

Abstract
Utilization of the cytochrome P450 3A4 (CYP3A4) inhibitor ritonavir as a pharmacoenhancer for anti-HIV drugs revolutionized the treatment of HIV infection. However, owing to ritonavir-related complications, there is a need for development of new CYP3A4 inhibitors with improved pharmacochemical properties, which requires a full understanding of the CYP3A4 inactivation mechanisms and the unraveling of possible inhibitor binding modes. We investigated the mechanism of CYP3A4 interaction with three desoxyritonavir analogues, containing the heme-ligating imidazole, oxazole, or pyridine group instead of the thiazole moiety (compounds 1, 2, and 3, respectively). Our data show that compound 3 is superior to ritonavir in terms of binding affinity and inhibitory potency owing to greater flexibility and the ability to adopt a conformation that minimizes steric clashing and optimizes protein–ligand interactions. Additionally, Ser119 was identified as a key residue assisting binding of ritonavir-like inhibitors, which emphasizes the importance of polar interactions in the CYP3A4–ligand association.
INTRODUCTION
Human cytochrome P450 3A4 (CYP3A4) is the major drug-metabolizing enzyme that oxidizes a wide range of structurally diverse xenobiotics and prescribed pharmaceuticals. Bioavailability and therapeutic efficiency of these administered drugs, therefore, depends on whether/how fast they are degraded by CYP3A4. CYP3A4 inhibition, on the other hand, leads to increased plasma levels of pharmaceuticals. CYP3A4-related drug–drug interactions are usually undesirable, as they may cause toxicity and other adverse complications. Carefully controlled CYP3A4 inactivation, however, can be clinically useful.
Treatment of HIV infection is one example of a beneficial CYP3A4 inactivation. HIV protease inhibitors, an important part of antiretroviral therapy, are rapidly metabolized primarily by liver and intestinal CYP3A4. Co-administration of low doses of ritonavir, an HIV protease inhibitor and a potent CYP3A4 inactivator (Figure 1), elevates plasma levels of HIV protease inhibitors, which enables more effective dosing regiments.1
Figure 1.
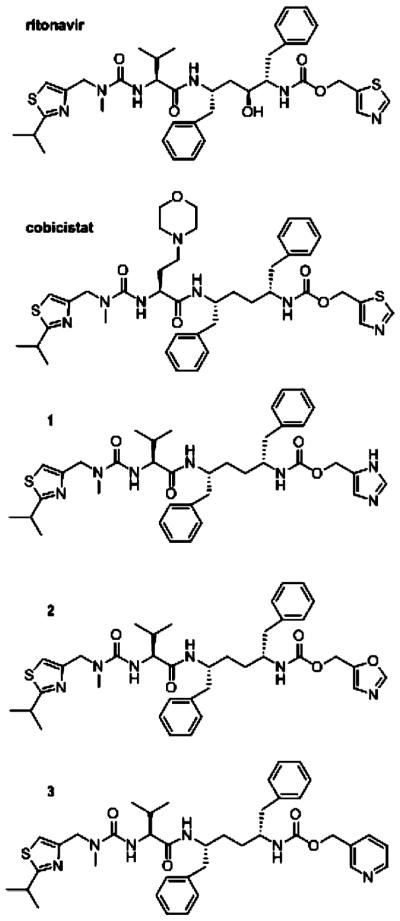
Chemical structures of ritonavir, cobicistat, and compounds 1–3.
Although ritonavir has been successfully utilized as a pharmacoenhancer (booster) for over a decade, there are several important complications associated with this drug. Because ritonavir was originally developed as an HIV protease inhibitor, its administration in subtherapeutic doses can promote the emergence of HIV drug resistance. Another disadvantage is the ability of ritonavir to affect activities of other CYPs, as well as the phase II conjugating enzymes and drug transporters.2,3 Ritonavir also activates xenobiotic-sensing receptors that regulate expression of CYP3A4,4 which can decrease the overall inhibitory potency of ritonavir upon its prolonged use. Additional side effects include lipid disorders and gastrointestinal disturbance. Finally, owing to poor physicochemical properties and, in particular, low aqueous solubility, ritonavir cannot be conveniently coformulated with other antiretroviral agents. Hence, it is highly desirable to develop boosters with more favorable pharmacological and physicochemical properties and fewer off-target effects.
Several companies are currently involved in the development of novel CYP3A4 inhibitors that could be used as pharmacoenhancers.1 One such compound, cobicistat, has been approved recently as part of a combination pill for treating HIV-1 infection in treatment-naïve patients. Cobicistat (Figure 1) is a ritonavir analogue lacking the key hydroxyl group required for the HIV protease binding and inhibition and is devoid of anti-HIV activity.5,6 Functional assays on human liver microsomes showed that cobicistat acts on the CYP3A enzymes more selectively than ritonavir and via a similar mechanism, although with somewhat lower effectiveness (kinact/Ki of 0.57 vs 0.90 min−1 μM−1 for ritonavir).6 Other advantages of the new booster include higher aqueous solubility, improved tolerability, and reduced off-target drug–drug interactions. No structural and functional information on the CYP3A4–cobicistat complex is currently available and, thus, the precise mechanism of binding and inhibition remain unknown.
Previously, we utilized spectroscopic, kinetic and structural approaches to investigate the mechanism of CYP3A4 inhibition by ritonavir and desthiazolylmethyloxycarbonyl ritonavir (DTMCR), an analogue that lacks the heme-ligating thiazole moiety.7,8 These in vitro studies demonstrated that heme ligation is essential for tight association of ritonavir-like compounds, whereas nonbonding interactions provided by the side groups define the binding mode. To further investigate the mechanism of CYP3A4 inactivation and to test whether the affinity and inhibitory potency of ritonavir can be further improved, we compared the properties of three desoxyritonavir analogues where the thiazole moiety was replaced with imidazole, oxazole, or pyridine (compounds 1, 2, and 3, respectively; Figure 1). On the basis of inhibitor binding affinity and association kinetics, as well as thermal denaturation, activity inhibition assays, mutagenesis, and X-ray data, we conclude that the pyridine-containing compound 3 is a stronger ligand and a more potent CYP3A4 inhibitor than ritonavir. In addition, Ser119 was shown to be a key residue that assists association of ritonavir-like inhibitors and stabilizes the ligand-bound form.
RESULTS
Binding Affinity and Association Kinetics of Compounds 1–3
All three desoxyritonavir analogues induced type II spectral changes in CYP3A4 (Figure 2), indicative of direct nitrogen ligation to the heme iron. The largest red-shift in the Soret band (416–424 nm) was caused by compound 1. According to the spectral dissociation constants (Ks) calculated from the equilibrium titrations (Insets (a) and (b) in Figure 2, Table 1), 1 and 3 have a 2-fold higher affinity for CYP3A4 than ritonavir, whereas the binding affinity of 2 is 10-fold lower.
Figure 2.

Spectral changes induced in CYP3A4 upon binding of 1 (A), 2 (B), and 3 (C). Absorbance spectra of ferric ligand-free (–), ligand-bound (---), ferrous ligand-bound (…), and ferrous CO complex (-·-·-·) were recorded at room temperature in 50 mM phosphate buffer, pH 7.4, containing 20% glycerol and 1 mM dithiothreitol. Insets (a) and (b) are absorbance changes observed during titration of CYP3A4 with 1–3 and plots of absorbance change vs ligand concentration, respectively.
Table 1.
Properties of Ritonavir and Compounds 1–3
| 442 nm band (Fe2+) | Ksa (nM) | Eo,7b (mV) | kfastc (s−1) | Tmd (°C) | IC50e (nM) | |
|---|---|---|---|---|---|---|
| ritonavir | +f | 51 ± 10f | −350 ± 5f | 1.4 ± 0.3f | 54.8 ± 0.1 | 550 ± 50 |
| 1 | − | 22 ± 4 | <<-370 | 1.9 ± 0.3 | 56.5 ± 0.2 | 280 ± 40 |
| 2 | ± | 570 ± 30 | ≈ −370 | 2.2 ± 0.4 | 54.8 ± 0.1 | 3400 ± 60 |
| 3 | + | 25 ± 5 | −350 ± 5 | 7.0 ± 0.5 | 57.2 ± 0.2 | 130 ± 15 |
Spectral dissociation constant.
Midpoint redox potential of the inhibitor-bound CYP3A4. Eo,7 of the ligand-free CYP3A4 is ~−300 mV.7
Rate constant of the fast phase of the ligand binding reaction.
Melting temperature of the CYP3A4-inhibitor complex. Tm for the ligand-free CYP3A4 in the absence and presence of 2% DMSO is 52.6 ± 0.1 and 52.1 ± 0.1 °C, respectively.
Concentration required for half-maximal inactivation of recombinant CYP3A4.
Determined previously.7
Similar to the ritonavir-ligated CYP3A4,7 the protein bound to compound 3 was fully converted to a 442 nm absorbing species upon reduction with sodium dithionite (Figure 2C). The conversion was partial for the CYP3A4–2 complex (~60%) and negligible for the 1-bound form (Figure 2A,B). Such spectral differences allowed us to test whether 1 and 2 can be displaced from the active site by stronger ligands. Displacement titrations showed that both ritonavir and 3 can substitute 1 and 2 and that the binding affinity of 3 for the 1- and 2-bound CYP3A4 was several-fold higher (Supporting Information Figure 1S). Compound 3 also has a higher affinity for CYP3A4 complexed with bromoergocryptine, a type I substrate (Ks of 5 vs 50 nM for ritonavir).7 Finally, 1 and 2 but not 3 dissociated from CYP3A4 during repetitive concen-tration/dilution cycles. Thus, the pyridine-containing 3 binds to CYP3A4 irreversibly and stronger than ritonavir.
It should be noted that ligation of compound 1 to CYP3A4 significantly slows down/impedes heme reduction, which could not be completed under the experimental conditions used. This could be the consequence of a large negative shift in the heme redox potential caused by the 1 ligation (>>70 mV, Table 1). A decrease in Eo,7 induced by 2 and 3 is ~50 and 70 mV, respectively.
Kinetic analyses showed that the reaction of CYP3A4 binding to 1, 2, and 3 was biphasic, with the rate constants for the fast and slow phases (kfast and kslow, respectively) increasing with an increase in ligand concentration (Figure 3). Although the kfast vs [ligand] dependence was hyperbolic for all compounds, the fits to the plots for 1 and 2 did not pass through the origin. This differs from the ritonavir binding reaction, which i s m ono phasic a t s ubeq u imolar inhibitor:CYP3A4 ratios and biphasic when the ligand is present in an excess.8 Most notably, the kfast vs [ritonavir] plot is V-shaped,8 with the breaking point at the inhibitor:CYP3A4 ratio reaching unity (Figure 3). Finally, ritonavir ligates to the heme slower than the investigated compounds, especially 3, kfast which is 5-fold higher (Table 1). For irreversible type II inhibitors of CYP3A4, kfast would be proportional to the inactivation rate constant (kinact) and, hence, can serve a measure of the inhibitory potency.
Figure 3.
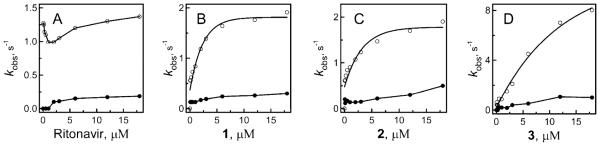
Plots of the observed rate constants for the fast (○) and slow (●) kinetic phases of the ritonavir and 1–3 binding to WT CYP3A4 vs ligand concentration.
Effect of Ritonavir-Like Molecules on CYP3A4 Stability
Tight-binding ligands often increase thermal stability of target proteins. To investigate how type II inhibitors affect stability of CYP3A4, we compared melting curves for different ligand-bound forms. Association of all investigated compounds led to an increase in the melting temperature (Tm) of CYP3A4, but the most pronounced stabilizing effect was caused by ligation of 3 (Figure 4A, Table 1). Thus, a simple and quick measurement of ligand-mediated shifts in Tm can be used as an additional tool for identification of high affinity CYP3A4 binders.
Figure 4.
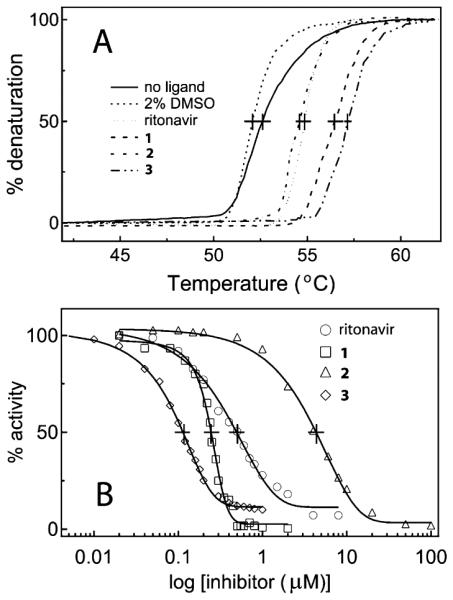
Effect of ritonavir and compounds 1–3 on thermal stability (A) and the BFC hydroxylation activity of CYP3A4 (B). The denaturation and inhibitory assays were conducted as described in the Experimental Section. The Tm and IC50 values derived from the plots (A) and (B), respectively, are given in Table 1.
Inhibitory Potency of Compounds 1–3
The inhibitory potency of the ritonavir analogues toward CYP3A4 was compared in vitro in a reconstituted system with the redox partner cytochrome P450 reductase (CPR) and 7-benzyloxy-4-(trifluoromethyl)coumarin (BFC) as a substrate. Formation of the hydroxylated product was monitored fluoroscopically, and the percentage of the activity remaining was plotted vs ligand concentration to determine a concentration required for half-maximal inactivation (IC50), a measure of the inhibitory effectiveness (Figure 4B). The derived IC50 values are in good agreement with the binding affinity (Table 1): compounds 1 and 3 inhibited the BFC hydroxylation 2–4-fold more effectively than ritonavir, whereas the inactivating potency of 2 was 6-fold lower. Further, the ratio between IC50s measured for ritonavir and its analogues under conditions of co- and preincubation with NADPH were close to unity (~1.1–1.3). This suggests that all compounds are very weak time-dependent inhibitors, inactivating CYP3A4 mainly through heme ligation rather than formation of a reactive metabolite(s). Taken together, the experimental data indicate that 3 is a stronger type II ligand and more potent inhibitor than ritonavir acting via a similar mechanism.
Crystal Structures of CYP3A4 Bound to Compounds 2 and 3
Although compound 1 promoted CYP3A4 crystallization, it did not remain in the active site of the crystalline protein or bind somewhere else. Instead, three water molecules were observed near the heme, one of which was directly coordinated to the iron (Figure 5A). This and an inward orientation of the F–F′ loop, characteristic for the ligand-free 1TQN and 1WOE structures,9,10 indicate that the protein is in a six-coordinated resting state. Compound 2 did cocrystallize with CYP3A4, but the electron density was seen only for the heme-ligating oxazole group (Figure 5B). Another indication of 2 present in the active site was a notable displacement of the I-helix, likely caused by steric hindrance with Phe304 (explained below).
Figure 5.
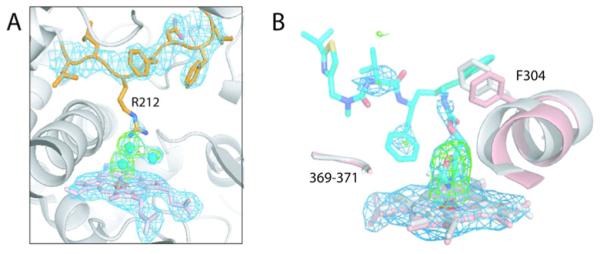
(A) The active site of CYP3A4 crystallized in the presence of 1. Compound 1 is not bound to the heme (shown in pink) or anywhere else. Instead, there are three water molecules (cyan spheres), one of which is directly coordinated to the heme iron. Residues comprising the F–F′ loop are rendered in orange. The side chain of Arg212 is disordered but can be modeled in an inward position as observed in the 1TQN model. (B) 2 remains bound to the heme during crystallization. However, only the heme-ligating oxazole moiety is well-defined in the X-ray structure, with the rest of the molecule being disordered. Compound 2 is modeled in an orientation similar to that of 3 (see Figure 6). Parts of the I-helix in the ligand-free and 2-bound CYP3A4 are displayed in gray and pink, respectively. A notable displacement in the I-helix is caused by steric hindrance between the ligand and Phe304. The 369–370 peptide, however, remains in place. 2Fo – Fc and Fo – Fc electron density maps contoured at 1σ and 3σ are colored in blue and green, respectively.
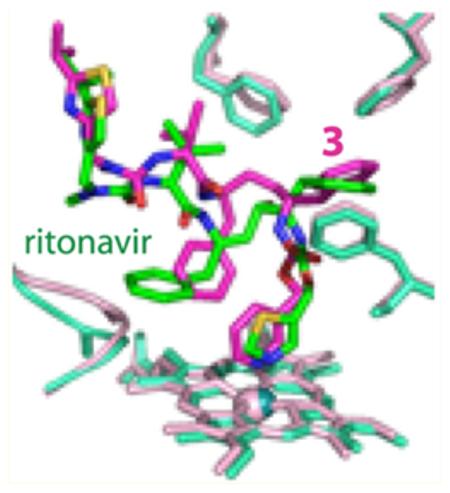
In contrast, in the CYP3A4–3 complex structure, only the isopropyl-thiazole of the ligand was undefined (Figure 6A). Compound 3 orients in the active site similarly to ritonavir but with a few notable differences. As observed for ritonavir,7 the phenyl group closest to the heme-ligating moiety in 3 (Phenyl-1) is imbedded into a hydrophobic pocket lined with the phenylalanine and leucine residues and, owing to steric clashing with Phe304, displaces the I-helix (Figure 6A). The second phenyl (Phenyl-2), however, does not protrude into a cavity above the heme plane as deeply as the respective group of ritonavir does. This eliminates steric hindrance with the Ile369-Ala370 peptide and prevents the heme shift observed in the 3NXU structure (Figure 6B). As a result, the Fe–N bond in the CYP3A4–3 complex is slightly shorter (2.1 vs 2.2–2.3 in the ritonavir-bound protein).
Figure 6.
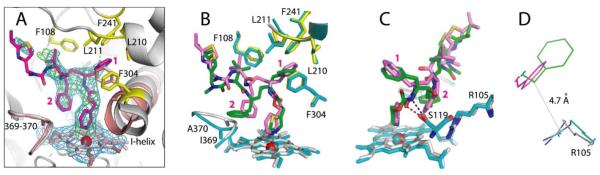
Crystal structure of the CYP3A4–3 complex. (A) Compound 3 (in magenta) binds to the heme via the pyridine nitrogen. Most of the ligand–protein interactions are provided by the phenyl side groups, labeled as (1) and (2). Phenyl-1 is imbedded into a hydrophobic pocket lined with Phe108, Leu210, Leu211, Phe241, and Phe304 (shown in yellow sticks). Steric clashing between Phenyl-1 and Phe304, which is also observed in the CYP3A4–ritonair complex, leads to the I-helix displacement. Phenyl-2 of 3, however, imposes no steric hindrance on the 369–370 peptide and, as a result, there is no heme shift. For comparison, the heme, 369–370 peptide, and I-helix of the superimposed ligand-free 1TQN structure are shown in pink. 2Fo – Fc and Fo – Fc electron density maps contoured at 1σ and 3σ are displayed in blue and green, respectively. (B,C) Relative orientation of 3 (magenta) and ritonavir (green). The heme, 369–370 peptide, and residues lining the hydrophobic pocket in the ritonavir-bound 3NXU structure are colored in cyan. Owing to higher flexibility, 3 binds in a conformation that eliminates clashing between Phenyl-2 and Ile369-Ala370 (B). Furthermore, Phenyl-2 orients near parallel to the pyridine ring and the guanidinium group of Arg105. This promotes π-stacking and cation–π interactions, stabilizing the complex. A peptide flip in 3 allows formation of a stronger hydrogen bond with Ser119 via the amide nitrogen (depicted as a blue dashed line in (C)). In contrast, ritonavir is H-bonded to Ser119 via the carbonyl oxygen (red dashed line). Such conformational differences could increase the binding affinity and inhibitory potency of compound 3. (D) A closer view at the Phe-2–Arg105 interaction site demonstrates that only the phenyl ring of 3 (magenta) can establish cation–π interactions with the Arg105 guanidinium group.
Another important difference is the relative orientation of Phenyl-2. In compound 3, this moiety is placed closer and near parallel to the heme-ligating pyridine and the guanidinium group of Arg105 (Figure 6B,C), thereby promoting π-stacking and cation–π interactions. The conformation of 3 is further stabilized by the inhibitor amide nitrogen donating a hydrogen atom to the Ser119 hydroxyl group. In contrast, the carbonyl oxygen of ritonavir accepts an H-bond from Ser119 (Figure 6C). Because 3 is in a more elevated position due to a larger pyridine ring, the peptide bond flip optimizes and strengthens the H-bond, whose length is 2.5 vs 2.8 Å in the ritonavir-bound structure. Altogether, such dissimilarities may increase the binding affinity and, consequently, the inhibitory potency of 3.
Effect of the S119A Substitution on the CYP3A4–Inhibitor Interaction
To elucidate the role of Ser119 in the ligand binding process, we investigated properties of the S119A mutant of CYP3A4. Elimination of the H-bond forming hydroxyl group in Ser119 significantly perturbed association of the ritonavir-like molecules. The mutation led to a several-fold decrease in the binding affinity of all investigated compounds, most notably of ritonavir (Table 2; Supporting Information Figure 2S). The more drastic effect was on the kinetics of inhibitor ligation. While all reactions remained biphasic, the kfast values were decreased by 6–22-fold (Figure 7 and Supporting Information Figure 2S, Table 2). Most strikingly, the kfast vs (compounds 1–3) plots were no longer hyperbolic (compare respective panels in Figures 3 and 7), in that the rate constants did not change or slightly decreased until the inhibitor:CYP3A4 ratio reached unity and then gradually increased. kfast for ritonavir, on the other hand, remained virtually unchanged at supra-equimolar ligand concentrations.
Table 2.
Effect of the S119A Mutation on the CYP3A4-Inhibitor Interaction
| Ks (nM) | kfast (s−1) | Tm (°C)a | |
|---|---|---|---|
| ritonavir | 240 ± 20 | 0.25 ± 0.03 | 55.2 ± 0.2 |
| 1 | 67 ± 5 | 0.17 ± 0.02 | 55.5 ± 0.2 |
| 2 | 970 ± 80 | 0.20 ± 0.03 | 53.2 ± 0.1 |
| 3 | 65 ± 9 | 0.32 ± 0.04 | 55.6 ± 0.2 |
Melting temperatures for the ligand-free CYP3A4 S119A in the absence and presence of 2% DMSO are 54.0 ± 0.1 and 53.5 ± 0.1 °C, respectively.
Figure 7.
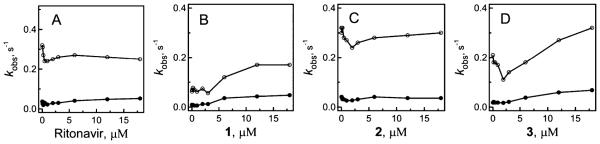
Plots of the observed rate constants for the fast (○) and slow (●) kinetic phases of the ritonavir and compounds 1–3 binding to CYP3A4 S119A vs ligand concentration.
Thermal stability of CYP3A4 was also affected by the S119A substitution in a ligand-dependent fashion. The melting temperature for the ligand-free and ritonavir-bound mutant was increased by 1.4 and 0.4 °C, respectively, indicative of some stabilization of the protein fold. In contrast, Tm for the 1-, 2-, and 3-bound CYP3A4 was 1–1.6 °C lower than the respective values derived for the wild type (WT; Tables 2 and 3). It can be concluded, therefore, that formation of the Ser119-mediated H-bond not only assists association of compounds 1–3 but also stabilizes the resulting complexes. The lower flexibility of ritonavir and its inability to optimally orient in the active site are the likely reason for the lack of the stabilizing effect of the Ser119-mediated interaction.
Table 3.
X-ray Data Collection and Refinement Statistics
| ligand |
|||
|---|---|---|---|
| water | 2 | 3 | |
| Data Statistics | |||
| space group | I222 | I222 | I222 |
| unit cell parameters | a = 76 Å, b = 99 Å, c = 125 Å; α,β,γ = 90° | a = 76 Å, b = 99 Å, c = 126 Å; α, β, γ = 90° | a = 78 Å, b = 100 Å, c = 131 Å; α, β, γ = 90° |
| resolution range | 65.3–2.60 (2.67–2.60)a | 49.8–2.72 (2.79–2.72) | 65.7–2.90 (2.98–2.90) |
| total reflections | 115923 | 114890 | 50075 |
| unique reflections | 16260 | 12236 | 11329 |
| redundancy | 7.1 (4.5) | 9.4 (5.4) | 4.4 (4.5) |
| completeness | 98.4 (97.5) | 99.9 (98.9) | 97.2 (99.0) |
| average I/σI | 10.3 (2.3) | 18.9 (3.5) | 10.9 (2.5) |
| R merge | 0.082 (0.516) | 0.072 (0.505) | 0.068 (0.548) |
| Refinement Statistics | |||
| R/R free b | 22.0/29.8 | 20.7/26.9 | 19.5/26.9 |
| rmsd | |||
| bond lengths (Å) |
0.011 | 0.009 | 0.011 |
| bond angles (deg) |
2.0 | 1.89 | 1.98 |
Values in brackets are for the highest resolution shell.
Rfree was calculated from a subset of 5% of the data that were excluded during refinement.
DISCUSSION
Inhibition of the drug-metabolizing CYP3A4 is usually undesired and considered as a liability.11 A successful utilization of CYP3A4 inhibitors in treatment of HIV infection,1 however, suggests that such compounds could help fighting other chronic conditions requiring administration of costly drugs that are predominantly cleared by CYP3A4. Therefore, there is a need for development of new boosters with improved pharmaco-logical and physicochemical properties, which is not possible without a full understanding of the CYP3A4 inhibitory mechanism.
In contrast to the prevalent notion that ritonavir is a mechanism-based inactivator of the CYP3A family of enzymes,12–16 our studies on highly purified recombinant CYP3A4 demonstrated that the key contribution to CYP3A4 inhibition is the ability of ritonavir to bind tightly and irreversibly, causing a negative shift in the heme redox potential which precludes an electron flux from CPR.7 Biochemical and structural comparisons of ritonavir and DTMCR binding, in turn, showed that tight association of ritonavir-like molecules to CYP3A4 requires heme coordination, while hydrophobic interactions provided by the side groups define the overall binding mode.8 The atypical (nonhyperbolic, V-shaped) ritonavir association kinetics was proposed to arise from the existence of a peripheral site, to which the drug may dock prior to moving into the active site cavity.8
In this work, we investigated how elimination of the ritonavir hydroxyl group, strictly required for the HIV protease inactivation, and substitution of the heme-ligating moiety influence the ability to bind and inhibit CYP3A4. The effect of thiazole-to-pyridine replacement in ritonavir has been previously investigated by assessing CYP-related spectral changes and IC50 for the CYP3A terfenadine hydroxylase activity in human liver microsomes.17 The magnitude of type II spectral changes induced by ritonavir and its pyridine-substituted derivative (Abbott laboratories compound A-83962) in micro-somal CYPs was found to be similar, but the inhibitory potency of A-83962 was 4-fold lower. Such a big difference in IC50 was explained by a greater liability of the pyridine moiety to chemical oxidation.
In contrast, here we show that the pyridine-substituted desoxyritonavir (compound 3) is superior to ritonavir in terms of binding affinity and inhibitory potency for CYP3A4 (Table 1). The extremely tight binding of 3 to CYP3A4 is likely due in part to a favorable interaction of the heme iron with the pyridine ring in the ligand, involving both σ-donation by the pyridine nitrogen lone pair and back-bonding of iron d-orbitals to antibonding orbitals of the aromatic π-system. An analogous mode of binding is possible with an sp2 lone pair of the imidazole-containing analogue, compound 1, which on the basis of σ-donating ability alone would be expected to form the tighter complex because of its greater intrinsic basicity. The reduced effectiveness of back-bonding from iron to imidazole, however, results in a lower overall binding affinity of 1. In addition, the imidazole ring has two tautomeric forms each with an sp2 lone pair that could in principle complex with the iron, and neither of which appears to map closely onto the corresponding nitrogen of the pyridine analogue. Thus, it is also possible that the stereoelectronic requirements for favorable binding play a significant role in controlling the binding affinities due to the constraints imposed by the tethered heteroatoms in each ligand. The magnitude of the 442 nm absorption of the ferrous species (negligible for the CYP3A4–1 complex; Figure 1) may reflect the Fe–N bonding strength and, hence, this parameter should be taken into consideration together with the Ks values during spectral evaluation of potential CYP3A4 inhibitors. The fact that compound 1 potently inhibits the BFC hydroxylating activity of CYP3A4 (Table 1) can be explained by a large negative shift in the heme redox potential, which may completely block an electron flow from CPR. Most likely, 1 gets expelled from the active site during crystallization because of the less favorable stereoelectronic properties of the imidazole nitrogen, leading to a weaker, dissociable Fe–N bond.
Our study also emphasizes the importance of two other factors that could control the CYP3A4–ligand interaction and define ligand binding mode. The first one is a structural flexibility of the ritonavir-like molecules, which increases upon elimination of the hydroxyl group. This allows 3 to adopt a conformation that minimizes steric hindrance imposed by Phenyl-2 (Figure 6B). Most importantly, Phenyl-2 can now orient parallel to the pyridine ring and the guanidinium group of Arg105 (Figure 6C,D) to form π-stacking and cation–π interactions, respectively, that stabilize the inhibitor-bound complex. Clashing between Phenyl-1 and Phe304 and the resulting displacement of the I-helix, however, remain significant (Figure 6A). This unfavorable interaction can be prevented if Phenyl-1 is substituted with a smaller hydrophobic moiety.
The second factor that can modulate both the rate of formation and stability of the CYP3A4–inhibitor complexes is the H-bonding interaction with Ser119 (Figure 6C). The active site Ser119 was reported to regulate substrate specificity and steroid hydroxylation activity of CYP3A4,18 as well as the binding/egress of type II inhibitors metyrapone and ketoconazole, and association of 4-aminopiperidine drugs.19–22 Here we show that elimination of the Ser119 hydroxyl group significantly lowers the binding affinity of ritonavir and compounds 1–3 and perturbs their ligation kinetics (Tables 1 and 2; Supporting Information Figure 2S). Furthermore, the S119A mutation transforms the hyperbolic character of the kfast vs [compound 1–3] dependence into V-shaped (Figures 3 and 7), and affects thermal stability of CYP3A4 in a ligand-dependent manner (Figure 4A, Tables 1 and 2). On the basis of these findings, we suggest that (i) Ser119 interacts with the inhibitor molecules at earlier stages of the binding process, (ii) deviations from the hyperbolic binding kinetics may, in part, be caused by conformational restraints affecting the Ser119-mediated protein–ligand interaction, and (iii) formation of the H-bond with Ser119 increases stability of the CYP3A4–inhibitor complex when conformational restraints in the ligand are minimized or eliminated. The notably decreased Tm and kfast and higher Ks values for the CYP3A4 S119A–3 complex (Tables 1 and 2) imply that the peptide flip, bringing the carbonyl oxygen of 3 closer to the Ser119 hydroxyl group, stabilizes the overall conformation and strengthens inhibitor binding.
That preincubation of 3 and other ritonavir analogues with NADPH does not significantly affect IC50 suggests an inhibitory mechanism that does not involve formation of a reactive metabolite. This agrees with our previous conclusions7 and a recent study on human microsomal CYP3A4.23 Although formation of reactive metabolites of ritonavir was proposed over a decade ago,12,13,15 their identity remains unknown. Comparative metabolomic screening of WT and Cyp3a-null mice led to a conclusion that CYP3A enzymes can bioactivate ritonavir via glycine conjugation and thiazole ring-opening pathways.24 Metabolism of compound 3 has not been investigated in this study, but the primary oxidation site is expected to be the terminal isopropyl group. This follows from the structural analogy of 3 to ritonavir12,24 and cobicistat, as well as biphasic binding kinetics (Figure 3), indicative of multiple orientation modes for ritonavir-like molecules within the active site of CYP3A4.
Owing to insufficient resolution or/and high thermal motion of the 3 isopropyl-thiazole, the role of this terminal group in the interaction with CYP3A4 could not be determined. It should be noted though that the CYP3A4 active site region is similar in both 3- and ritonavir-bound structures (the rms deviation for the 457 Cα atoms is only 0.53 Å), and the isopropyl-thiazole of 3 can be modeled in a nearly identical conformation (Figure 6B). This means that a water-mediated bridge between the isopropyl-thiazole nitrogen of 3 and “polar umbrella” (residues Asp61, Asp76, Arg106, Arg372, and Glu374), analogous to that observed in the ritonavir-bound structure,7 can be easily established. In either case, the data presented here is sufficient to conclude that compound 3 is a higher affinity ligand and a more potent CYP3A4 inhibitor than ritonavir and, hence, it can serve as a better template for developing novel pharmacoen-hancers.
CONCLUSIONS
The CYP3A4 inhibitor ritonavir has been successfully used as a booster for anti-HIV drugs for over a decade. However, there is presently a search for new CYP3A4 inhibitors that have improved pharmacochemical properties and cause fewer side effects. In this study, we investigated whether the CYP3A4 inhibitory potency of ritonavir can be further improved by comparing the properties of three desoxyritonavir analogues that contain the imidazole, oxazole, or pyridine group instead of the heme-ligating thiazole (compounds 1, 2, and 3, respectively). On the basis of the binding affinity, association and inhibitory kinetics, stabilization effect, and the binding mode of the investigated compounds, we conclude that 3 is a stronger ligand and a more potent CYP3A4 inhibitor than ritonavir. This, in part, is due to the favorable stereoelectronic properties of the pyridine nitrogen and increased structural flexibility of the desoxyritonavir backbone, which allows 3 to establish a stronger Fe–N bond and adopt a conformation that minimizes steric clashing and optimizes protein–ligand interactions, in particular, with Ser119. Our mutagenesis data show that the active site Ser119 is a key residue that not only assists binding of ritonavir-like inhibitors but also stabilizes the ligand-bound form. Thus, polar interactions are important in the CYP3A4–ligand association and need to be taken into consideration during the drug design process. Compound 3 may serve as a starting template for the development of a new generation of potent CYP3A4 inhibitors. One way for its structural optimization could be replacement of the Phenyl-1 side group with a smaller hydrophobic moiety in order to prevent steric clashing with Phe304 and the I-helix displacement.
EXPERIMENTAL SECTION
Synthesis of compounds 1–3 is described in the Supporting Information. The purity of 1–3 was >95% as determined by HPLC analysis.
Protein Expression and Purification
C-terminal His-tagged WT and S119A CYP3A4Δ3-22 were expressed in Escherichia coli, purified, and quantified as reported previously.7
Spectral Binding Titrations
Ligand binding to WT and S119A CYP3A4 was monitored spectrophotometrically. Proteins were titrated with small aliquots of dimethyl sulfoxide (DMSO) solutions of ritonavir (Toronto Research Chemicals), 1, 2, or 3 in 50 mM phosphate, pH 7.4, 20% glycerol, and 1 mM dithiothreitol (buffer A). The final solvent concentration did not exceed 2%. Spectral dissociation constants (Ks) were determined as described in detail elsewhere.7
Kinetics of Ligand Binding
Kinetics of ligand binding to CYP3A4 was monitored at room temperature in a SX.18MV stopped flow apparatus (Applied Photophysics, UK). Protein solutions (2 μM) were mixed with various concentrations of inhibitors in 50 mM phosphate, pH 7.4, and absorbance changes were followed at 426 nm for ritonavir and 2, and at 430 and 425 nm for 1 and 3, respectively. Owing to low solubility, the maximal concentration of compounds 1–3 used in the stopped flow experiments was 36 µM. Kinetic data were analyzed using the program IgorPro (WaveMetrics, Inc.).
Redox Potential Measurement
Redox potentials of the inhibitor-bound CYP3A4 were estimated spectroscopically as previously described7 using benzyl viologen (−359 mV) as a redox dye.
Thermal Denaturation
CYP3A4 (2 µM) in the absence and presence of 10 µM inhibitors or 2% DMSO was heated in 100 mM phosphate buffer, pH 7.4, in a controlled way using a Cary 3 spectrophotometer, with a ramp rate of 0.5 °C/min and the 37–65 °C temperature range. Protein denaturation was monitored at 280 nm. A denaturation midpoint (melting temperature, Tm) was determined as a temperature at which the folded and unfolded states are equally populated.
Inhibitory Potency Assays
The inhibitory potency of ritonavir and compounds 1–3 on the BFC hydroxylase activity of CYP3A4 was evaluated fluorometrically in a reconstituted system with CPR. The reaction was carried out at room temperature in 25 mM phosphate buffer, pH 7.4, containing 3 mM MgCl2. CYP3A4 (0.3 µM) and rat CPR (0.6 µM) were incubated for 5 min with various concentrations of inhibitors in the absence and presence of 100 µM NADPH and then mixed with 50 µM BFC. Formation of 7-hydroxy-4-trifluoromethyl-coumarin was followed in a Hitachi F100 fluorimeter with λex = 430 nm and λem = 500 nm. A concentration required for half-maximal inactivation (IC50) was derived from the [% activity] vs [inhibitor] plots.
Crystallization and Determination of the X-ray Structures of the CYP3A4–Inhibitor Complexes
CYP3A4 was crystallized in the presence of inhibitors by a microbatch method under oil. Ligand-bound CYP3A4 (0.6 µL, 54–56 mg/mL, with a 2–3-fold ligand excess) in buffer A was mixed with 0.6 µL of 10–11% polyethylene glycol 3350 and 90–100 mM sodium malonate, pH 5.9–6.0, and covered with paraffin oil. Crystals grew within several days at room temperature and belonged to the I222 space group, with one molecule per asymmetric unit. X-ray diffraction data were collected at the Stanford Synchrotron Radiation Laboratory beamline 7–1, using Paratone-N as a cryoprotectant. Crystal structures were solved by molecular replacement with PHASER25 and the 1TQN or 3NXU structure as a search model. Only water molecules were present in the active site of CYP3A4 crystallized in the presence of compound 1. The water-, 2-, and 3-bound models were refined to 2.60, 2.72, and 2.9 Å resolution with COOT26 and REFMAC,25 with the final R/Rfree values of 22.0/29.8, 20.7/26.9, and 19.5/26.9, respectively. Data collection and refinement statistics are summarized in Table 3.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant GM33688, Gilead Sciences, Inc., and the California Center for Antiviral Drug Discovery and involves research carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. We thank Dr. R. Chamberlin for helpful discussions and critical reading of the manuscript and Lianhong Xu and Hong Ye of Gilead Sciences for synthesizing and providing the compounds used in this study.
ABBREVIATIONS USED
- CYP
cytochrome P450
- CYP3A4
3A4 isoform of cytochrome P450
- CPR
cytochrome P450 reductase
- DMSO
dimethyl sulfoxide
- DTMCR
desthiazolylmethyloxycarbonyl ritonavir
- WT
wild type
Footnotes
Supporting Information
Synthesis of compounds 1–3, experimental evidence for reversibility of the 1 and 2 binding to CYP3A4, and spectral and kinetic data on association of ritonavir, 1, 2, and 3 to CYP3A4 S119A. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Atomic coordinates and structure factors for the water-, 2-, and 3-bound CYP3A4 have been deposited to the Protein Data Bank with the ID codes 4I3Q, 4I4G, and 4I4H, respectively.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Xu L, Desai MC. Pharmacokinetic enhancers for HIV drugs. Curr. Opin. Invest. Drugs. 2009;10:775–786. [PubMed] [Google Scholar]
- (2).Wyen C, Fuhr U, Frank D, Aarnoutse RE, Klaassen T, Lazar A, Seeringer A, Doroshyenko O, Kirchheiner JC, Abdulrazik F, Schmeisser N, Lehmann C, Hein W, Schomig E, Burger DM, Fatkenheuer G, Jetter A. Effect of an antiretroviral regimen containing ritonavir boosted lopinavir on intestinal and hepatic CYP3A, CYP2D6 and P-glycoprotein in HIV-infected patients. Clin. Pharmacol. Ther. 2008;84:75–82. doi: 10.1038/sj.clpt.6100452. [DOI] [PubMed] [Google Scholar]
- (3).Foisy MM, Yakiwchuk EM, Hughes CA. Induction effects of ritonavir: implications for drug interactions. Ann. Pharmacother. 2008;42:1048–1059. doi: 10.1345/aph.1K615. [DOI] [PubMed] [Google Scholar]
- (4).Dixit V, Hariparsad N, Li F, Desai P, Thummel KE, Unadkat JD. Cytochrome P450 enzymes and transporters induced by anti-human immunodeficiency virus protease inhibitors in human hepatocytes: implications for predicting clinical drug interactions. Drug Metab. Dispos. 2007;35:1853–1859. doi: 10.1124/dmd.107.016089. [DOI] [PubMed] [Google Scholar]
- (5).Mathias AA, German P, Murray BP, Wei L, Jain A, West S, Warren D, Hui J, Kearney BP. Pharmacokinetics and pharmacodynamics of GS-9350: a novel pharmacokinetic enhancer without anti-HIV activity. Clin. Pharmacol. Ther. 2010;87:322–329. doi: 10.1038/clpt.2009.228. [DOI] [PubMed] [Google Scholar]
- (6).Xu L, Liu H, Murray B, Callebaut C, Lee MS, Hong A, Strickley RG, Tsai L, Stray K, Wang KM, Rhodes Y, Desai GR, Cobicistat MC. (GS-9350): A potent and selective inhibitor of human CYP3A as a novel pharmacoenhancer. ACS Med. Chem. Lett. 2010;1:209–213. doi: 10.1021/ml1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sevrioukova IF, Poulos TL. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. U. S. A. 2010;107:18422–18427. doi: 10.1073/pnas.1010693107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sevrioukova IF, Poulos TL. Interaction of human cytochrome P4503A4 with ritonavir analogs. Arch. Biochem. Biophys. 2012;520:108–116. doi: 10.1016/j.abb.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05 Å resolution. J. Biol. Chem. 2004;279:38091–38094. doi: 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- (10).Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- (11).Zhou S, Yung Chan S, Cher Goh B, Chan E, Duan W, Huang M, McLeod HL. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin. Pharmacokinet. 2005;44:279–304. doi: 10.2165/00003088-200544030-00005. [DOI] [PubMed] [Google Scholar]
- (12).Koudriakova T, Iatsimirskaia E, Utkin I, Gangl E, Vouros P, Storozhuk E, Orza D, Marinina J, Gerber N. Metabolism of the human immunodeficiency virus protease inhibitors indinavir and ritonavir by human intestinal microsomes and expressed cytochrome P4503A4/3A5: mechanism-based inactivation of cytochrome P4503A by ritonavir. Drug Metab. Dispos. 1998;26:552–561. [PubMed] [Google Scholar]
- (13).von Moltke LL, Durol AL, Duan SX, Greenblatt DJ. Potent mechanism-based inhibition of human CYP3A in vitro by amprenavir and ritonavir: comparison with ketoconazole. Eur. J. Clin. Pharmacol. 2000;56:259–261. doi: 10.1007/s002280000125. [DOI] [PubMed] [Google Scholar]
- (14).Luo G, Lin J, Fiske WD, Dai R, Yang TJ, Kim S, Sinz M, LeCluyse E, Solon E, Brennan JM, Benedek IH, Jolley S, Gilbert D, Wang L, Lee FW, Gan LS. Concurrent induction and mechanism-based inactivation of CYP3A4 by an L-valinamide derivative. Drug Metab. Dispos. 2003;31:1170–1175. doi: 10.1124/dmd.31.9.1170. [DOI] [PubMed] [Google Scholar]
- (15).Ernest CS, II, Hall SD, Jones DR. Mechanism-based inactivation of CYP3A by HIV protease inhibitors. J. Pharmacol. Exp. Ther. 2005;312:583–591. doi: 10.1124/jpet.104.075416. [DOI] [PubMed] [Google Scholar]
- (16).Obach RS, Walsky RL, Venkatakrishnan K. Mechanism-based inactivation of human cytochrome p450 enzymes and the prediction of drug–-drug interactions. Drug Metab. Dispos. 2007;35:246–255. doi: 10.1124/dmd.106.012633. [DOI] [PubMed] [Google Scholar]
- (17).Kempf DJ, Marsh KC, Kumar G, Rodrigues AD, Denissen JF, McDonald E, Kukulka MJ, Hsu A, Granneman GR, Baroldi PA, Sun E, Pizzuti D, Plattner JJ, Norbeck DW, Leonard JM. Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob. Agents Chemother. 1997;41:654–660. doi: 10.1128/aac.41.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Roussel F, Khan KK, Halpert JR. The importance of SRS-1 residues in catalytic specificity of human cytochrome P450 3A4. Arch. Biochem. Biophys. 2000;374:269–278. doi: 10.1006/abbi.1999.1599. [DOI] [PubMed] [Google Scholar]
- (19).Park H, Lee S, Suh J. Structural and dynamical basis of broad substrate specificity, catalytic mechanism, and inhibition of cytochrome P450 3A4. J. Am. Chem. Soc. 2005;127:13634–13642. doi: 10.1021/ja053809q. [DOI] [PubMed] [Google Scholar]
- (20).Ekroos M, Sjogren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U. S. A. 2006;103:13682–13687. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Krishnamoorthy N, Gajendrarao P, Thangapandian S, Lee Y, Lee KW. Probing possible egress channels for multiple ligands in human CYP3A4: a molecular modeling study. J. Mol. Model. 2009;16:607–614. doi: 10.1007/s00894-009-0571-5. [DOI] [PubMed] [Google Scholar]
- (22).Sun H, Scott DO. Metabolism of 4-aminopiperidine drugs by cytochrome P450s: molecular and quantum mechanical insights into drug design. ACS Med. Chem. Lett. 2011;2:638–643. doi: 10.1021/ml200117z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sekiguchi N, Higashida A, Kato M, Nabuchi Y, Mitsui T, Takanashi K, Aso Y, Ishigai M. Prediction of drug–drug interactions based on time-dependent inhibition from high throughput screening of cytochrome P450 3A4 inhibition. Drug Metab. Pharmacokinet. 2009;24:500–510. doi: 10.2133/dmpk.24.500. [DOI] [PubMed] [Google Scholar]
- (24).Li F, Lu J, Ma X. Metabolomic screening and identification of the bioactivation pathways of ritonavir. Chem. Res. Toxicol. 2011;24:2109–2114. doi: 10.1021/tx2004147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).CCP4 Collaborative computational project number 4. The CCP4 suite programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- (26).Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.