

Abstract
Mass spectrometry (MS) is becoming increasingly popular in the field of structural biology for analyzing protein three-dimensional-structures and for mapping protein–protein interactions. In this review, the specific contributions of chemical crosslinking and native MS are outlined to reveal the structural features of proteins and protein assemblies. Both strategies are illustrated based on the examples of the tetrameric tumor suppressor protein p53 and multisubunit vinculin-Arp2/3 hybrid complexes. We describe the distinct advantages and limitations of each technique and highlight synergistic effects when both techniques are combined. Integrating both methods is especially useful for characterizing large protein assemblies and for capturing transient interactions. We also point out the future directions we foresee for a combination of in vivo crosslinking and native MS for structural investigation of intact protein assemblies.
Keywords: chemical cross-linking, native mass spectrometry, protein 3D structure, protein-protein interactions
Introduction
The aim of structural biology is to elucidate the three-dimensional (3D) structure of proteins and protein complexes to understand the reason why and how they acquire the structures they have, how it facilitates their cellular activity, and how structural alterations affect function. Therefore, the gold standard in structural characterization is to elucidate protein 3D structures at atomic resolution, using either X-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy.1,2 However, many proteins or protein assemblies are not amenable to analysis with these high-resolution techniques owing to their low solubility, weak stability, large size, or inability to form crystals. As such, membrane protein complexes are still underrepresented in structural data sets despite their outstanding biological importance.3 This gap in structural information can be bridged by low-resolution methods that provide complementary structural data. Interacting protein partners, for instance, can be identified by yeast two-hybrid screens,4 fluorescence resonance energy transfer,5 native mass spectrometry (MS),6–8 and chemical crosslinking.9 Electron microscopy (EM),10 small-angle X-ray scattering (SAXS),11 and ion-mobility (IM) MS12 can reveal the overall architecture of a protein complex. Residues that mediate protein–protein interactions can be identified by hydrogen–deuterium exchange,13 chemical crosslinking,9,14 and chemical footprinting,15 whereas the relative orientations of subunits can be inferred from cryo-EM,10 SAXS,11 and IM-MS.12
In this review, we will specifically focus on the contributions made by chemical crosslinking9,14 and native MS 6–8 for revealing the structural features of proteins and protein assemblies. Native MS maintains protein complexes intact within the mass spectrometer, by preserving weak noncovalent interactions between protein subunits and associated biomolecules, whereas crosslinking delivers distance information of specific residues within protein assemblies. Although each method has its own advantages and limitations, we will highlight their synergistic value when used in combination (Fig. 1). We begin by describing the different aspects of information that can be gained through using each individual technique, and then outline the additional capabilities that emerge when the two methods are integrated. We will also point out the challenges that lie ahead as well as the future directions we foresee for this hybrid approach.
Figure 1.
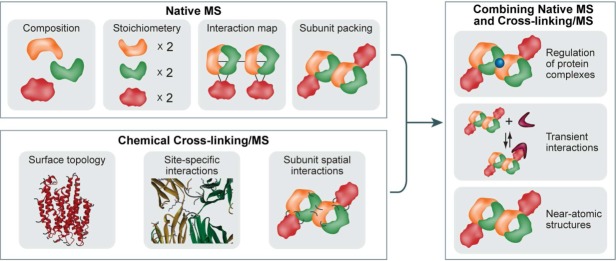
Advancing structural investigation of large protein assemblies by integrating native MS and chemical crosslinking MS. The analysis of the intact protein complexes by native MS can provide information on the composition, stoichiometry, subunit architecture, and topological organization of an assembly. Chemical crosslinking/MS can probe protein surface topology, reveal specific protein–protein interaction sites, and map the intermolecular subunit contacts of protein complexes. Consequently, “marrying” these two complementary approaches and using the structural data provided by each method will facilitate investigations of challenging protein systems: for example, dynamic assemblies or proteins that are not amenable to high-resolution structural techniques such as X-ray crystallography or NMR. Here, this combination is demonstrated for studying interaction between a large multisubunit complex and a relatively small protein, for characterizing labile protein interactions, and for generating 3D, near-atomic models, when combined with computational modeling.
General Aspects of Protein MS
MS is a method in which the mass-to-charge ratio (m/z) of an ion is measured. The analytes are first ionized and transferred into the gas phase before they are separated according to their m/z ratios in the mass analyzer. Finally, ions that emerge from the mass analyzer are sensed by the detector. One of the most prominent ways to ionize a protein sample is electrospray ionization (ESI),16 whereby voltage is applied to a liquid to create an aerosol. Although ESI is the method of choice for the applications we describe here, a complementary soft ionization technique is matrix-assisted laser desorption/ionization (MALDI), which involves the cocrystallization of the analyte with a dedicated matrix and ionization and desorption with a pulsed laser beam.17–20 In fact, there are a few reports where MALDI-MS has been used in combination with chemical crosslinking to analyze intact protein complexes.21,22
To date, multiple types of mass analyzers (e.g., ion trap, time-of-flight [TOF], quadrupole, orbitrap) are available and as a result, a number of configurations of mass spectrometers, each of which vary in their capabilities and limitations, can be designed for different purposes. Hybrid instruments that are based on a combination of two or more mass analyzers allow to perform tandem MS (MS/MS) experiments.23 In this type of analysis, a specific m/z ratio is isolated from the rest of the ions, and activated within a collision cell to induce their dissociation into smaller components. Various dissociation methods which yield complementary information may be applied, as collision-induced dissociation (CID), electron-transfer dissociation, electron-capture dissociation, and surface-induced dissociation (SID).24–26 The ions thus produced are then analyzed in the second mass analyzer yielding structural and sequential data as described below.
The analysis by both chemical crosslinking/MS and native MS require prior purification of the protein or protein complex of interest. This can be achieved by overexpression and purification of a recombinant version of the studied system. In case of investigating protein assemblies, the individual components have to be purified to reconstitute the whole complex in vitro; alternatively, in vivo reconstitution can take place by coexpressing the various subunits.27 Although reconstituted systems usually benefit from large yields that facilitate the structural analysis, they are often employed using bacteria (in most cases Escherichia coli) as the host system. As a consequence, post-translational modifications, and associations with interacting protein partners, may be lost. Therefore, various biochemical approaches have been developed that enable the isolation of endogenous protein complexes directly from cells or tissues. For example, one of the subunits is labeled with a tag (e.g., FLAG, His, glutathione-S-transferase, tandem affinity purification), which is then used as a bait for isolating the entire protein complex by affinity chromatography applying conditions that maintain the integrity of the protein complex.28 Notably, the degree to which the sample is purified will dictate the quality of the data that are obtained in the subsequent MS analysis.
The Principles of Chemical Crosslinking/MS
During the last 15 years, chemical crosslinking, combined with enzymatic digestion and MS analysis of the reaction products, has evolved into an alternative strategy to structurally resolve protein–protein interactions.29 Crosslinking enables the establishment of a set of structurally defined interactions by covalently connecting pairs of functional groups within a protein. From the constraints obtained by the chemical crosslinks, distance maps can be created within a protein or a protein complex, which will serve as the basis for deducing low-resolution 3D structures.30–32 The strengths of the crosslinking/MS strategy include the theoretically unlimited size of the protein or protein complex under investigation, and the minimal sample requirements in the femto- to attomole range, owing to the high sensitivity of MS analysis. Recently, the chemical crosslinking/MS approach has generated interest among system biologists and highly promising results indicate that the technique may be applied to map complex protein networks directly in cells.33–35
Types of crosslinkers
Nearly 40 years ago, N-hydroxysuccinimide (NHS) esters were introduced as homobifunctional, highly amine-reactive, crosslinking reagents.36,37 To date, NHS esters are the reagents most widely used to conduct protein–protein crosslinking, with bis(sulfosuccinimidyl)glutarate (BS2G) as one of the most prominent examples (Scheme 2). NHS esters react with nucleophiles to release the NHS or sulfo-NHS group and to create covalent amide and imide bonds with primary or secondary amines, such as the N-terminus and ε-amino groups in lysine side chains of proteins. It has further been shown that NHS esters react with hydroxyl groups in serines, threonines, and tryrosines, albeit only at about 20% of lysine reactivity.38,39
Scheme 1.
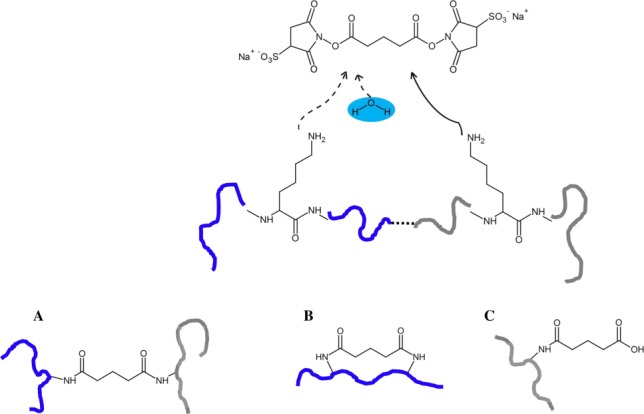
Reaction of amine-reactive NHS esters, exemplified for BS2G (upper panel). Reaction products with proteins include (A) interpeptide crosslinks, (B) intrapeptide crosslinks, and (C) “dead-end” crosslinks, that is, peptides that are modified by a partially hydrolyzed crosslinker.
The length of the carbon chain connecting both NHS groups determines the distance between the two reactive amino acid side chains that the crosslinker can bridge. In the case of bis(sulfosuccinimidyl)suberate (BS3), Cα–Cα distances between 26 and 30 Å have been found to be connected by the linker.40 To facilitate the identification of crosslinks in the mass spectra, isotope-labeled, that is deuterated crosslinkers are employed to facilitate the identification of the crosslinked peptides. However, it is impossible to ignore the retention time shift of deuterated compounds in reversed phase liquid chromatography (RP-LC), which prevents crosslinked species from coeluting.38 Therefore, using 13C-labeled reagents should be preferred over the application of deuterated reagents. In addition to targeting amine groups, crosslinkers are available that react with sulfhydryl groups in cysteines41 or with acidic residues.42,43
An additional class of compounds, known as photoreactive crosslinkers, holds great promise for conducting chemical crosslinking under in vivo conditions.44 The lower specificity of the crosslinking reaction also allows membrane-spanning regions in proteins—containing a high number of hydrophobic amino acids—to be targeted. Also, the potential to conduct crosslinking reactions in a two-step fashion, make photoreactive crosslinkers an attractive choice for mapping protein–protein interactions. The photoreactive group is induced to react with the target molecules by exposure to long-wavelength UV light. Photoreactive reagents include azides, diazirines, diazo compounds, and benzophenones.29 Most of the photoreactive crosslinkers are heterobifunctional reagents, which in addition, possess an amine-reactive group. From our experience, we consider diazirine- and benzophenone-based crosslinkers to be most useful for protein 3D structure analysis and for mapping protein–protein interactions.
MS/MS cleavable crosslinkers
Crosslinkers that dissociate under CID conditions in the mass spectrometer may be utilized to facilitate the identification of crosslinked products based on the characteristic fragment ions and constant neutral losses in MS/MS spectra.45–47 Out of the array of available MS/MS cleavable linkers, two highly promising examples are shown in Scheme 3. Both crosslinkers contain CID-labile groups, that is, a urea moiety as in the “urea-linker”46 or a sulfoxide as in the disuccinimidyl sulfoxide (DSSO) crosslinker.47 The unique feature of both crosslinkers is their ability to create characteristic marker ions, which is the basis for discriminating inter- and intrapeptide crosslinks from peptides that are modified by a partially hydrolyzed crosslinker (“dead-end” crosslink) (Scheme 2). MS/MS cleavable linkers are especially advantageous in the study of large protein assemblies where the mass spectra are highly complex and one has to sift through large data sets. The DSSO crosslinker has been successfully employed in a recently published study to investigate the Cmr complex by an integrative structural biology approach.48 The molecular architecture of the Pyrococcus furiosus Cmr complex, which is an RNA-guided endonuclease that cleaves foreign RNA targets as part of the CRISPR prokaryotic defense system, was investigated by combining a number of different structural biology techniques. The crystal structures of Cmr1, Cmr2, Cmr4, and Cmr6 were combined with known structural information to interpret the cryo-EM map of the complex. To support the determination of structure, chemical crosslinking with the MS/MS cleavable DSSO linker (Scheme 3) was employed, resulting in a pseudoatomic model of the complex.
Scheme 2.
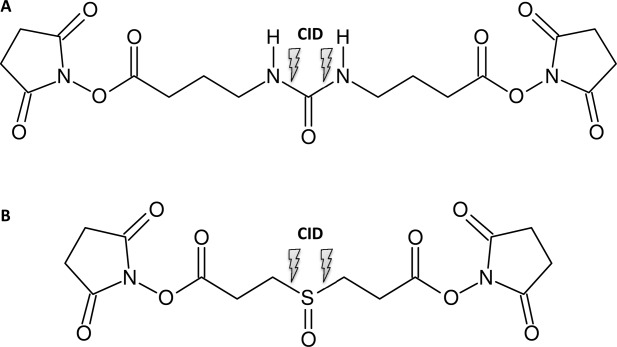
Structure of MS/MS cleavable crosslinkers. (A) “urea-linker” and (B) DSSO.
How to conduct chemical crosslinking experiments
For each protein system under investigation, the optimum crosslinking conditions, in respect to reaction time and excess of crosslinker, have to be carefully evaluated. Over-crosslinking has to be avoided to keep the protein's native conformation intact. After the crosslinking reaction, the enzymatic digestion may be conducted with a mixture of proteases, such as AspN or GluC (cleaving N- or C-terminally of acidic amino acids) and trypsin (cleaving C-terminally of arginines and lysines). In most crosslinking applications where lysines are targeted, one has to consider a reduced digestion efficiency of trypsin at modified lysines resulting in larger peptides. When choosing the optimum protease for digestion, one should aim for crosslinked products with a maximum size of about 4 kDa, which is in most cases best achieved by using a mixture of proteases.
Data analysis
Identifying crosslinked peptides poses additional difficulties for data analysis as the number of potential crosslinks grows quadratically, with increasing sample complexity. Thus, novel bioinformatic tools are required to examine the large data sets generated during MS and MS/MS analyses of the peptide mixtures. Considerable efforts have been made to develop specific software tools that are capable of analyzing these complex data sets; nevertheless, software that enables the fully automated analysis of MS and MS/MS data created from crosslinked product mixtures is still lacking. When doing in vivo crosslinking studies inside the cell without prior purification of the protein, calculation times can be very long as all database entries for one organism have to be screened for crosslinked products. The current bottleneck in data analysis must be resolved if chemical crosslinking is to evolve into a generally applicable and rapid method for global structural proteomic studies, underscoring the need to develop novel, powerful bioinformatics strategies. To date, nearly every group working on chemical crosslinking/MS has developed their own software, making it hard to keep track of the most recent developments.49 An attempt of a compilation of currently available crosslinking software is provided in Table1. In our lab, we rely on our in-house developed software StavroX, which is freely accessible.63 To screen data originating from MS/MS cleavable crosslinkers for the presence of characteristic fragment ions, the MeroX software has been introduced recently.64
Table 1.
Summary of Existing Crosslinking Softwarea
The names of the programs as well as the web addresses are given.
Analysis of full-length p53 tetramer
One recent example for the application of crosslinking/MS is the structural analysis of the tumor suppressor protein p53. This homotetramer contains a high percentage of intrinsically disordered regions, making it inherently difficult to study the full-length protein by conventional methods for protein 3D structure analysis. Full-length wild-type human p53, that is, without the stabilizing mutations M133L/V203A/N239Y/N268 that are generally employed for studying the structural properties of this assembly, was subjected to chemical crosslinking with a homobifunctional amine-reactive compound.70 To discriminate between crosslinks that are created between two p53 monomers (intermolecular crosslinks) and crosslinks that are created within one p53 monomer (intramolecular crosslinks), a 1:1 mixture of 14N- and 15N-labeled p53 was used.60,70,72 The crosslinking data of full-length p53 in the absence of DNA were in overall agreement with the full-length model of p53 that was derived based on the SAXS experiments,73 but they also revealed a great flexibility in the unstructured C-terminal region of p53 (Fig. 4). To date, structural data of full-length p53 the in absence of DNA are rare, with two existing structures (SAXS and EM data) that exhibit drastic differences in the molecular organization of p53.73,74 Based on the crosslinking/MS data, it was determined that the regulatory domains of different p53 monomers are much closer to each other than perceived by the existing SAXS model. Particularly, crosslinks involving residues of the tetramerization and regulatory domains can be explained only by a more compact arrangement of full-length p53 tetramer. In contrary, the EM structure cannot be brought in agreement with our crosslinking data. This study demonstrates that chemical crosslinking/MS proves valuable for deriving 3D structural information of proteins containing a large amount of intrinsically disordered regions where high-resolution structural methods usually fail.
Figure 4.
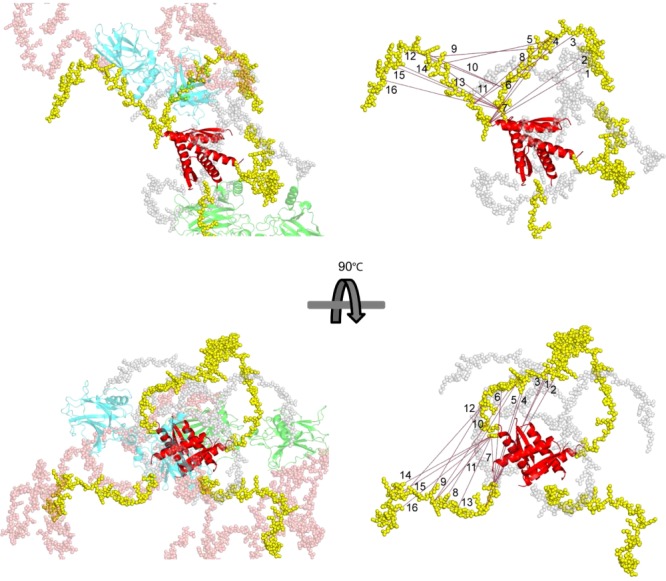
The presentation of full-length p53 in DNA-free state in a crossshaped structure according to a previously published SAXS model.73 DNA-binding domain (DBD; cyan and light green) and tetramerization domain (Tet.; red) are displayed in cartoon representation, connecting linkers (gray), N-termini (salmon), and C-terminal regulatory domains (yellow) are presented in spacefill mode. The crosslinking distances (in Å) are given for the most likely crosslinked residues. It should be noted that crosslinks (numbered 1–16) are presented for both monomers M1 and M2. Right-hand side: intermolecular crosslinks (red dotted lines) in simplified representation; left-hand side: in full represenation. Reprinted with permission from Arlt C, et al., Proteomics, 2015.
The Basics of Native MS
The field of native MS is based on the ability to maintain protein complexes intact within the mass spectrometer, by preserving weak noncovalent interactions between protein subunits and associated biomolecules such as DNA, cofactors, and ligands.6–8 Although mass is a one-dimensional property, combining information from analysis of the intact complex with that of the smaller subcomplexes generated can yield 3D structural information on the protein assembly. The details on the composition, stoichiometry, and subunit network of the protein complex as well as insights regarding its heterogeneity and dynamic interactions can be obtained.6–8,13,75 In addition, coupling native MS with ion-mobility (IM) provides valuable information on the overall topology and shape of the assembly.76 Below, we describe the steps taken to derive this structural information and demonstrate the application of the method for the membrane-cytoskeletal protein vinculin and the hybrid complexes it forms with Arp2/3, a complex that nucleates actin polymerization and branching77 (Fig. 5).
Figure 5.
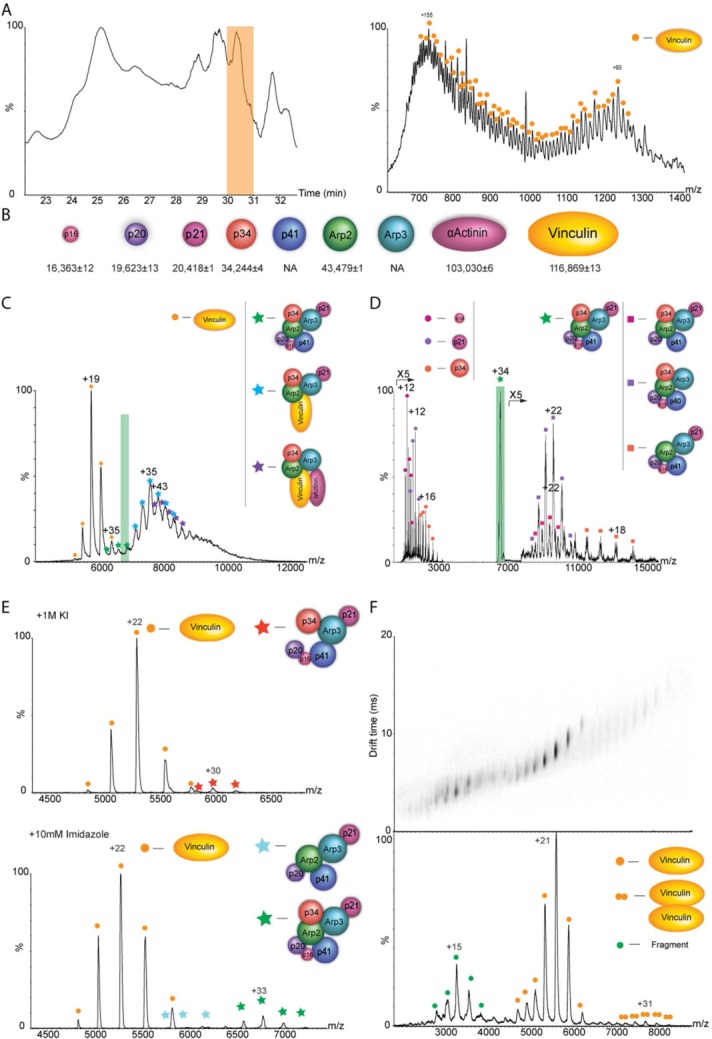
The cytoskeleton protein vinculin forms hybrid complexes with components of the Arp2/3 actin polymerization complex as revealed by native MS analysis. Cytoskeleton-related complexes from chicken gizzard smooth muscle were subjected to purification by stepwise ammonium sulfate precipitation, gel filtration, and anion exchange columns. The fraction that contained vinculin, α-actinin, and all seven subunits of the Arp2/3 protein complex, based on proteomic MS analysis, was further characterized by native MS. (A) Initially, the precise mass of protein components was determined by using a monolithic column-based approach in which protein constituents were separated prior to MS on an LC system using a monolithic column under denaturing conditions. A small portion of the flow was directed online into the mass spectrometer, for mass measurements of intact proteins and the remaining flow was fractionated for subsequent proteomic analysis. A representative chromatogram of the LC separation is shown on the left in which the peak representing the elution of vinculin is highlighted in yellow. The resulting ESI-QTOF mass spectrum of vinculin is shown on the right. (B) This approach together with MS/MS analysis enabled us to relate the specific subunit sequences with intact masses of almost all proteins within the sample as shown in the diagram. (C) Mass spectrum of the selected fraction recorded under native conditions. The predominant species detected, within the m/z range 5000–6800, is assigned to monomeric vinculin. Additional charge states, which correspond in mass to three coexisting protein complexes, are observed between m/z of 7000 and 11,000. The masses of these complexes indicate the presence of intact Arp2/3, a vinculin-associated Arp2/3 complex, and a vinculin-α actinin-associated Arp2/3 complex. (D) Validation of the complexes’ composition was undertaken by means of MS/MS; an example of a selected charge states is shaded in purple (D). The isolated peak is labeled with a star. Circles denote the released subunits and squares correspond to the remaining stripped complex. The MS/MS spectrum shows the dissociation products of ions isolated at m/z of 6917. Charge states above m/z of 7000 correspond to “stripped” Arp2/3 complexes. Individual Arp2/3 subunits, p16, p21, and p34 are observed at the low m/z values. (E) To enhance the structural characterization, subcomplexes were generated by manipulating the pH, increasing the ionic strength, or adding organic solvents to the buffer and subsequently acquiring MS and MS/MS data. Here, we show the effects of adding either 1M of KCl (top panel) or 10 mM of imidazole (bottom panel) to the sample and measuring the corresponding MS data. (F) To examine the topology of vinculin dimers, the recombinant protein was subjected to IM analysis. The figure shows the two-dimensional IM-mass spectrum with the corresponding line-projections in the m/z dimension. Adjusted with permission from Chorev DS, et al., Nat Commun, 2014, 5, 3758.
Instrumentation for high-molecular-weight analysis
Typically, native MS analysis has mostly been performed using a nano-electrospray ionization source (nESI) coupled to a quadrupole-time-of-flight (Q-TOF) mass analyzer, modified for high mass analysis.78,79 In essence, nESI is a miniaturized version of the ESI source,80 which enables lower flow rates (nL/min) of the ionized solution, in comparison to conventional ESI (µL/min). Consequently, the desolvatation process is more efficient, thus requiring smaller amounts of sample. Typically, spectra are produced by using 1–3 µL of sample in the micromolar range, which is loaded into gold-coated borosilicate or quartz capillaries with a tip diameter on the order of 1 µm.81 To maintain the noncovalent interactions of large complexes within the Q-TOF mass spectrometer, higher pressures are often used in the initial vacuum stages of the instrument to stabilize the macromolecular protein complexes.79 In addition, low-frequency quadrupoles are employed to broaden the mass-resolving capabilities to include species with m/z values exceeding that of 6000. This, however, comes at the expense of resolution.78
Recently, the analyses of large protein complexes have also become feasible by using a modified Orbitrap instrument.82 Unlike the TOF mass analyzer, which measures flight times, the Orbitrap analyzer measures the axial frequency of oscillation of trapped ions along a central electrode.83 It is estimated that the upper limit of the instrument for intact protein complexes is currently at m/z of 25,000–30,000, corresponding to a mass of roughly 5 MDa in positive ion mode nESI.84 Furthermore, the ability to select a precursor ion for MS/MS analysis was lately incorporated within the Orbitrap instrument.85 Overall, although the extended mass range Orbitrap analyzer is still in its early days, it holds great promise to advance the field of native MS owing to its enhanced resolution and impressive sensitivity, features that will likely assist in broadening the scope of applications of this method to less abundant protein assemblies.
Mass determination of protein complex subunits
In general, the investigation of protein assemblies by means of native MS is a multistep process. First, the unique mass of each of the protein components in the sample is defined. This is a crucial stage as assigning a mass to a specific subunit based on information contained in a protein sequence database is often not reliable. Many proteins derived from eukaryotic sources are truncated and post-translationally modified to such an extent that there is no correlation between the final product and the theoretical mass. This scenario is especially prominent when studying uncharacterized endogenous protein complexes isolated directly from cells.
One way to define the exact mass of each of the individual subunits is through MS analysis of a denatured solution of the protein complex.86 Although this method enables accurate mass measurements of the individual components in a single step, overlapping charge states may often complicate the analysis. To overcome this challenge, a LC separation method prior to MS analysis may be utilized. Such an approach has recently been demonstrated by using a monolithic column87 in which the constituent subunits of a protein complex were separated on the column, based on their size and chemical properties. The eluate was then split into two parts: One fraction was sprayed directly into the mass spectrometer to accurately determine the mass of the individual subunits, whereas the second fraction was collected into a 96-well plate for subsequent identification by proteomic analysis. Based on the elution profile, the identities of subunits can be accurately correlated with their mass.87 In the case of the vinculin-associated protein complexes, we determined the precise masses of all protein components within a purified fraction isolated from smooth muscle tissue by a combination of MS/MS analysis and the monolithic column-based approach. The chromatography pattern of the monolithic column and the online ESI mass spectrum of vinculin are shown in Figure 5(A), alongside with a summary of all protein components that coexisted within the sample [Fig. 5(B)].
Structural characterization of intact protein complexes
Once the unique mass of each subunit has been assigned, the structural properties of the intact complex can be investigated. To this end, the sample is dissolved in nondenaturing MS-compatible volatile buffers that preserve the integrity of multisubunit complex. In addition, conditions within the mass spectrometer are optimized to preserve noncovalent interactions and mass spectra of the intact complex are then recorded. By measuring the mass of the protein assembly, taking into consideration the masses of the individual subunits, the composition and stoichiometry of subunits are tentatively assigned. In cases of multicomponent assemblies, in which multiple subunit combinations are possible, it is often beneficial to use algorithms that are capable of calculating all possible compositions within a given error range based on the molecular masses of the subunits.88 MS/MS analysis can then be used to validate the assignment (as described below).
One powerful advantage of MS is that all coexisting protein complexes as well as subpopulations of a single assembly can be detected within a single spectrum. As a consequence, the dynamic nature, heterogeneity, and structural plasticity of protein complexes can be revealed. For example, when the sample of vinculin-associated proteins was analyzed, three coexisting protein complexes corresponding to vinculin-Arp2-Arp3-p21-p34, vinculin-α actinin-Arp2-Arp3-p34, and the intact Arp2/3 were identified [Fig. 5(C)]. Despite the overlapping charge states, the composition of the complexes could be unambiguously determined based on the assigned unique masses of each of the proteins [Fig. 5(B)] and the use of the “SUMMIT” algorithm, which calculates all possible protein compositions that add up to the target mass within a given error range.88 Notably, several additional software packages have been developed to assist in the analysis of such complicated spectra.89–91 Furthermore, different volatile buffers that are compatible with MS (e.g., ammonium acetate, ethylenediammonium diacetate, and triethylammonium acetate) or reagents that can modulate the number of charges per molecule, can be screened for their ability to enhance the separation between sample components, thereby enabling an unambiguous assignment of signals.92–95
The ability to isolate a discrete m/z value and induce its dissociation within the mass spectrometer as it is achieved in MS/MS experiments, is a critical step in the structural characterization process. The MS/MS pattern enables confirmation of the composition of the intact complex or its subcomplexes [Fig. 5(D)]. This is typically done by CID (see above), by colliding the ions with neutral gas atoms or molecules.96 These multiple collisions lead to charge-driven unfolding of a monomer, which may then be expelled from the complex.97 The pattern of MS/MS spectra generated by this method allows distinguishing between core and peripheral subunits of the assembly. Peripheral subunits characterized by a relatively small contact interface with the surrounding subunits will be the first ones to be expelled from the complex after an increase in collision energies, whereas core subunits will be protected from such exclusion.98
An alternative activation method is SID in which the selected ions collide with an inert surface in a single fast and high-energy event.99 This gives rise to direct dissociation of the complex into folded subunits and subcomplexes without large-scale structural rearrangements. Particularly, SID tends to selectively disrupt weak protein–protein interfaces. As a consequence, this activation method provides information on the quaternary structure of the assembly by releasing substructures representing the native topology.
By generating an array of smaller subcomplexes, a subunit interaction map of the complex can be deciphered86,100 Such subcomplexes can be produced either in solution, by manipulating the pH, increasing the ionic strength, or adding organic solvents to the buffer [Fig. 5(E)]. Alternatively, complexes can be created within the mass spectrometer by CID or SID activation as outlined above.99,101 Based on the overlapping components within the subcomplexes, the connectivity network between subunits is built.88
To better understand how subunits are packed, and what is the 3D arrangement of a protein complex, IM-MS can be used.12,102,103 This method measures the time it takes for an ion to travel through a dense environment of gas molecules of neutral charge under the influence of a weak electric field, thus yielding a drift time value for each ion [Fig. 5(F)]. The drift time is dependent not only on the ion's mass, but also on the overall shape of the analyzed protein complex: An assembly with a large volume will experience more collisions with the background gas, and consequently travel more slowly than a complex with the same mass, but a more compact structure. The measured drift time can then be converted to collision cross-section (Ω) values, which reflect the 3D shape of the ion.12,102,103 However, experimental conditions have to be carefully evaluated to maintain not only the noncovalent interactions, but also the native structural properties of the ions and avoid conformational collapse or unfolding.104 Overall, collision cross-section values derived from IM-MS data have been extremely valuable in providing information on the overall size and 3D organization of protein assemblies, especially when combined with subunit interaction maps and homology modeling approaches.105–107
A comprehensive list of achievements reflecting the diverse capabilities of native MS would encompass assemblies as large as virus capsids,84,108 multicomponent and asymmetric assemblies, such as the COP9 signalosome complex,86 as complicated as poly-disperse systems,91 and as challenging as membrane protein complexes.109 Based on the MS data, atomic models of protein assemblies have been generated,88 and insights into the molecular mechanism of action of protein complexes were obtained.95 In addition, the speed of analysis and separation afforded by MS place the method in a respectable position to capture dynamic processes such as protein folding,95 assembly,110,111 and subunit exchange reactions.112 Nevertheless, like any experimental technique, native MS has its own limitations, which are discussed below. Therefore, the combination with a complementary experimental technique, such as chemical crosslinking/MS, will not only offer a more comprehensive structural description of protein assemblies, but will also enable the examination of protein systems that, until now, have been resistant to characterization by native MS alone.
When the Whole is More than the Sum of its Parts: Integrating Chemical Crosslinking/MS with Native MS
Each of the two MS-based methods described above—chemical crosslinking/MS and native MS—has its specific advantages. Native MS combined with IM measurements is a powerful approach for determining the stoichiometry and connectivity between subunits and for defining the topological arrangement and overall architecture of protein assemblies. Yet, this method cannot identify the direct binding region within the complex, localize specific binding interfaces, or detect fleeting protein interactions. These features, however, can be resolved by chemical crosslinking analysis—a method that may be challenged when forced to reveal the structural organization of multicomponent complexes, which lack any prior structural knowledge. Consequently, integrating these two complementary techniques would be a natural step toward a more complete characterization of protein assemblies.
In the following section, we will discuss the added value that we believe is achieved when native MS and chemical crosslinking/MS are combined. We specifically focus on challenging scenarios that can be resolved by integrating the two approaches, such as the case when studying the interaction between a large multisubunit complex and a relatively small protein, or when probing transient interactions. We then describe a hybrid approach in which the two MS-based methods join forces with other structural biology approaches to provide a comprehensive analysis of difficult-to-study biological systems. Finally, challenges and future directions are presented.
Unraveling the Regulatory Mechanisms of Protein Complexes
Large protein complexes orchestrating fundamental cellular activities are often activated or deactivated through interactions with individual proteins that are much smaller in size. For example, the folding activity of the ∼1-MDa eukaryotic Chaperonin Containing Tcp1 (CCT) complex, responsible for maintaining protein folding homeostasis in cells, is modulated by interactions with phosducin-like protein 2 (PLP2, 38 kDa).113 Similarly, protein degradation by the ∼750 kDa 20S proteasome complex is inhibited by interactions with small homodimeric proteins as NAD(P)H:quinone-oxidoreductase-1 (NQO1, 31kDa) and DJ-1 (20 kDa).114,115 Unraveling the molecular mechanism underlying the action of such regulators requires the structural characterization of their interactions with the complex they control. This task, however, is not simple, considering the relatively small size of the regulator, and the large dimensions of their associated protein assembly.
Native MS analysis of a sample containing a mixture of the protein complex of interest and its regulator can primarily provide information on the existence of such an interaction and the affinity of the composing parts.116 A combination of MS and MS/MS experiments makes it possible to define the stoichiometry of the interaction; that is, the number of regulators that are bound to the protein complex. Coupling IM might even enable the identification of the area in which the regulatory protein binds. However, the specific sites of association between the regulator and the molecular machine, which would yield clues to the mechanism of its action, will remain elusive. This lack of information could be filled by chemical crosslinking results.
To map the exact sites where the regulator interacts with the protein complex, crosslinking experiments can be performed using either a homobifunctional or heterobifunctional crosslinker. Reactions with a homobifunctional crosslinker involve a single-step reaction, which yields both inter- and intramolecular crosslinked peptides (Scheme 2). One advantage of this type of analysis is that if part or all of the protein components have known high-resolution structures, the quality of the results can be assessed by validating the applicability of the identified intrasubunit crosslinked peptides in the crystal structures.117,118 The drawback, on the other hand, lies in the generation of a complex mixture of peptides, comprising a high background of unmodified linear peptides. As a consequence, the signal intensities of crosslinked peptides are low and data interpolation is challenging. The use of a heterobifunctional crosslinker can ease these difficulties.119 In this type of reaction, two steps are involved. First, one reactive site of the crosslinker is reacted with the small regulator. After the removal of excess crosslinker, a different type of chemical reaction, preferably a photoreaction, is performed, using the second functional group connecting the regulator and its associated protein complex. This two-step approach reduces the formation of high-molecular-weight aggregates and eliminates the formation of intramolecular crosslinks within the larger complex, thus simplifying data analysis.
Functional regulation of a protein complex can also be achieved by post-translational modifications, especially by phosphorylation.120 The addition of multiple phosphate groups, which add both negative charge and volume, can reversibly alter the conformation and stability of the assembly, resulting in the modulation of intrinsic biological activity. This was recently demonstrated for the membrane motor F1FO-ATPase complex, in a study that effectively combined native MS and chemical crosslinking/MS.121 Mass spectra of the intact ATPase complex clarified the number of nucleotides bound to the complex. A dephosphorylated enzyme, however, displayed reduced nucleotide occupancy and decreased stability. Using the lysine-specific deuterated and nondeuterated crosslinker BS3 and comparing the crosslinking patterns of untreated and dephosphorylated ATPase, it was possible to probe the conformational changes that occur at the catalytic interface, providing a rationale for reduced nucleotide occupancy. Thus, the synergy of approaches uncovered the effect of phosphorylation on the dynamic subunit interactions within a membrane-embedded protein complex.
Stabilizing Transient and Dynamic Protein Complexes
Transient or dynamic protein–protein interactions, which form and break easily, are important to many aspects of cellular function, including protein folding, modification, transport, intracellular signaling, and regulation of the cell cycle.122 Therefore, characterizing these interactions is critical to the understanding of various biological processes; yet, owing to their labile nature, they are technically difficult to study and harder to detect than more stable interactions. For example, the yeast initiation factor 3 (eIF3), which is a hexameric subunit assembly, has so far resisted high-resolution structural determination owing to its dynamic nature. However, an integrated approach combining both native MS and chemical crosslinking/MS provided a 3D topological model of the complex in association with eIF5, an additional initiation factor.123 Specifically, a list of interacting subcomplexes was derived by performing controlled dissociation of activated complexes in the gas phase or by disruption in solution and determining the composition of the complexes by native MS. Additional interactions were extracted by chemical crosslinking using both deuterated and nondeuterated BS3. Collectively, this information was encoded as subunit connectivity restrains, which were included in a scoring function for building 3D topological models of the protein assemblies.
An additional crosslinking technique for comprehensive profiling of protein interactions network, including weak and/or transient components, is QTAX (quantitative analysis of tandem affinity purified in vivo crosslinked protein complexes).124 In this method, a snapshot of the stable and transient interactions is formed in vivo, using formaldehyde. The captured protein complexes and their interacting proteins are isolated by a two-step affinity purification procedure under fully denaturing conditions, using a histidine–biotin–histidine-tag. The purified proteins are then identified by LC/MS/MS, and the protein–protein interactions can be differentiated from background proteins based on their stable isotope labeling of amino acids in cell culture (SILAC) ratios. If a protein is a background protein, then it is purified in equal amounts from both the tagged and the control strains, and all peptides representing that protein will elute as a pair with a SILAC ratio of ≈1. In contrast, protein interaction partners will be enriched in the tagged sample. This strategy was used to identify the 26S proteasome interaction network, which involves hundreds of proteins intertwined in several critical cellular processes.124
We envision an alternative approach for combining in vivo chemical crosslinking/MS with native MS (Fig. 6). To identify the interactions within the cellular environment and reveal their spatio-temporal regulation in response to various different biological stimuli, cells will be cultured in different isotope-labeled media, containing combinations of 13C- and 15N-labeling. Differential incorporation of the stable isotopes will enable distinguishing the different forms of the same peptide owing to their mass differences. In addition, diazirine-containing photoreactive amino acids, such as photo-methionine and photo-leucine, may be used.125 These photoreactive amino acids are known to be endogenously incorporated into the sequence of proteins during translation, substituting for methionine and leucine. Crosslinks are formed between spatially close amino acids upon activation with UV-A irradiation of the cell culture. This approach enables efficient control of covalent bond formation compared to the formaldehyde crosslinking used in the QTAX approach described above.124 Protein complexes can be detected by incorporating an affinity tag into the protein of interest which is used as bait for pulling down interacting proteins. Isolation of the tagged protein can then be performed under either native or denaturing conditions, before and after photoactivation, respectively. Pull-down experiments under native conditions enable characterization of the complex stoichiometry, heterogeneity, and architecture, by means of native MS. This step does not require the isotopic labeling; however, by harnessing the same experimental setup insight into the composition, stoichiometries and interactions of the protein complex components can be gained. In addition, validating the integrity of the isolated protein assembly will reduce false positives; that is, copurified components, which are a major source of error in high-throughput affinity purification, followed by MS. Pull-down experiments of the bait protein under denaturing conditions will maintain interactions with neighboring proteins fixing even weakly associated components, which will be identified after enzymatic digestion by LC/MS/MS analysis. The latter will provide interaction contacts at the individual peptide level. The different combinations of 13C- and 15N-labeling will allow quantifying different crosslinked products in respect to the conditions used for cell growth, giving insights into altered protein interactions. The strength of this integrated strategy is the coupling of two complementary MS approaches in a single biological setup.
Figure 6.
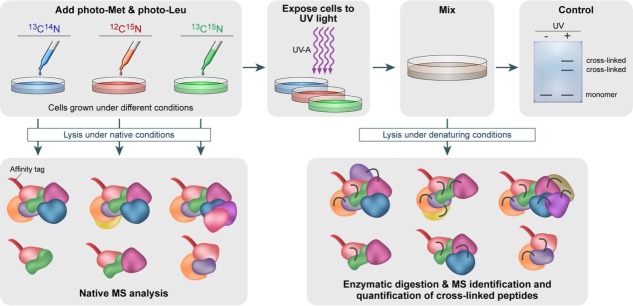
Schematic representation of an in vivo strategy for identifying protein–protein interactions. Cells are grown in media that are devoid of leucine and methionine. Photoreactive leucine and methionine analogues substitute the naturally occurring amino acids. To determine how multiple growth conditions influence the composition of a protein complex, cell cultures treated under different conditions will be cultured in different isotope-labeled media, containing the combinations of 13C- and 15N-labeling. Chemical crosslinking is then induced by UV-A illumination, forming a covalent bond with nearby protein side chains. Protein–protein interactions are identified by combining denaturing and native lysis conditions with pull-down experiments, using an affinity tag. Pull-down experiments under native conditions, before the formation of crosslinks, will enable to define subunit stoichiometries and characterization of all interacting proteins within the complex, using native MS. This step can also be performed in the absence of isotope labeling. Illuminating the sample with UV-A light will produce crosslinks capable of capturing even transient and weak interactions. These will be identified after pull-down experiments under denaturing conditions and analysis by both native MS and peptide MS profiling. The former provides a direct identification of the protein–protein interaction sites. MS/MS analysis of crosslinked products allows quantifying altered protein complex formation upon altered biological conditions based on 13C- and 15N-labeling.
From Low-Resolution Structures to High-Resolution Models
Native MS as well as crosslinking/MS are low-resolution 3D protein structural techniques. However, by integrating these data with information produced by complementary structural biology techniques and computational molecular modeling approaches, atomic resolution structures can be generated.88,117,118,126 This is exemplified by two recent studies combining EM, native IM-MS, and computational modeling to determine a 3D structural model of the multisubunit CRISPR complex.127,128 Another excellent study integrated cryo-EM and crosslinking/MS data to determine the architecture of the 39S large subunit of the mammalian mitochondrial ribosome.129 The cryo-EM structure at 4.9 Å resolution, combined with data from chemical crosslinking/MS, revealed structural features at near-atomic resolution and provided detailed insight into the architecture of the polypeptide exit site. The model of the 26S proteasome complex is yet another example of a structure that benefited from a hybrid approach, combining chemical crosslinking, cryo-EM, X-ray crystallography, and proteomic approaches.118 Thus, the synergy between MS-based techniques, and structural methods such as X-ray crystallography, NMR, and EM, gives rise to a powerful approach for structural investigations of protein assemblies, especially for the examination of complex systems that are resistant to characterization by any single technique.
Conclusions
In view of the impact that integrative approaches have had on structural biology, we trust that combining chemical crosslinking/MS with native MS will yield mutual benefits. As highlighted in this review article, both techniques are complementary on many levels.121,130 The two methods can handle large, heterogeneous, and dynamic protein complexes and tolerate protein impurities. Both methods can identify protein interactions, with chemical crosslinking/MS providing the specific interaction sites at peptide resolution, and native MS defining the interaction network and stoichiometry on the subunit level. Similarly, the spatial arrangement of subunits can be determined at the local and global levels by chemical crosslinking/MS and IM-MS, respectively. Thus, the attributes obtained from each method can be merged into a comprehensive structural representation that, if supplemented with computational approaches, may even yield near-atomic level models. Though the number of studies combining these two MS-based approaches is thus far exceedingly low, we expect that the urge to tackle more challenging biological systems will trigger the ever-increasing use of this integrative mode. In this respect, one hurdle that must yet be overcome is that each MS method demands its own specialized mass spectrometer. With the advent of modern Orbitrap mass spectrometers,82 capable of analyzing small peptides as well as very large protein complexes at high resolution, the instrumental gap between crosslinking/MS and native MS methods will be bridged.
Glossary
- BS2G
bis(sulfosuccinimidyl)glutarate
- BS3
bis(sulfosuccinimidyl)suberate
- CID
collision-induced dissociation
- DSSO
disuccinimidyl sulfoxide
- EM
electron microscopy
- ESI
electrospray ionization
- IM
ion-mobility
- LC
liquid chromatography
- MALDI
matrix-assisted laser desorption/ionization
- MS
mass spectrometry
- MS/MS
tandem MS
- nESI
nano-electrospray ionization
- NHS
N-hydroxysuccinimide
- NMR
nuclear magnetic resonance
- QTAX
quantitative analysis of tandem affinity purified in vivo crosslinked protein complexes
- Q-TOF
quadrupole time-of-flight
- RP
reversed phase
- SAXS
small-angle X-ray scattering
- SID
surface-induced dissociation
- SILAC
stable isotope labeling of amino acids in cell culture
- 3D
three-dimensional
- TOF
time-of-flight.
References
- Tugarinov V, Hwang PM, Kay LE. Nuclear magnetic resonance spectroscopy of high-molecular-weight proteins. Annu Rev Biochem. 2004;73:107–146. doi: 10.1146/annurev.biochem.73.011303.074004. [DOI] [PubMed] [Google Scholar]
- Ilari A, Savino C. Protein structure determination by x-ray crystallography. Methods Mol Biol. 2008;452:63–87. doi: 10.1007/978-1-60327-159-2_3. [DOI] [PubMed] [Google Scholar]
- Walian P, Cross TA, Jap BK. Structural genomics of membrane proteins. Genome Biol. 2004;5:215. doi: 10.1186/gb-2004-5-4-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish JR, Gulyas KD, Finley RL., Jr Yeast two-hybrid contributions to interactome mapping. Curr Opin Biotechnol. 2006;17:387–393. doi: 10.1016/j.copbio.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Sun Y, Hays NM, Periasamy A, Davidson MW, Day RN. Monitoring protein interactions in living cells with fluorescence lifetime imaging microscopy. Methods Enzymol. 2012;504:371–391. doi: 10.1016/B978-0-12-391857-4.00019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch JL, Ruotolo BT, Simmons DA, Robinson CV. Protein complexes in the gas phase: technology for structural genomics and proteomics. Chem Rev. 2007;107:3544–3567. doi: 10.1021/cr068289b. [DOI] [PubMed] [Google Scholar]
- Heck AJ. Native mass spectrometry: a bridge between interactomics and structural biology. Nat Methods. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- Sharon M. How far can we go with structural mass spectrometry of protein complexes? J Am Soc Mass Spectrom. 2010;21:487–500. doi: 10.1016/j.jasms.2009.12.017. [DOI] [PubMed] [Google Scholar]
- Sinz A. Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom Rev. 2006;25:663–682. doi: 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- Milne JL, Borgnia MJ, Bartesaghi A, Tran EE, Earl LA, Schauder DM, Lengyel J, Pierson J, Patwardhan A, Subramaniam S. Cryo-electron microscopy—a primer for the non-microscopist. FEBS J. 2013;280:28–45. doi: 10.1111/febs.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL. Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, Yang SJ, Scott JW, Dillard BD, Adams MW, Tainer JA. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetrecht C, Rose RJ, van Duijn E, Lorenzen K, Heck AJ. Ion mobility mass spectrometry of proteins and protein assemblies. Chem Soc Rev. 2010;39:1633–1655. doi: 10.1039/b914002f. [DOI] [PubMed] [Google Scholar]
- Konermann L, Pan J, Liu YH. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev. 2011;40:1224–1234. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- Leitner A, Walzthoeni T, Kahraman A, Herzog F, Rinner O, Beck M, Aebersold R. Probing native protein structures by chemical cross-linking, mass spectrometry, and bioinformatics. Mol Cell Proteomics. 2010;9:1634–1649. doi: 10.1074/mcp.R000001-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gau BC, Sharp JS, Rempel DL, Gross ML. Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal Chem. 2009;81:6563–6571. doi: 10.1021/ac901054w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Waki H, Ido Y, Akita S, Yoshida Y, Yohida T. Protein and polymer analyses up to m/z 100,000 by laser ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 1988;2:151–153. [Google Scholar]
- Charvat A, Abel B. How to make big molecules fly out of liquid water: applications, features and physics of laser assisted liquid phase dispersion mass spectrometry. Phys Chem Chem Phys. 2007;9:3335–3360. doi: 10.1039/b615114k. [DOI] [PubMed] [Google Scholar]
- Bich C, Zenobi R. Mass spectrometry of large complexes. Curr Opin Struct Biol. 2009;19:632–639. doi: 10.1016/j.sbi.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Bich C, Bovet C, Rochel N, Peluso-Iltis C, Panagiotidis A, Nazabal A, Moras D, Zenobi R. Detection of nucleic acid-nuclear hormone receptor complexes with mass spectrometry. J Am Soc Mass Spectrom. 2010;21:635–645. doi: 10.1016/j.jasms.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Chen F, Gerber S, Korkhov VM, Mireku S, Bucher M, Locher KP, Zenobi R. On the efficiency of NHS ester cross-linkers for stabilizing integral membrane protein complexes. J Am Soc Mass Spectrom. 2015;26:493–498. doi: 10.1007/s13361-014-1035-4. [DOI] [PubMed] [Google Scholar]
- Glish GL, Vachet RW. The basics of mass spectrometry in the twenty-first century. Nat Rev Drug Discov. 2003;2:140–150. doi: 10.1038/nrd1011. [DOI] [PubMed] [Google Scholar]
- Wysocki VH, Joyce KE, Jones CM, Beardsley RL. Surface-induced dissociation of small molecules, peptides, and non-covalent protein complexes. J Am Soc Mass Spectrom. 2008;19:190–208. doi: 10.1016/j.jasms.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JR, Ruse CI, Nakorchevsky A. Proteomics by mass spectrometry: approaches, advances, and applications. Annu Rev Biomed Eng. 2009;11:49–79. doi: 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]
- Jones AW, Cooper HJ. Dissociation techniques in mass spectrometry-based proteomics. Analyst. 2011;136:3419–3429. doi: 10.1039/c0an01011a. [DOI] [PubMed] [Google Scholar]
- Demartino GN. Reconstitution of PA700, the 19S regulatory particle, from purified precursor complexes. Methods Mol Biol. 2012;832:443–452. doi: 10.1007/978-1-61779-474-2_31. [DOI] [PubMed] [Google Scholar]
- Oeffinger M. Two steps forward—one step back: advances in affinity purification mass spectrometry of macromolecular complexes. Proteomics. 2012;12:1591–1608. doi: 10.1002/pmic.201100509. [DOI] [PubMed] [Google Scholar]
- Sinz A. The advancement of chemical cross-linking and mass spectrometry for structural proteomics: from single proteins to protein interaction networks. Expert Rev Proteomics. 2014;11:733–743. doi: 10.1586/14789450.2014.960852. [DOI] [PubMed] [Google Scholar]
- Rappsilber J. The beginning of a beautiful friendship: cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J Struct Biol. 2011;173:530–540. doi: 10.1016/j.jsb.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce JE. In vivo protein complex topologies: sights through a cross-linking lens. Proteomics. 2012;12:1565–1575. doi: 10.1002/pmic.201100516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpa JJ, Parker CE, Petrotchenko EV, Han J, Pan J, Borchers CH. Mass spectrometry-based structural proteomics. Eur J Mass Spectrom. 2012;18:251–267. doi: 10.1255/ejms.1178. [DOI] [PubMed] [Google Scholar]
- Chavez JD, Weisbrod CR, Zheng C, Eng JK, Bruce JE. Protein interactions, post-translational modifications and topologies in human cells. Mol Cell Proteomics. 2013;12:1451–1467. doi: 10.1074/mcp.M112.024497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisbrod CR, Chavez JD, Eng JK, Yang L, Zheng C, Bruce JE. In vivo protein interaction network identified with a novel real-time cross-linked peptide identification strategy. J Proteome Res. 2013;12:1569–1579. doi: 10.1021/pr3011638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buncherd H, Roseboom W, de Koning LJ, de Koster CG, de Jong L. A gas phase cleavage reaction of cross-linked peptides for protein complex topology studies by peptide fragment fingerprinting from large sequence database. J Proteomics. 2014;108:65–77. doi: 10.1016/j.jprot.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Bragg PD, Hou C. Subunit composition, function, and spatial arrangement in Ca2+-activated and Mg2+-activated adenosine triphosphatases of Escherichia coli and Salmonella typhimurium. Arch Biochem Biophys. 1975;167:311–321. doi: 10.1016/0003-9861(75)90467-1. [DOI] [PubMed] [Google Scholar]
- Lomant AJ, Fairbanks G. Chemical probes of extended biological structures—synthesis and properties of cleavable protein cross-linking reagent [dithiobis(succinimidyl-S-35 propionate) J Mol Biol. 1976;104:243–261. doi: 10.1016/0022-2836(76)90011-5. [DOI] [PubMed] [Google Scholar]
- Kalkhof S, Sinz A. Chances and pitfalls of chemical cross-linking with amine-reactive N-hydroxysuccinimide esters. Anal Bioanal Chem. 2008;392:305–312. doi: 10.1007/s00216-008-2231-5. [DOI] [PubMed] [Google Scholar]
- Madler S, Bich C, Touboul D, Zenobi R. Chemical cross-linking with NHS esters: a systematic study on amino acid reactivities. J Mass Spectrom. 2009;44:694–706. doi: 10.1002/jms.1544. [DOI] [PubMed] [Google Scholar]
- Merkley ED, Rysavy S, Kahraman A, Hafen RP, Daggett V, Adkins JN. Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Prot Sci. 2014;23:747–759. doi: 10.1002/pro.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanson GT. 2008. Bioconjugate Techniques 2nd Edition, Elsevier Ltd, Oxford.
- Novak P, Kruppa GH. Intra-molecular cross-linking of acidic residues for protein structure studies. Eur J Mass Spectrom. 2008;14:355–365. doi: 10.1255/ejms.963. [DOI] [PubMed] [Google Scholar]
- Leitner A, Joachimiak LA, Unverdorben P, Walzthoeni T, Frydman J, Forster F, Aebersold R. Chemical cross-linking/mass spectrometry targeting acidic residues in proteins and protein complexes. Proc Natl Acad Sci USA. 2014;111:9455–9460. doi: 10.1073/pnas.1320298111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn M, Ihling CH, Golbik R, Sawers RG, Sinz A. Mapping cell envelope and periplasm protein interactions of Escherichia coli respiratory formate dehydrogenases by chemical cross-linking and mass spectrometry. J Proteome Res. 2014;13:5524–5535. doi: 10.1021/pr5004906. [DOI] [PubMed] [Google Scholar]
- Lu YL, Tanasova M, Borhan B, Reid GE. Ionic reagent for controlling the gas-phase fragmentation reactions of cross-linked peptides. Anal Chem. 2008;80:9279–9287. doi: 10.1021/ac801625e. [DOI] [PubMed] [Google Scholar]
- Muller MQ, Dreiocker F, Ihling CH, Schafer M, Sinz A. Cleavable cross-linker for protein structure analysis: reliable identification of cross-linking products by tandem MS. Anal Chem. 2010;82:6958–6968. doi: 10.1021/ac101241t. [DOI] [PubMed] [Google Scholar]
- Kao A, Randall A, Yang YY, Patel VR, Kandur W, Guan SH, Rychnovsky SD, Baldi P, Huang L. Mapping the structural topology of the yeast 19S proteasomal regulatory particle using chemical cross-linking and probabilistic modeling. Mol Cell Proteomics. 2012;11:1566–1577. doi: 10.1074/mcp.M112.018374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benda C, Ebert J, Scheltema RA, Schiller HB, Baumgartner M, Bonneau F, Mann M, Conti E. Structural model of a CRISPR RNA-silencing complex reveals the RNA-target cleavage activity in Cmr4. Mol Cell. 2014;56:43–54. doi: 10.1016/j.molcel.2014.09.002. [DOI] [PubMed] [Google Scholar]
- Mayne SLN, Patterton HG. Bioinformatics tools for the structural elucidation of multi-subunit protein complexes by mass spectrometric analysis of protein-protein cross-links. Brief Bioinformatics. 2011;12:660–671. doi: 10.1093/bib/bbq087. [DOI] [PubMed] [Google Scholar]
- Tang Y, Chen YF, Lichti CF, Hall RA, Raney KD, Jennings SF. CLPM: a cross-linked peptide mapping algorithm for mass spectrometric analysis. BMC Bioinformatics. 2005;6:S9. doi: 10.1186/1471-2105-6-S2-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain S, Draghicescu P, Singh P, Goodlett DR, Noble WS. Detecting cross-linked peptides by searching against a database of cross-linked peptide pairs. J Proteome Res. 2010;9:2488–2495. doi: 10.1021/pr901163d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Zhang L, Freitas MA. Identification and characterization of disulfide bonds in proteins and peptides from tandem MS data by use of the MassMatrix MS/MS search engine. J Proteome Res. 2008;7:138–144. doi: 10.1021/pr070363z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen MI, Refsgaard JC, Peng L, Houen G, Hojrup P. CrossWork: software-assisted identification of cross-linked peptides. J Proteomics. 2011;74:1871–1883. doi: 10.1016/j.jprot.2011.04.019. [DOI] [PubMed] [Google Scholar]
- Clauser KR, Baker P, Burlingame AL. Role of accurate mass measurement (+/− 10 ppm) in protein identification strategies employing MS or MS MS and database searching. Anal Chem. 1999;71:2871–2882. doi: 10.1021/ac9810516. [DOI] [PubMed] [Google Scholar]
- Fenyo D. A software tool for the analysis of mass spectrometric disulfide mapping experiments. Comput Appl Biosci. 1997;13:617–618. doi: 10.1093/bioinformatics/13.6.617. [DOI] [PubMed] [Google Scholar]
- Gao Q, Xue S, Doneanu CE, Shaffer SA, Goodlett DR, Nelson SD. Pro-CrossLink. Software tool for protein cross-linking and mass spectrometry. Anal Chem. 2006;78:2145–2149. doi: 10.1021/ac051339c. [DOI] [PubMed] [Google Scholar]
- Peri S, Steen H, Pandey A. GPMAW—a software tool for analyzing proteins and peptides. Trends Biochem Sci. 2001;26:687–689. doi: 10.1016/s0968-0004(01)01954-5. [DOI] [PubMed] [Google Scholar]
- Nielsen T, Thaysen-Andersen M, Larsen N, Jorgensen FS, Houen G, Hojrup P. Determination of protein conformation by isotopically labelled cross-linking and dedicated software: application to the chaperone, calreticulin. Int J Mass Spectrom. 2007;268:217–226. [Google Scholar]
- de Koning LJ, Kasper PT, Back JW, Nessen MA, Vanrobaeys F, Van Beeumen J, Gherardi E, de Koster CG, de Jong L. Computer-assisted mass spectrometric analysis of naturally occurring and artificially introduced cross-links in proteins and protein complexes. FEBS J. 2006;273:281–291. doi: 10.1111/j.1742-4658.2005.05053.x. [DOI] [PubMed] [Google Scholar]
- Taverner T, Hall NE, O'Hair RA, Simpson RJ. Characterization of an antagonist interleukin-6 dimer by stable isotope labeling, cross-linking, and mass spectrometry. J Biol Chem. 2002;277:46487–46492. doi: 10.1074/jbc.M207370200. [DOI] [PubMed] [Google Scholar]
- Anderson GA, Tolic N, Tang XT, Zheng CX, Bruce JE. Informatics strategies for large-scale novel cross-linking analysis. J Proteome Res. 2007;6:3412–3421. doi: 10.1021/pr070035z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinner O, Seebacher J, Walzthoeni T, Mueller LN, Beck M, Schmidt A, Mueller M, Aebersold R. Identification of cross-linked peptides from large sequence databases. Nat Methods. 2008;5:315–318. doi: 10.1038/nmeth.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotze M, Pettelkau J, Schaks S, Bosse K, Ihling CH, Krauth F, Fritzsche R, Kuhn U, Sinz A. StavroX—a software for analyzing crosslinked products in protein interaction studies. J Am Soc Mass Spectrom. 2012;23:76–87. doi: 10.1007/s13361-011-0261-2. [DOI] [PubMed] [Google Scholar]
- Gotze M, Pettelkau J, Fritzsche R, Ihling CH, Schafer M, Sinz A. Automated assignment of MS/MS cleavable cross-links in protein 3D-structure analysis. J Am Soc Mass Spectrom. 2015;26:83–97. doi: 10.1007/s13361-014-1001-1. [DOI] [PubMed] [Google Scholar]
- Jaiswal M, Crabtree NM, Bauer MA, Hall R, Raney KD, Zybailov BL. XLPM: efficient algorithm for the analysis of protein-protein contacts using chemical cross-linking mass spectrometry. BMC Bioinformatics. 2014;15:S16. doi: 10.1186/1471-2105-15-S11-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Chowdhury SM, Manes NP, Wu S, Mayer MU, Adkins JN, Anderson GA, Smith RD. Xlink-identifier: an automated data analysis platform for confident identifications of chemically cross-linked peptides using tandem mass spectrometry. J Proteome Res. 2011;10:923–931. doi: 10.1021/pr100848a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg CAG, Lambert W, Kjellstrom S, Wiegandt A, Wulff RP, Mansson C, Rutsdottir G, Emanuelsson C. Detection of crosslinks within and between proteins by LC-MALDI-TOFTOF and the Software FINDX to reduce the MSMS-data to acquire for validation. Plos One. 2012;7:e38927. doi: 10.1371/journal.pone.0038927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchaud A, Singh P, Shaffer SA, Goodlett DR. xComb: a cross-linked peptide database approach to protein-protein interaction analysis. J Proteome Res. 2010;9:2508–2515. doi: 10.1021/pr9011816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer L, Chen ZA, Rappsilber J. Quantitative cross-linking/mass spectrometry using isotope-labelled cross-linkers. J Proteomics. 2013;88:120–128. doi: 10.1016/j.jprot.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt C, Ihling CH, Sinz A. 2015. Structure of full-length p53 tumor suppressor probed by chemical crosslinking and mass spectrometry. PMID: 25728495 [Medline] [DOI] [PubMed]
- Merkley ED, Baker ES, Crowell KL, Orton DJ, Taverner T, Ansong C, Ibrahim YM, Burnet MC, Cort JR, Anderson GA, Smith RD, Adkins JN. Mixed-isotope labeling with LC-IMS-MS for characterization of protein-protein interactions by chemical cross-linking. J Am Soc Mass Spectrom. 2013;24:444–449. doi: 10.1007/s13361-012-0565-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettelkau J, Thondorf I, Theisgen S, Lilie H, Schroder T, Arlt C, Ihling CH, Sinz A. Structural analysis of guanylyl cyclase-activating protein-2 (GCAP-2) homodimer by stable isotope-labeling, chemical cross-linking, and mass spectrometry. J Am Soc Mass Spectrom. 2013;24:1969–1979. doi: 10.1007/s13361-013-0734-6. [DOI] [PubMed] [Google Scholar]
- Tidow H, Melero R, Mylonas E, Freund SM, Grossmann JG, Carazo JM, Svergun DI, Valle M, Fersht AR. Quaternary structures of tumor suppressor p53 and a specific p53 DNA complex. Proc Natl Acad Sci USA. 2007;104:12324–12329. doi: 10.1073/pnas.0705069104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okorokov AL, Sherman MB, Plisson C, Grinkevich V, Sigmundsson K, Selivanova G, Milner J, Orlova EV. The structure of p53 tumour suppressor protein reveals the basis for its functional plasticity. EMBO J. 2006;25:5191–5200. doi: 10.1038/sj.emboj.7601382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Nissan G, Sharon M. Capturing protein structural kinetics by mass spectrometry. Chem Soc Rev. 2011;40:3627–3637. doi: 10.1039/c1cs15052a. [DOI] [PubMed] [Google Scholar]
- Lanucara F, Holman SW, Gray CJ, Eyers CE. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat Chem. 2014;6:281–294. doi: 10.1038/nchem.1889. [DOI] [PubMed] [Google Scholar]
- Chorev DS, Moscovitz O, Geiger B, Sharon M. Regulation of focal adhesion formation by a vinculin-Arp2/3 hybrid complex. Nat Commun. 2014;5:3758. doi: 10.1038/ncomms4758. [DOI] [PubMed] [Google Scholar]
- Sobott F, Hernandez H, McCammon MG, Tito MA, Robinson CV. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal Chem. 2002;74:1402–1407. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- Chernushevich IV, Thomson BA. Collisional cooling of large ions in electrospray mass spectrometry. Anal Chem. 2004;76:1754–1760. doi: 10.1021/ac035406j. [DOI] [PubMed] [Google Scholar]
- Wilm M, Mann M. Analytical properties of the nanoelectrospray ion source. Anal Chem. 1996;68:1–8. doi: 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]
- Kirshenbaum N, Michaelevski I, Sharon M. Analyzing large protein complexes by structural mass spectrometry. J Vis Exp. 2010 doi: 10.3791/1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose RJ, Damoc E, Denisov E, Makarov A, Heck AJ. High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat Methods. 2012;9:1084–1086. doi: 10.1038/nmeth.2208. [DOI] [PubMed] [Google Scholar]
- Makarov A. Electrostatic axially harmonic orbital trapping: a high-performance technique of mass analysis. Anal Chem. 2000;72:1156–1162. doi: 10.1021/ac991131p. [DOI] [PubMed] [Google Scholar]
- Snijder J, van de Waterbeemd M, Damoc E, Denisov E, Grinfeld D, Bennett A, Agbandje-McKenna M, Makarov A, Heck AJ. Defining the stoichiometry and cargo load of viral and bacterial nanoparticles by Orbitrap mass spectrometry. J Am Chem Soc. 2014;136:7295–7299. doi: 10.1021/ja502616y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov ME, Damoc E, Denisov E, Compton PD, Horning S, Makarov AA, Kelleher NL. From protein complexes to subunit backbone fragments: a multi-stage approach to native mass spectrometry. Anal Chem. 2013;85:11163–11173. doi: 10.1021/ac4029328. [DOI] [PubMed] [Google Scholar]
- Sharon M, Mao H, Boeri Erba E, Stephens E, Zheng N, Robinson CV. Symmetrical modularity of the COP9 signalosome complex suggests its multifunctionality. Structure. 2009;17:31–40. doi: 10.1016/j.str.2008.10.012. [DOI] [PubMed] [Google Scholar]
- Rozen S, Tieri A, Ridner G, Stark AK, Schmaler T, Ben-Nissan G, Dubiel W, Sharon M. Exposing the subunit diversity within protein complexes: a mass spectrometry approach. Methods. 2013;59:270–277. doi: 10.1016/j.ymeth.2012.12.013. [DOI] [PubMed] [Google Scholar]
- Taverner T, Hernandez H, Sharon M, Ruotolo BT, Matak-Vinkovic D, Devos D, Russell RB, Robinson CV. Subunit architecture of intact protein complexes from mass spectrometry and homology modeling. Acc Chem Res. 2008;41:617–627. doi: 10.1021/ar700218q. [DOI] [PubMed] [Google Scholar]
- van Breukelen B, Barendregt A, Heck AJ, van den Heuvel RH. Resolving stoichiometries and oligomeric states of glutamate synthase protein complexes with curve fitting and simulation of electrospray mass spectra. Rapid Commun Mass Spectrom. 2006;20:2490–2496. doi: 10.1002/rcm.2620. [DOI] [PubMed] [Google Scholar]
- Morgner N, Robinson CV. Massign: an assignment strategy for maximizing information from the mass spectra of heterogeneous protein assemblies. Anal Chem. 2012;84:2939–2948. doi: 10.1021/ac300056a. [DOI] [PubMed] [Google Scholar]
- Stengel F, Baldwin AJ, Bush MF, Hilton GR, Lioe H, Basha E, Jaya N, Vierling E, Benesch JL. Dissecting heterogeneous molecular chaperone complexes using a mass spectrum deconvolution approach. Chem Biol. 2012;19:599–607. doi: 10.1016/j.chembiol.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomeli SH, Peng IX, Yin S, Loo RR, Loo JA. New reagents for increasing ESI multiple charging of proteins and protein complexes. J Am Soc Mass Spectrom. 2010;21:127–131. doi: 10.1016/j.jasms.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling HJ, Daly MP, Feld GK, Thoren KL, Kintzer AF, Krantz BA, Williams ER. Effects of supercharging reagents on noncovalent complex structure in electrospray ionization from aqueous solutions. J Am Soc Mass Spectrom. 2010;21:1762–1774. doi: 10.1016/j.jasms.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeja SG, Tipton JD, Emmett MR, Marshall AG. New reagents for enhanced liquid chromatographic separation and charging of intact protein ions for electrospray ionization mass spectrometry. Anal Chem. 2010;82:7515–7519. doi: 10.1021/ac1016858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyachenko A, Gruber R, Shimon L, Horovitz A, Sharon M. Allosteric mechanisms can be distinguished using structural mass spectrometry. Proc Natl Acad Sci USA. 2013;110:7235–7239. doi: 10.1073/pnas.1302395110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcluckey SA. Principles of collisional activation in analytical mass spectrometry. J Am Soc Mass Spectrom. 1992;3:599–614. doi: 10.1016/1044-0305(92)85001-Z. [DOI] [PubMed] [Google Scholar]
- Benesch JL. Collisional activation of protein complexes: picking up the pieces. J Am Soc Mass Spectrom. 2009;20:341–348. doi: 10.1016/j.jasms.2008.11.014. [DOI] [PubMed] [Google Scholar]
- Sharon M, Taverner T, Ambroggio XI, Deshaies RJ, Robinson CV. Structural organization of the 19S proteasome lid: insights from MS of intact complexes. PLoS Biol. 2006;4:e267. doi: 10.1371/journal.pbio.0040267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Wysocki VH. Surface induced dissociation: dissecting noncovalent protein complexes in the gas phase. Acc Chem Res. 2014;47:1010–1018. doi: 10.1021/ar400223t. [DOI] [PubMed] [Google Scholar]
- Hernandez H, Dziembowski A, Taverner T, Seraphin B, Robinson CV. Subunit architecture of multimeric complexes isolated directly from cells. EMBO Rep. 2006;7:605–610. doi: 10.1038/sj.embor.7400702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez H, Robinson CV. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat Protoc. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- Jurneczko E, Barran PE. How useful is ion mobility mass spectrometry for structural biology? The relationship between protein crystal structures and their collision cross sections in the gas phase. Analyst. 2011;136:20–28. doi: 10.1039/c0an00373e. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Hyung SJ, Ruotolo BT. Ion mobility-mass spectrometry for structural proteomics. Expert Rev Proteomics. 2012;9:47–58. doi: 10.1586/epr.11.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelevski I, Eisenstein M, Sharon M. Gas-phase compaction and unfolding of protein structures. Anal Chem. 2010;82:9484–9491. doi: 10.1021/ac1021419. [DOI] [PubMed] [Google Scholar]
- Teplow DB, Lazo ND, Bitan G, Bernstein S, Wyttenbach T, Bowers MT, Baumketner A, Shea JE, Urbanc B, Cruz L, Borreguero J, Stanley HE. Elucidating amyloid beta-protein folding and assembly: a multidisciplinary approach. Acc Chem Res. 2006;39:635–645. doi: 10.1021/ar050063s. [DOI] [PubMed] [Google Scholar]
- van Duijn E, Barendregt A, Synowsky S, Versluis C, Heck AJ. Chaperonin complexes monitored by ion mobility mass spectrometry. J Am Chem Soc. 2009;131:1452–1459. doi: 10.1021/ja8055134. [DOI] [PubMed] [Google Scholar]
- Politis A, Park AY, Hyung SJ, Barsky D, Ruotolo BT, Robinson CV. Integrating ion mobility mass spectrometry with molecular modelling to determine the architecture of multiprotein complexes. PLoS One. 2010;5:e12080. doi: 10.1371/journal.pone.0012080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd DA, Ariza A, Edwards TA, Barr JN, Stonehouse NJ, Ashcroft AE. Probing bunyavirus N protein oligomerisation using mass spectrometry. Rapid Commun Mass Spectrom. 2014;28:793–800. doi: 10.1002/rcm.6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganowsky A, Reading E, Allison TM, Ulmschneider MB, Degiacomi MT, Baldwin AJ, Robinson CV. Membrane proteins bind lipids selectively to modulate their structure and function. Nature. 2014;510:172–175. doi: 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon M, Witt S, Glasmacher E, Baumeister W, Robinson CV. Mass spectrometry reveals the missing links in the assembly pathway of the bacterial 20 S proteasome. J Biol Chem. 2007;282:18448–18457. doi: 10.1074/jbc.M701534200. [DOI] [PubMed] [Google Scholar]
- Liu J, Konermann L. Assembly of hemoglobin from denatured monomeric subunits: heme ligation effects and off-pathway intermediates studied by electrospray mass spectrometry. Biochemistry. 2013;52:1717–1724. doi: 10.1021/bi301693g. [DOI] [PubMed] [Google Scholar]
- Aquilina JA, Shrestha S, Morris AM, Ecroyd H. Structural and functional aspects of hetero-oligomers formed by the small heat shock proteins alphaB-crystallin and HSP27. J Biol Chem. 2013;288:13602–13609. doi: 10.1074/jbc.M112.443812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling PC, Srayko M, Takhar KS, Pozniakovsky A, Hyman AA, Leroux MR. Functional interaction between phosducin-like protein 2 and cytosolic chaperonin is essential for cytoskeletal protein function and cell cycle progression. Mol Biol Cell. 2007;18:2336–2345. doi: 10.1091/mbc.E07-01-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G, Tsvetkov P, Kahana C, Shaul Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005;19:316–321. doi: 10.1101/gad.319905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscovitz O, Ben-Nissan G, Fainer I, Pollack P, Mizrachi L, Sharon M. The Parkinson's-associated protein DJ-1 regulates the 20S proteasome. PMID: 25833141 [Medline] (in press) [DOI] [PubMed]
- Moscovitz O, Tsvetkov P, Hazan N, Michaelevski I, Keisar H, Ben-Nissan G, Shaul Y, Sharon M. A mutually inhibitory feedback loop between the 20S proteasome and its regulator, NQO1. Mol Cell. 2012;47:76–86. doi: 10.1016/j.molcel.2012.05.049. [DOI] [PubMed] [Google Scholar]
- Herzog F, Kahraman A, Boehringer D, Mak R, Bracher A, Walzthoeni T, Leitner A, Beck M, Hartl FU, Ban N, Malmstrom L, Aebersold R. Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science. 2012;337:1348–1352. doi: 10.1126/science.1221483. [DOI] [PubMed] [Google Scholar]
- Lasker K, Forster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, Beck F, Aebersold R, Sali A, Baumeister W. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci USA. 2012;109:1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauth F, Ihling CH, Ruttinger HH, Sinz A. Heterobifunctional isotope-labeled amine-reactive photo-cross-linker for structural investigation of proteins by matrix-assisted laser desorption/ionization tandem time-of-flight and electrospray ionization LTQ-Orbitrap mass spectrometry. Rapid Commun Mass Spectrom. 2009;23:2811–2818. doi: 10.1002/rcm.4188. [DOI] [PubMed] [Google Scholar]
- Cohen P. The regulation of protein function by multisite phosphorylation-a 25 year update. Trends Biochem Sci. 2000;25:596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- Schmidt C, Zhou M, Marriott H, Morgner N, Politis A, Robinson CV. Comparative cross-linking and mass spectrometry of an intact F-type ATPase suggest a role for phosphorylation. Nat Commun. 2013;4:1985. doi: 10.1038/ncomms2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins JR, Diboun I, Dessailly BH, Lees JG, Orengo C. Transient protein-protein interactions: structural, functional, and network properties. Structure. 2010;18:1233–1243. doi: 10.1016/j.str.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Politis A, Schmidt C, Tjioe E, Sandercock AM, Lasker K, Gordiyenko Y, Russel D, Sali A, Robinson CV. Topological models of heteromeric protein assemblies from mass spectrometry: application to the yeast eIF3:eIF5 complex. Chem Biol. 2015;22:117–128. doi: 10.1016/j.chembiol.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero C, Milenkovic T, Przulj N, Kaiser P, Huang L. Characterization of the proteasome interaction network using a QTAX-based tag-team strategy and protein interaction network analysis. Proc Natl Acad Sci USA. 2008;105:13333–13338. doi: 10.1073/pnas.0801870105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchanek M, Radzikowska A, Thiele C. Photo-leucine and photo-methionine allow identification of protein-protein interactions in living cells. Nat Methods. 2005;2:261–267. doi: 10.1038/nmeth752. [DOI] [PubMed] [Google Scholar]
- Kalisman N, Adams CM, Levitt M. Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc Natl Acad Sci USA. 2012;109:2884–2889. doi: 10.1073/pnas.1119472109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Duijn E, Barbu IM, Barendregt A, Jore MM, Wiedenheft B, Lundgren M, Westra ER, Brouns SJ, Doudna JA, van der Oost J, Heck AJ. Native tandem and ion mobility mass spectrometry highlight structural and modular similarities in clustered-regularly-interspaced shot-palindromic-repeats (CRISPR)-associated protein complexes from Escherichia coli and Pseudomonas aeruginosa. Mol Cell Proteomics. 2012;11:1430–1441. doi: 10.1074/mcp.M112.020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillon C, Zhou M, Zhang J, Politis A, Beilsten-Edmands V, Cannone G, Graham S, Robinson CV, Spagnolo L, White MF. Structure of the CRISPR interference complex CSM reveals key similarities with cascade. Mol Cell. 2013;52:124–134. doi: 10.1016/j.molcel.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber BJ, Boehringer D, Leitner A, Bieri P, Voigts-Hoffmann F, Erzberger JP, Leibundgut M, Aebersold R, Ban N. Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature. 2014;505:515–519. doi: 10.1038/nature12890. [DOI] [PubMed] [Google Scholar]
- Stengel F, Aebersold R, Robinson CV. Joining forces: integrating proteomics and cross-linking with the mass spectrometry of intact complexes. Mol Cell Proteomics. 2012;11:R111.014027. doi: 10.1074/mcp.R111.014027. [DOI] [PMC free article] [PubMed] [Google Scholar]