

Abstract
Here we study the intact stoichiometry and top-down fragmentation behavior of three integral membrane proteins which were natively reconstituted into detergent micelles: the mechano-sensitive ion channel of large conductance (MscL), the Kirbac potassium channel and the p7 viroporin from the hepatitis C virus. By releasing the proteins under nondenaturing conditions inside the mass spectrometer, we obtained their oligomeric sizes. Increasing the ion activation (collision energy) causes unfolding and subsequent ejection of a highly charged monomer from the membrane protein complexes. Further increase of the ion activation then causes collision-induced dissociation (CID) of the ejected monomers, with fragments observed which were predominantly found to stem from membrane-embedded regions. These experiments show how in a single experiment, we can probe the relation between higher-order structure and protein sequence, by combining the native MS data with fragmentation obtained from top-down MS.
Keywords: mass spectrometry, ion channels, top-down sequencing, integral membrane proteins, collision-induced dissociation
Introduction
The development of electrospray ionization (ESI)1 and matrix-assisted laser desorption ionization (MALDI)2 has led to an explosion of mass spectrometry (MS)-based proteomics approaches. Central to these approaches is the detection and/or quantification of proteins under denaturing conditions, usually after enzymatic digestion, by fragmenting them to obtain sequence-specific information. At the same time the development of a miniaturized version of Fenn's electrospray (nano-ESI) by Wilm and Mann has made it possible to transfer proteins and protein complexes intact and in their folded state into the gas-phase.3,4 Especially in combination with ion mobility, native mass spectrometry is rapidly becoming an invaluable tool for structural biology, as it yields not only information on mass, ligand binding and oligomeric size, but also allows for separation of copopulated conformations of a single protein.5–9
In recent years, native mass spectrometry has been extended to the study of integral membrane proteins (IMP) in the gas-phase. The realization that micelles—the formation of which is to a large extent hydrophobically driven—could survive transfer to the ultimate hydrophobic environment,10 the vacuum of the mass spectrometer, enabled studies on detergent micelles with IMPs inside.11 The apparent protective capabilities of suitable detergent micelles were found to be sufficient in most cases to observe oligomeric membrane protein complexes in their native global fold.12,13 More recent developments in the field show how native MS can reveal lipid binding,14 probe conformational dynamics of membrane proteins,15,16 and even assist with finding the ideal crystallization conditions for X-ray diffraction.17
One of the strengths of mass spectrometry is its ability to provide information ranging from sequence information of proteins (e.g., mutations, truncations, post-translational modifications) to small molecule binding and the global folding or oligomerization state of complexes, without averaging over all different species which may be present in a sample.18–20 Bottom-up approaches, where a protein is digested in solution prior to analysis by MS, are very effective in sequencing globular and membrane proteins with suitable enzymatic cleavage sites, based on peptide fragmentation and subsequent comparison to a database of theoretical fragments. Sequence coverage for IMPs however that consist of extensive membrane spanning regions can be poor with bottom-up approaches. Infrequent prevalence of cleavage sites in membrane spanning regions makes sequencing difficult, due to large precursor peptides.21
Another approach that is currently drawing renewed interest is the sequencing of proteins using top-down methods, for example with the aim to characterize individual protein isoforms. Top-down approaches use collision-induced dissociation (CID) of intact proteins—often denatured—to produce fragments, yielding sequence information. Sequencing by CID can even be performed starting from native proteins and protein complexes, as reported previously.22–24 In CID, ions are accelerated by a voltage offset (“collision energy”) and undergo multiple, energetic collisions with an inert gas (e.g., argon). The activation of the ions, their internal energy, is controlled via the kinetic energy of the ions and the frequency of collisions with the inert gas. Low acceleration voltages and high gas-pressures reduce the internal energy of the ion (“cooling”), whereas lower gas-pressures and high acceleration voltages allow the ion to accelerate more between collisions, resulting in an energy transfer that augments the internal energy of the ions (“heating”).25 As CID is a rather slow, multi-step process, the internal energy of the ions increases slowly until dissociation occurs. As such, CID is an ergodic (quasi-thermal) process, which preferentially leads to fragmentation of the weakest interaction in the protein or complex—usually a noncovalent bond.
When using top-down approaches, time-consuming steps such as enzymatic digestion which are commonly used in bottom-up approaches, are no longer necessary. Apart from (partial) primary sequence data which can be used to investigate the identity of an unknown protein, top-down sequencing can also provide information on the full complement of post-translational modifications present.26 The interest for top-down MS has been instigated by the results of the human genome project which led to the realization that we have fewer genes than initially anticipated.27 As a consequence, the complexity of our biological machinery must stem, amongst others, from protein sequence variations.28 Recently the term “proteoform” has been coined to encompass such variations of primary structure, and linked with top-down MS.29
IMPs have previously been studied using top-down MS, but so far always in combination with a denaturing LC/MS setup.18,30–32 Membrane proteins were solubilized using organic solvents, leading to loss of their oligomeric state and unfolding of the protein.19 Using this approach, the use of detergents can be largely circumvented and the denaturing conditions for the protein make top-down fragmentation an efficient tool for probing sequential heterogeneity and post-translational modifications. Although yielding information on proteoforms, such approaches do not maintain the link to their higher-order structure, which adds yet another level of complexity allowing for an even greater diversity in our biological machinery.33 Notable examples which underline this relationship are phosphorylation of a kinase to regulate its activity,34 and the alternative splicing of the FAS receptor which either generates a membrane bound form that promotes apoptosis, or a soluble isoform that prevents programmed cell death.35
Here we show how—by combining native MS with top-down CID fragmentation—a single experiment can reveal both the higher-order structure (oligomeric distribution of a membrane protein) as well as proteoforms (accurate mass and amino acid sequence) of a membrane protein. By increasing the collision energy past the threshold for release of membrane proteins from detergent micelles, highly charged monomer subunits are generated which upon further activation fragment, yielding sequence information.
Results and Discussion
Three integral membrane proteins were natively reconstituted into detergent micelles and studied by native top-down MS. The gating mechanism of the pentameric mechanosensitive ion channel of large conductance (MscL)15 and the oligomeric state of the tetrameric Kirbac3.1 ion channel have been studied previously,36 whereas the hepatitis C Virus (HCV) p7 viroporin had not been investigated before. As detergent micelles are quite heterogeneous in size, native MS analysis of embedded membrane proteins is not possible without releasing them, by using collision-induced dissociation (CID) to remove the attached detergent. For the commonly used nonionic detergent dodecyl-maltoside (DDM), collision energies of 100–200 V are required to remove the detergent molecules (Fig. 1), but values can vary depending on protein size, surface area of the membrane-embedded regions and the strength of the protein-detergent interaction (see below).
Figure 1.
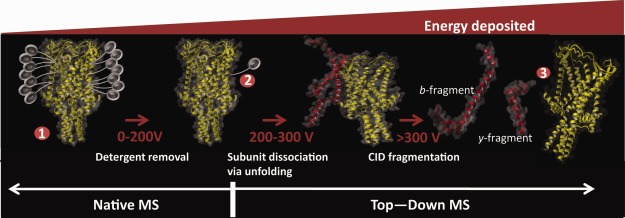
Effect of different energy regimes used in mass spectrometry of membrane proteins under native conditions when using sugar based detergents, such as the commonly used detergent DDM. The apparent protective ability of detergent micelles in the gas-phase allow noncovalent interactions to be maintained for membrane proteins at levels (up to 200 V) at which soluble protein complexes would already undergo significant dissociation. Increasing the collision energy above the threshold for membrane protein release leads to collision induced unfolding, followed by dissociation and even fragmentation, yielding sequence information. Collision energies reported are typical values for DDM, but will vary based on the protein and detergents used.
The ability of detergent micelles to protect natively reconstituted membrane protein complexes from gas-phase dissociation is believed to be due to absorbing the collision energy, leading to ejection of detergent molecules instead. Indeed, it has been shown that the stability of oligomeric membrane proteins towards dissociation in the gas-phase can be correlated to the extent of their membrane spanning regions.36 Furthermore, especially when stripping the last remaining detergent molecules, the strength of interaction between detergent and IMP becomes another factor to take into account. When the interactions between the subunits of an IMP are weaker than those of the subunits with the detergent, dissociation of the complex will precede detergent stripping. Such effects can be observed for example when subunit interactions are hydrophobically driven. Although a strong stabilizing factor in solution, such interactions are absent in the gas-phase due to the loss of the hydrophobic effect in the vacuum of the mass spectrometer.
Another factor defining membrane protein stability, the net charge carried by the ion, is believed to be correlated to the soluble area of the membrane protein. Membrane proteins reconstituted in detergents appear to carry as much charge after release from the detergent micelles as would be predicted for a soluble protein the size of the soluble region of the IMP.37 As a consequence IMPs typically yield charge states that are lower than observed for soluble proteins of a similar size. The increase in internal energy in CID is dependent on both the acceleration offset (collision energy) as well as the net charge the ion carries. Thus, although the settings typically used for IMP release from detergent micelles are seemingly harsh, the lower charge states in combination with the protective capabilities of the detergent often allow for the release of intact membrane protein complexes. Increasing the collision energy beyond the threshold for the release of IMPs yields dissociation similar to that observed for soluble proteins.36 Dissociation of a complex with CID proceeds in an asymmetric fashion, via unfolding of a single subunit. Unfolding coincides with redistribution of charges, yielding a highly charged monomer and a compact lowly charged oligomer, missing a single subunit.38 Generation of an unfolded, highly charged monomer is crucial to the ability to sequence the membrane protein, as it facilitates efficient fragmentation.
Figure 2(A) shows a mass spectrum for the ion channel Kirbac3.1 under conditions optimized for maximal sequence coverage obtained by top-down fragmentation (see methods and materials). There is still tetrameric Kirbac3.1 remaining, but the spectrum is dominated by the charge state distribution of the ejected monomer (indicated by red circles). Detergent release, subunit dissociation and subsequent top-down fragmentation follow the scheme shown in Figure 1, but the actual collision energies required for these steps depend on detergent, charge and mass of the complex. The energy threshold, where detergent removal and protein release occur, strongly depends on the strength of interaction between protein subunits, protein and detergent and the nature of the detergent itself. At higher energies, unfolded monomers are ejected (complex dissociation), which yields well-resolved peaks in the low m/z region that can be used to obtain accurate masses for the protein. For Kirbac subunits a mass of 33,586 ± 30 Da was found, which is in reasonable agreement with the theoretical mass of 33,577 Da (Table1). An additional advantage of such high acceleration voltages is the disassembly of detergent clusters to monomers and dimers, thus leading to minimal interference with the fragment identification.
Figure 2.

Mass spectra for the oligomeric membrane proteins (A) Kirbac3.1 solubilized in tridecyl-maltoside (3DM) at the maximal operating voltages of the instrument and (B) P7 in triton X-100 at an elevated collision energy level (275 V) yielded monomer and oligomer information as well as sequence-specific fragments. The oligomer size is indicated by colored circles, and areas highlighted in blue indicate the regions where CID fragments are observed. (C) The 17+ charge state of MscL G22C was isolated prior to fragmentation to abolish interference from remaining detergents with the fragment identification. Remaining detergent peaks in any spectra are denoted as D.
Table 1.
Masses Observed for the IMPs p7, MscL, and Kirbac3.1
| Theoretical monomer mass | Observed monomer mass | Theoretical oligomer mass | Observed oligomer mass | |
|---|---|---|---|---|
| P7 (6-mer) | 6952.39 | 6954.39 ± 3 | 41714.34 | 41717 ± 157 |
| MscL (5-mer) | 15694.51 | 15695.02 ± 6 | 78485.55 | 78882 ± 42 |
| Kirbac (4-mer) | 33577.57 | 33586.48 ± 30 | 134310.28 | 134371 ± 142 |
Native MS of p7 solubilized in triton X-100 showed predominantly monomer, with mainly hexameric and small amounts of pentameric p7 present [Fig. 2(b)]. Triton X-100 is a prominent example of a detergent which is unable to form an extensive network of hydrogen bonds, and it has been shown that MscL pentamers are released at much lower collisional activation.15,17 Nevertheless, with p7 in triton x-100 we still require collision energies of 175 V to observe oligomers, although monomers are already seen at lower collisional energies (Supporting Information Fig. S1). These observations can be explained in two ways: either p7 is present both as oligomeric as well as monomeric species within the triton X-100 detergent micelles in solution, where monomeric p7 is more easily removed from its detergent cover in the gas-phase. This would match our experience with p7 as a polydisperse protein which forms assemblies of varying size. On the other hand, the rather weak subunit interaction in p7 might suggest that the observed p7 monomers are already dissociating from the oligomeric complex upon increasing collision energies. The collision energy required to rid a membrane protein from its detergent cover in the gas-phase may well already disrupt protein-protein interactions, if they are weak; with electrostatic interactions and hydrogen bonds acting as stabilizing factors in vacuo whereas hydrophobic interactions are lost. If the interaction between detergent and protein is stronger than between protein subunits, this might lead to preferential dissociation of the complex before stripping of detergents is complete. Although the latter could explain why such broad peaks are observed for p7 oligomers (due to remaining detergents attached), our current experiments cannot distinguish between these possible explanations. In comparison, MscL (G22C mutant) reconstituted in triton X-100 detergent was previously shown to release as a pentameric complex already at very low collision energy (10V)15. For fragmentation of MscL the 17+ charge state was isolated in the quadrupole, prior to dissociation of subunits and subsequent fragmentation. As CID fragmentation of MscL was obtained at rather low collision energies, significant signal from detergent clusters survives, which due to the heterogeneous composition of triton X-100 led to spectral overlap with MscL top-down CID fragments. By using mass selection prior to CID, the interference of detergent was circumvented, allowing facile assignment of fragments in the lower m/z regions [Fig. 2(c)].
Increasing the acceleration voltages well past the thresholds for detergent removal and subunit dissociation yielded CID fragments for all three ion channels (Fig. 2 blue boxes). As already mentioned, the energy required to induce top-down CID fragmentation depends on the detergent, but also on the mass and charge of the protein. To facilitate comparison of acceleration voltages between different systems, the laboratory frame energy (in eV) is often used which can be obtained by multiplying the ion charge with the acceleration (collision) voltage. The energies required for CID fragmentation differ drastically between proteins, with laboratory frame energies between 2.20 and 2.75 keV for p7, whereas for Kirbac the values range from 9.2 to 11.2 keV [for the charge states in Fig. 2(a,b)]. One factor for these different fragmentation thresholds in CID, which is an ergodic process, is the mass of the protein, as larger proteins have more degrees of freedom over which the increase of internal energy can be distributed. For membrane proteins however such a relationship is more difficult to assess, as the stripping of detergent in the gas-phase is the crucial step prior to fragmentation. Although both p7 and MscL were reconstituted in triton x-100, they require similar energy levels for fragmentation (2.20-2.75 keV for p7 and 2.89 keV for MscL 17+), even though pentameric MscL is almost double the mass of the p7 hexamer. Another complicating factor for sequencing of membrane proteins by CID is that membrane proteins tend to have lower average charge states than soluble proteins, most likely as a result of being partially embedded in micelles at the beginning of the electrospray process37. Fortunately, this effect is compensated by the asymmetric dissociation induced by CID which produces highly charged monomers and top-down fragments (Fig. 3). In hetero-oligomeric complexes though, typically only the smallest exposed subunit dissociates while taking a disproportionate amount of charge with it – thereby limiting the use of top-down CID sequencing to this subunit. In such cases it might be beneficial to first produce subcomplexes in solution by destabilization with chaotropic agents, prior to asymmetric dissociation and sequencing39,40. The 17+ MscL precursor displayed b-fragments with a charge of predominantly 3+ and 4+, although some 2+ fragments were observed as well, and a small series of 6+ y-fragments [Fig. 3(a−c)]. Kirbac shows some singly charged C-terminal y-fragments, but otherwise mainly b- fragments with charge states ranging from 1+ up to 7+ [Fig. 3(b)]. The high prevalence of b-ions for these spectra is remarkable, as they are considered to be much more labile than y-ions due to the possibility to undergo subsequent rearrangements, resulting in a-fragments.41 It should be noted though that conditions here were somewhat less energetic than in standard Q-TOF CID, as the voltages used are near the maximum possible on the instrument and contribute to a large part to detergent removal.
Figure 3.
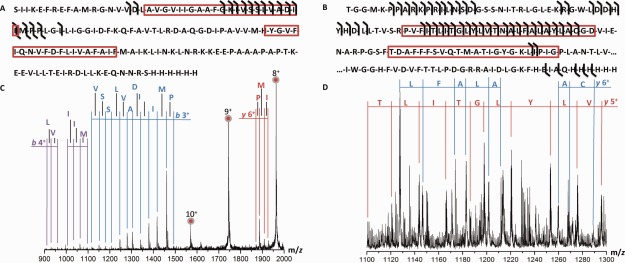
Primary structure of membrane proteins investigated using top-down CID. (A) Sequence of MscL G22C with b- and y-fragments indicated and (B) Excerpt (1–112 and 265–300) from the sequence of Kirbac3.1. Predicted membrane spanning regions are indicated with a red box. (C) Part of the spectra observed for MscL and (D) Kirbac3.1 displaying CID fragments.
The top-down CID fragments of different native charge states were found to be clustered around limited stretches of the protein, with a sequence coverage for MscL and Kirbac of 11% and 18%, respectively [Fig. 3(a,b)]. Fragmentation for both these proteins appears mostly in the N-terminal part of the protein sequence, with little to no fragments from the middle or end of the sequence. Such limited fragmentation efficiency is quite common in top-down studies and has also been reported for larger IMPs before. It has been postulated that even under denaturing conditions these proteins maintain some residual structure,42 which results in the limited fragmentation efficiency for larger IMPs. The p7 viroporin on the other hand, which is much smaller than the other two proteins, showed more intense CID fragments (Fig. 4). The sequence coverage was 73%, which would indicate the absence of residual structure due to its smaller size. Fragmentation was neither observed in the N- or C-terminus nor in amino acids 36−44 in this case.
Figure 4.
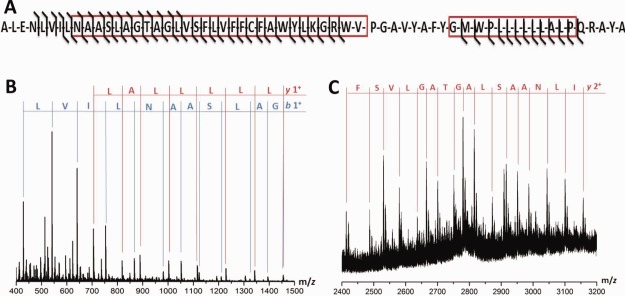
Top-down CID fragments observed for the viral porin p7 at 275 V collision energy. (A) Fragmentation observed for the sequence of p7 with membrane spanning regions indicated by a red box. Fragments observed in the (B) low m/z region and (C) high m/z region of p7.
Recent top-down proteomics work by Kelleher et al. on 126 denatured membrane proteins has shown that CID has a four-fold preference for transmembrane domains.43 As all membrane proteins studied here were known to have membrane-spanning alpha-helices, we used the sequences of the proteins to predict which regions where embedded in the lipid bilayer.44,45 Such approaches are often used to identify membrane regions when no X-ray or NMR structure or homology model is available. Although high-resolution structures of Kirbac3.1 and a homologues structure of MscL are available, the exact structure of the oligomeric p7 viroporin is still under debate.46–48 Comparing the observed fragmentation sites with the predicted transmembrane domains, we note that all three proteins displayed fragmentation within their transmembrane-spanning domains [Fig. 5(a)]. For MscL and p7, cleavage seems to occur predominantly in membrane regions, whereas Kirbac displays sites in soluble regions as well. As transmembrane prediction approaches should be used with caution, we validated our observations by plotting the observed CID fragments onto the available high-resolution structure of Kirbac3.1. To provide a similar graphical representation for p7 and MscL, we used homologous structures, as there are currently no high-resolution structures available for these IMPs [Fig. 5(b)].
Figure 5.

Top-down CID of membrane proteins (detergent micelles; native ESI) leads to fragmentation predominantly in membrane-embedded regions. A: Transmembrane prediction for each of the three proteins reveals potential membrane-embedded areas (red curves); comparison with observed CID fragmentation sites (black bars) shows a preference for membrane regions. B: Mapping the observed fragmentation sites onto the 3D-structures of the three membrane proteins, confirms the results obtained by TMH prediction. Peptide fragments observed are mapped (in red) onto the structures of the ion (in gray). PDB IDs 2M6X, 2OAR and 3ZRS, were respectively used for p7, MscL, and Kirbac3.1 visualization.
Top-down fragmentation of these membrane proteins delivers extensive sequence tags, which is quite remarkable as CID of highly charged proteins tends to favor cleavage sites which are specific to certain combinations of amino acids. Preferential C-terminal cleavage after an aspartic acid for lowly charged, and N-terminal cleavage before a proline residue for highly charged proteins have been reported.49 Such preferred cleavages often limit the sequence information obtained and make the spectra difficult to interpret, due to the considerable differences in fragment intensity. This does not seem to be the case however for the three IMPs studied here, where a more equal fragmentation propensity was observed. As CID strongly depends on charge localization and side-chain charging is expected to be absent in the transmembrane helices, this would suggest that protons can move towards these regions when becoming mobile during the CID process. It has been postulated that rather than interacting with basic side chains as expected for soluble proteins, the mobile protons are primarily attached to the amide backbone in such hydrophobic stretches of the amino acid sequence.43,50 As such interactions are less specific than interactions with a basic side chain, top-down CID of membrane proteins yields not only a greater number of backbone cleavages, but also over a larger number of backbone positions.43
Here we have shown how a single set of experiments yields the oligomeric state, accurate subunit mass and (partial) sequence information on membrane proteins, possibly even indicating membrane-embedded parts of the sequence. The main advantage of such an experimental approach is that the interdependency between amino-acid sequence (proteoform) and higher-order structure can be investigated simultaneously, even in heterogeneous samples. In comparison to the reported sequence coverage of 9–22% for top-down CID of denatured membrane proteins with multiple transmembrane domains on a Q-TOF instrument,42 we obtain similar values in top-down CID of native, oligomeric IMPs embedded in detergent micelles. Although currently the experimental approach is not yet suitable for high-throughput studies, recent developments in coupling size exclusion chromatography with electrospray ionization51 or automated sampling52 might significantly increase the viability of such attempts in the context of structural proteomics. As the experimental strategy applied here is easily integrated with the current methodology used when studying IMPs by native MS, it opens up new opportunities to study the effect of PTMs and sequence variations on the higher-order structure of membrane proteins and their complexes.
Materials and Methods
Sample preparation
Kirbac3.1 was expressed overnight at 19°C in E. coli BL21 CodonPlus (DE3) RP (Agilent) and purified according to the protocol described previously,53 with the exception of the gel-filtration step that was performed in 200 mM ammonium acetate and 0.5 mM 3DM detergent. The protein was subsequently concentrated to 0.8 mg/mL using Vivaspin concentrators with molecular weight cutoff of 50 kDa. In these experiments, we used the W46R mutant as it shows an increased stability compared to the wild-type Kirbac. P7 samples were prepared by dissolving 0.1 mg lyophilized p7 (H77 strain with the H17A mutation) in 100 µL triton X-100 (10x CMC), vortexed, and subsequently spun down to remove any large remaining particles prior to analysis. Pentameric MscL (G22C5 mutant) reconstituted in triton X-100 was prepared as reported earlier.15
Mass spectrometry
All samples were introduced into the vacuum of the mass spectrometer without further desalting using nanoESI with gold-coated borosilicate capillaries at capillary voltages of 1.2−1.6 kV. Spectra were recorded in positive ion mode on a commercially available travelling wave ion mobility mass spectrometer (Synapt G2 HDMS, Waters) in TOF-mode. The instrument was fitted with a 32 k quadrupole and settings optimized for transmission of large complexes. Gas pressures were 6.0−9.0 mbar, 1.5 e−2 mbar and 4.6 e−2 mbar for backing, source and trap cell, respectively. Collision energies were optimized for each membrane protein specifically in order to obtain maximum CID fragmentation, whilst maintaining some of the original oligomeric protein present. Voltages used were 150/150/10/125 for p7, 125/10/10/160 for MscL and 200/200/10/200 for sampling cone, trap CE, trap DC bias and transfer CE, respectively. The m/z scale of all mass spectra was externally calibrated using a 10 mg/mL CsI solution. The spectra were processed using Masslynx 4.1 (Waters). Transmembrane predictions were performed using the TMHMM server, as described elsewhere.39,40
Acknowledgments
The authors would like to thank N. Zitzmann (Oxford, UK) for the donation of the HCV p7 (protein and the Hercules foundation for funding of the Synapt G2 instrument.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supplementary Information Figure 1.
References
- Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Waki H, Ido Y, Akita S, Yoshida Y, Yoshida T, Matsuo T. Protein and polymer analyses up tom/z 100 000 by laser ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 1988;2:151–153. [Google Scholar]
- Wilm M, Mann M. Analytical properties of the nanoelectrospray ion source. Anal Chem. 1996;68:1–8. doi: 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]
- Wilm MS, Mann M. Electrospray and Taylor-Cone theory, Dole's beam of macromolecules at last? Int J Mass Spectrom Ion Process. 1994;136:167–180. [Google Scholar]
- Hilton GR, Benesch JLP. Two decades of studying non-covalent biomolecular assemblies by means of electrospray ionization mass spectrometry. J R Soc Interface. 2012;9:801–816. doi: 10.1098/rsif.2011.0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch JLP, Ruotolo BT. Mass spectrometry: come of age for structural and dynamical biology. Curr Opin Struct Biol. 2011;21:641–649. doi: 10.1016/j.sbi.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konijnenberg A, Butterer A, Sobott F. Native ion mobility-mass spectrometry and related methods in structural biology. Biochim Biophys Acta - Proteins Proteomics. 2013;1834:1239–1256. doi: 10.1016/j.bbapap.2012.11.013. [DOI] [PubMed] [Google Scholar]
- Loo Ja. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev. 1997;16:1–23. doi: 10.1002/(SICI)1098-2787(1997)16:1<1::AID-MAS1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Heck AJR, Van Den Heuvel RHH. Investigation of intact protein complexes by mass spectrometry. Mass Spectrom Rev. 2004;23:368–389. doi: 10.1002/mas.10081. [DOI] [PubMed] [Google Scholar]
- Sharon M, Ilag LL, Robinson CV. Evidence for micellar structure in the gas phase. J Am Chem Soc. 2007;129:8740–8746. doi: 10.1021/ja067820h. [DOI] [PubMed] [Google Scholar]
- Ilag LL, Ubarretxena-Belandia I, Tate CG, Robinson CV. Drug binding revealed by tandem mass spectrometry of a protein-micelle complex. J Am Chem Soc. 2004;126:14362–14363. doi: 10.1021/ja0450307. [DOI] [PubMed] [Google Scholar]
- Barrera NP, Di Bartolo N, Booth PJ, Robinson CV. Micelles protect membrane complexes from solution to vacuum. Science. 2008;321:243–246. doi: 10.1126/science.1159292. [DOI] [PubMed] [Google Scholar]
- Borysik AJ, Hewitt DJ, Robinson CV. Detergent release prolongs the lifetime of native-like membrane protein conformations in the gas-phase. J Am Chem Soc. 2013;135:6078–6083. doi: 10.1021/ja401736v. [DOI] [PubMed] [Google Scholar]
- Laganowsky A, Reading E, Hopper JTS, Robinson CV. Mass spectrometry of intact membrane protein complexes. Nat Protoc. 2013;8:639–651. doi: 10.1038/nprot.2013.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konijnenberg A, Yilmaz D, Ingólfsson HI, Dimitrova A, Marrink SJ, Li Z, Vénien-Bryan C, Sobott F, Koçer A. Global structural changes of an ion channel during its gating are followed by ion mobility mass spectrometry. Proc Natl Acad Sci USA. 2014;111:17170–17175. doi: 10.1073/pnas.1413118111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcoux J, Wang SC, Politis A, Reading E, Ma J, Biggin PC, Zhou M, Tao H, Zhang Q, Chang G, N Morgner, CV Robinson. Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc Natl Acad Sci USA. 2013;110:9704–9709. doi: 10.1073/pnas.1303888110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganowsky A, Reading E, Allison TM, Ulmschneider MB, Degiacomi MT, Baldwin AJ, Robinson CV. Membrane proteins bind lipids selectively to modulate their structure and function. Nature. 2014;510:172–175. doi: 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangaraj B, Ryan CM, Souda P, Krause K, Faull KF, Weber APM, Fromme P, Whitelegge JP. Data-directed top-down Fourier-transform mass spectrometry of a large integral membrane protein complex: photosystem II from Galdieria sulphuraria. Proteomics. 2010;10:3644–3656. doi: 10.1002/pmic.201000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitelegge JP, Gundersen CB, Faull KF. Electrospray-ionization mass spectrometry of intact intrinsic membrane proteins. Protein Sci. 1998;7:1423–1430. doi: 10.1002/pro.5560070619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catherman AD, Li M, Tran JC, Durbin KR, Compton PD, Early BP, Thomas PM, Kelleher NL. Top down proteomics of human membrane proteins from enriched mitochondrial fractions. Anal Chem. 2013;85:1880–1888. doi: 10.1021/ac3031527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll J, Altman MC, Fearnley IM, Walker JE. Identification of membrane proteins by tandem mass spectrometry of protein ions. Proc Natl Acad Sci USA. 2007;104:14330–14335. doi: 10.1073/pnas.0706817104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch JLP, Ruotolo BT, Sobott F, Wildgoose J, Gilbert A, Bateman R, Robinson CV. Quadrupole-time-of-flight mass spectrometer modified for higher-energy dissociation reduces protein assemblies to peptide fragments. Anal Chem. 2009;81:1270–1274. doi: 10.1021/ac801950u. [DOI] [PubMed] [Google Scholar]
- Belov ME, Damoc E, Denisov E, Compton PD, Horning S, Makarov AA, Kelleher NL. From protein complexes to subunit backbone fragments: a multi-stage approach to native mass spectrometry. Anal Chem. 2013;85:11163–11173. doi: 10.1021/ac4029328. [DOI] [PubMed] [Google Scholar]
- Rathore D, Dodds ED. Collision-induced release, ion mobility separation, and amino acid sequence analysis of subunits from mass-selected noncovalent protein complexes. J Am Soc Mass Spectrom. 2014;25:1600–1609. doi: 10.1007/s13361-014-0946-4. [DOI] [PubMed] [Google Scholar]
- Chernushevich IV, Thomson BA. Collisional cooling of large ions in electrospray mass spectrometry. Anal Chem. 2004;76:1754–1760. doi: 10.1021/ac035406j. [DOI] [PubMed] [Google Scholar]
- Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridriksson EK, McLafferty FW. Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J Am Chem Soc. 1999;121:806–812. [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter H, Apweiler R, Holzhütter H-G, Jungblut PR. Finding one's way in proteomics: a protein species nomenclature. Chem Cent J. 2009;3:11. doi: 10.1186/1752-153X-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LM, Kelleher NL. Proteoform: a single term describing protein complexity. Nat Methods. 2013;10:186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitelegge JP. Thylakoid membrane proteomics. Photosynth Res. 2003;78:265–277. doi: 10.1023/B:PRES.0000006828.65688.0d. [DOI] [PubMed] [Google Scholar]
- Berridge G, Chalk R, D'Avanzo N, Dong L, Doyle D, Kim JI, Xia X, Burgess-Brown N, Deriso A, Carpenter EP, et al. High-performance liquid chromatography separation and intact mass analysis of detergent-solubilized integral membrane proteins. Anal Biochem. 2011;410:272–280. doi: 10.1016/j.ab.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnley IM, Skehel JM, Walker JE. Electrospray ionization mass spectrometric analysis of subunits of NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Biochem Soc Trans. 1994;22:551–555. doi: 10.1042/bst0220551. [DOI] [PubMed] [Google Scholar]
- Robinson C, V, Sali A, Baumeister W. The molecular sociology of the cell. Nature. 2007;450:973–982. doi: 10.1038/nature06523. [DOI] [PubMed] [Google Scholar]
- Yeo MG, Oh HJ, Cho H-S, Chun JS, Marcantonio EE, Song WK. Phosphorylation of Ser 21 in Fyn regulates its kinase activity, focal adhesion targeting, and is required for cell migration. J Cell Physiol. 2011;226:236–247. doi: 10.1002/jcp.22335. [DOI] [PubMed] [Google Scholar]
- Izquierdo JM, Majós N, Bonnal S, Martínez C, Castelo R, Guigó R, Bilbao D, Valcárcel J. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell. 2005;19:475–484. doi: 10.1016/j.molcel.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Wang SC, Politis A, Di Bartolo N, Bavro VN, Tucker SJ, Booth PJ, Barrera NP, Robinson CV. Ion mobility mass spectrometry of two tetrameric membrane protein complexes reveals compact structures and differences in stability and packing. J Am Chem Soc. 2010;132:15468–15470. doi: 10.1021/ja104312e. [DOI] [PubMed] [Google Scholar]
- Barrera NP, Zhou M, Robinson CV. The role of lipids in defining membrane protein interactions: insights from mass spectrometry. Trends Cell Biol. 2013;23:1–8. doi: 10.1016/j.tcb.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Benesch JLP, Aquilina JA, Ruotolo BT, Sobott F, Robinson CV. Tandem mass spectrometry reveals the quaternary organization of macromolecular assemblies. Chem Biol. 2006;13:597–605. doi: 10.1016/j.chembiol.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Levy ED, Boeri Erba E, Robinson CV, Teichmann SA. Assembly reflects evolution of protein complexes. Nature. 2008;453:1262–1265. doi: 10.1038/nature06942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh JA, Hernández H, Hall Z, Ahnert SE, Perica T, Robinson CV, Teichmann SA. Protein complexes are under evolutionary selection to assemble via ordered pathways. Cell. 2013;153:461–470. doi: 10.1016/j.cell.2013.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki VH, Resing KA, Zhang Q, Cheng G. Mass spectrometry of peptides and proteins. Methods. 2005;35:211–222. doi: 10.1016/j.ymeth.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Carroll J, Altman MC, Fearnley IM, Walker JE. Identification of membrane proteins by tandem mass spectrometry of protein ions. Proc Natl Acad Sci USA. 2007;104:14330–14335. doi: 10.1073/pnas.0706817104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner OS, Catherman AD, Early BP, Thomas PM, Compton PD, Kelleher NL. Fragmentation of integral membrane proteins in the gas phase. Anal Chem. 2014;86:4627–4634. doi: 10.1021/ac500864w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, von Heijne G, Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol. 1998;6:175–182. [PubMed] [Google Scholar]
- Luik P, Chew C, Aittoniemi J, Chang J, Wentworth P, Dwek RA, Biggin PC, Vénien-Bryan C, Zitzmann N. The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc Natl Acad Sci USA. 2009;106:12712–12716. doi: 10.1073/pnas.0905966106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OuYang B, Xie S, Berardi MJ, Zhao X, Dev J, Yu W, Sun B, Chou JJ. Unusual architecture of the p7 channel from hepatitis C virus. Nature. 2013;498:521–525. doi: 10.1038/nature12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TL, Thompson GS, Kalverda AP, Kankanala J, Bentham M, Wetherill LF, Thompson J, Barker AM, Clarke D, Noerenberg M, et al. Structure-guided design affirms inhibitors of hepatitis C virus p7 as a viable class of antivirals targeting virion release. Hepatology. 2014;59:408–422. doi: 10.1002/hep.26685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton KA, Pitteri SJ, Laskowski M, McLuckey SA. Effects of single amino acid substitution on the collision-induced dissociation of intact protein ions: Turkey ovomucoid third domain. J Proteome Res. 2004;3:1033–1041. doi: 10.1021/pr049910w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki VH, Tsaprailis G, Smith LL, Breci LA. Mobile and localized protons: a framework for understanding peptide dissociation. J Mass Spectrom. 2000;35:1399–1406. doi: 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Muneeruddin K, Thomas JJ, Salinas PA, Kaltashov IA. Characterization of small protein aggregates and oligomers using size exclusion chromatography with online detection by native electrospray ionization mass spectrometry. Anal Chem. 2014;86:10692–10699. doi: 10.1021/ac502590h. [DOI] [PubMed] [Google Scholar]
- Wachs T, Henion J. Electrospray device for coupling microscale separations and other miniaturized devices with electrospray mass spectrometry. Anal Chem. 2001;73:632–638. doi: 10.1021/ac000935y. [DOI] [PubMed] [Google Scholar]
- Bavro VN, De Zorzi R, Schmidt MR, Muniz JRC, Zubcevic L, Sansom MSP, Vénien-Bryan C, Tucker SJ. Structure of a KirBac potassium channel with an open bundle crossing indicates a mechanism of channel gating. Nat Struct Mol Biol. 2012;19:158–163. doi: 10.1038/nsmb.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information Figure 1.