Yang S, Liu R, Simpkins JW. Increased β-secretase activity and expression in rats following transient cerebral ischemia. Brain Res. 2004;1009:1–8. doi: 10.1016/j.brainres.2003.09.086. [DOI] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the β-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Yamada K, Yabuki C, Seubert P, Schenk D, Hori Y, Ohtsuki S, Terasaki T, Hashimoto T, Iwatsubo T. Aβ immunotherapy: intracerebral sequestration of Aβ by an anti-Aβ monoclonal antibody 266 with high affinity to soluble Aβ. J Neurosci. 2009;29:11393–11398. doi: 10.1523/JNEUROSCI.2021-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Furuhata M, Kamiya S, Hisada M, Miyaji H, Magami Y, Yamamoto K, Fujiwara H, Mizuguchi J. Positive modulation of IL-12 signaling by sphingosine kinase 2 associating with the IL-12 receptor beta 1 cytoplasmic region. J Immunol. 2003;171:1352–1359. doi: 10.4049/jimmunol.171.3.1352. [DOI] [PubMed] [Google Scholar]
- Zemann B, Kinzel B, Müller M, Reuschel R, Mechtcheriakova D, Urtz N, Bornancin F, Baumruker T, Billich A. Sphingosine kinase type 2 is essential for lymphopenia induced by the immunomodulatory drug FTY720. Blood. 2006;107:1454–1458. doi: 10.1182/blood-2005-07-2628. [DOI] [PubMed] [Google Scholar]