

Significance
Phototrophic organisms worldwide produce estimated 10 gigatons of sulfoquinovose (SQ) per year; hence, complete degradation of SQ by bacteria is an important part of the biogeochemical sulfur cycle. Here, we show that Pseudomonas putida SQ1 catabolizes SQ to 3-sulfolactate (SL) in analogy to the Entner–Doudoroff pathway for glucose-6-phosphate, involving five newly discovered reactions, enzymes, and genes, and three newly discovered organosulfur intermediates. The SL can be mineralized by other bacteria, thus closing the sulfur cycle within a bacterial community. The genes for the SQ Entner–Doudoroff pathway can be found in genomes of a wide range of Proteobacteria, which shows that SQ utilization is a widespread and important, but still underrecognized, trait of bacteria in all environments where SQ is produced and degraded.
Keywords: bacterial biodegradation, organosulfonate, sulfolipid, 6-deoxy-6-sulfoglucose, sulfur cycle
Abstract
Sulfoquinovose (SQ; 6-deoxy-6-sulfoglucose) is the polar head group of the plant sulfolipid SQ-diacylglycerol, and SQ comprises a major proportion of the organosulfur in nature, where it is degraded by bacteria. A first degradation pathway for SQ has been demonstrated recently, a “sulfoglycolytic” pathway, in addition to the classical glycolytic (Embden–Meyerhof) pathway in Escherichia coli K-12; half of the carbon of SQ is abstracted as dihydroxyacetonephosphate (DHAP) and used for growth, whereas a C3-organosulfonate, 2,3-dihydroxypropane sulfonate (DHPS), is excreted. The environmental isolate Pseudomonas putida SQ1 is also able to use SQ for growth, and excretes a different C3-organosulfonate, 3-sulfolactate (SL). In this study, we revealed the catabolic pathway for SQ in P. putida SQ1 through differential proteomics and transcriptional analyses, by in vitro reconstitution of the complete pathway by five heterologously produced enzymes, and by identification of all four organosulfonate intermediates. The pathway follows a reaction sequence analogous to the Entner–Doudoroff pathway for glucose-6-phosphate: It involves an NAD+-dependent SQ dehydrogenase, 6-deoxy-6-sulfogluconolactone (SGL) lactonase, 6-deoxy-6-sulfogluconate (SG) dehydratase, and 2-keto-3,6-dideoxy-6-sulfogluconate (KDSG) aldolase. The aldolase reaction yields pyruvate, which supports growth of P. putida, and 3-sulfolactaldehyde (SLA), which is oxidized to SL by an NAD(P)+-dependent SLA dehydrogenase. All five enzymes are encoded in a single gene cluster that includes, for example, genes for transport and regulation. Homologous gene clusters were found in genomes of other P. putida strains, in other gamma-Proteobacteria, and in beta- and alpha-Proteobacteria, for example, in genomes of Enterobacteria, Vibrio, and Halomonas species, and in typical soil bacteria, such as Burkholderia, Herbaspirillum, and Rhizobium.
Sulfoquinovose (SQ; 6-deoxy-6-sulfoglucose) is the polar head group of the plant sulfolipids sulfoquinovosyl diacylglycerols (SQDGs), which were discovered more than 60 y ago (1). The sulfolipids are located in the photosynthetic (thylakoid) membranes of all higher plants, mosses, ferns, and algae, as well as in most photosynthetic bacteria. They are also present in some nonphotosynthetic bacteria, and SQ is in the surface layer of some archaea (2–4). SQ is continuously degraded in all environments where SQ is produced, or it would enrich in these environments. The complete degradation of SQ to, ultimately, CO2, concomitant with a recycling of the bound sulfur in the form of inorganic sulfate, is dominated by microbes (e.g., by soil bacteria) (2, 5–9). However, a more detailed exploration of SQ-degrading microbes, of their SQ-degradation pathways, and of the enzymes and genes involved has only recently been initiated (10–12). The common view on SQ degradation is based on the consideration that SQ is a structural analog of glucose-6-phosphate (GP) (10) and, hence, that SQ degradation may proceed in analogy to the Embden–Meyerhof (glycolysis) pathway, or by analogy to the Entner–Doudoroff pathway or the pentose–phosphate (phosphoketolase) pathway.
We recently discovered that Enterobacterium Escherichia coli K-12 is able to use SQ for growth (12). E. coli K-12 catalyzes a glycolytic breakdown of SQ, that is, “sulfoglycolysis” in addition to “normal” glycolysis. Sulfoglycolysis (EcoCyc pathway PWY-7446) involves three newly discovered intermediates, 6-deoxy-6-sulfofructose (SF), 6-deoxy-6-sulfofructose-1-phosphate (SFP), and 3-sulfolactaldehyde (SLA), and it is catalyzed by four newly discovered enzymes, SQ isomerase, SF kinase, SFP aldolase, and SLA reductase (12). The reactions yield dihydroxyacetone phosphate (DHAP), which supports energy conservation and growth of E. coli, and the C3-organosulfonate SLA as an analog of glyceraldehyde-3-phosphate (GAP). The SLA is reduced to 2,3-dihydroxypropane sulfonate (DHPS) by the SLA reductase, and the DHPS is excreted (12). Hence, E. coli K-12 can use only half of the carbon in SQ and is not able to catalyze a desulfonation reaction, whereas other bacteria can degrade DHPS [or the 3-sulfolactate (SL) formed from SQ, as discussed below] completely, including desulfonation and release of sulfate, via defined pathways (MetaCyc pathway PWY-6616) (13–15). Thus, the sulfur cycle for SQ can be closed within bacterial communities (11, 12 and cf. 5, 7).
Several Pseudomonas strains that can use SQ for growth have been isolated from soil (8, 9), and the excretion of another C3-organosulfonate, SL, instead of DHPS or sulfate, has been reported (9, 10). We also isolated an SQ-utilizing member of the genus Pseudomonas, Pseudomonas putida strain SQ1, from a freshwater sediment (Lake Constance, Germany). This strain also excretes stoichiometric amounts of SL during growth with SQ, but not DHPS or sulfate (11). In addition, we found high activity of an NAD+-dependent SQ dehydrogenase in cell-free extracts of P. putida SQ1 cells (12). Thus, we had experimental access to a second catabolic pathway for SQ, in addition to sulfoglycolysis in E. coli K-12, apparently via an initial oxidation of SQ this time.
Most pseudomonads use glucose exclusively via the Entner–Doudoroff pathway enzymes and do not encode fructose-6-phosphate kinase for glycolytic breakdown of glucose (e.g., the well-studied isolate of P. putida, strain KT2440) (16–20). Its major cytosolic pathway for glucose (Fig. 1A) is relevant as an analogy to a possible SQ degradation pathway: Glucose is taken up into the cell and phosphorylated to GP, the analog of SQ (compare Fig. 1 A and B). Then, GP is oxidized by an NAD(P)+-dependent GP dehydrogenase to 6-phosphogluconolactone (PGL), and the PGL is hydrolyzed to 6-phosphogluconate (PG), the PG is dehydrated to 2-keto-3-deoxy-6-phosphogluconate (KDPG), and the KDPG is cleaved into pyruvate and GAP (Fig. 1A). The five enzymes [i.e., glucose kinase (Glk), GP dehydrogenase (Zwf), PGL lactonase (Pgl), PG dehydratase (Edd), and KDPG aldolase (Eda)] are encoded in a single gene cluster (not shown; refer to ref. 20), which is highly conserved in P. putida species, and also in P. putida strain SQ1 (21). Even though it is conceivable that the classical Entner–Doudoroff pathway enzymes for GP might also catalyze the analogous reactions for the substrate SQ (Fig. 1B), the well-studied P. putida strains KT2440 and F1, for example, are unable to grow with SQ, in contrast to strain SQ1 (11).
Fig. 1.

Core enzyme reactions of the classical Entner–Doudoroff pathway for GP in Pseudomonas species in comparison to the newly discovered Entner–Doudoroff-type pathway for SQ, with its corresponding genes in Pseudomonas putida SQ1. (A) The cytosolic part of the Entner–Doudoroff pathway for glucose (19) is an alternative to classical glycolysis and generates two molecules of pyruvate from GP via PGL, PG, and KDPG, which are cleaved to pyruvate and GAP; the GAP is converted to pyruvate, as indicated. (B) Entner–Doudoroff pathway for SQ, which is a structural analog of GP, involves analogous enzyme reactions and the intermediates SGL, SG, and KDSG, which are cleaved to pyruvate and SLA, as demonstrated in this study. The pyruvate is used for growth, and the SLA is oxidized to SL, which is excreted. (C) Identified genes for the five core enzymes of the Entner–Doudoroff pathway for SQ (color-coded in B) are encoded in one gene cluster in P. putida SQ1, together with predicted genes for transport, for regulation, and presumably for funneling of other SQ derivatives into the pathway (main text). Note that the gene identifiers in this figure refer to IMG locus tags in the IMG annotation of the draft genome sequence of P. putida SQ1 (IMG Project ID Gp0039102) and that gene functions are specified according to their original IMG annotation in this figure.
For this study, we had SQ available as a substrate through chemical synthesis (11) and the analytical chemistry established for detection of SQ, its sulfosugar and C3-intermediates, and thus its enzyme activities (12), and we established a draft-genome sequence of strain SQ1 for proteomics (21), which set the stage for an exploration of its SQ degradation pathway. Here, we demonstrate that P. putida SQ1 uses SQ via what we term the “Sulfo–Entner–Doudoroff” pathway, in addition to the classical Entner–Doudoroff pathway for glucose, via five newly discovered enzymes and genes, and via three newly identified organosulfur intermediates.
Results
Inducible NAD+-Dependent SQ Dehydrogenase Activity and Conversion of SQ to SL in Cell-Free Extracts of SQ-Grown Cells.
The NAD+-dependent SQ dehydrogenase activity was inducibly and highly produced during growth with SQ. The specific activity determined in cell-free extracts (soluble protein fraction) of SQ-grown cells (633 ± 18 mU/mg of protein) was more than 90-fold higher than the specific activity determined in extracts of glucose- or succinate-grown cells (each <7 mU/mg of protein), as determined in a photometrical assay by formation of the coproduct NADH (recorded at 365 nm). For comparison, GP caused similar high NAD+-dependent dehydrogenase activity in extracts of glucose-grown cells (576 ± 19 mU/mg of protein), and this activity was 12-fold decreased in extracts of SQ-grown cells (45 ± 1 mU/mg of protein) and 25-fold decreased in succinate-grown cells (24 ± 7 mU/mg of protein). When NADP+ was used as an electron acceptor, the SQ dehydrogenase activity was only 4% of the SQ dehydrogenase activity with NAD+, whereas the GP dehydrogenase activity was 60% of the GP dehydrogenase activity with NAD+. We concluded that an inducible SQ dehydrogenase in SQ-grown cells had a strong preference for NAD+ and was poor at catalyzing a reaction with GP as a substrate, and that, vice versa, the NAD(P)+-dependent GP dehydrogenase in glucose-grown cells was poor at catalyzing a reaction with SQ.
The SQ dehydrogenase reaction in soluble protein fractions of SQ-grown cells was followed by HPLC-MS analysis, when samples were taken at intervals from reactions with SQ and NAD+ in excess (Fig. 2). The chromatograms confirmed a complete disappearance of SQ during the reaction. In addition, formation of four sulfonated intermediates was detected, as indicated by four novel peaks with different retention times, molecular masses, and MS/MS fragmentation patterns, which, however, all exhibited fragment ions that are characteristic of organosulfonates (diagnostic ions from a loss of the sulfonate group, m/z = 81; Figs. S1 and S2). One of the novel peaks represented SL, as identified by an identical retention time, molecular mass, and MS/MS fragmentation in comparison to authentic SL standard. The three other peaks exhibited molecular masses and MS/MS fragmentation patterns (Fig. S1) that corresponded to 6-deoxy-6-sulfogluconolactone (SGL), 6-deoxy-6-sulfogluconate (SG), and 2-keto-3,6-dideoxy-6-sulfogluconate (KDSG), respectively, as transient intermediates (structures are shown in Fig. 1B). In comparison, in reactions under the same conditions with extracts of glucose-grown cells, only a minor disappearance of SQ, concomitant with formation of trace amounts of SGL and SG, but no formation of KDSG and SL, was detectable by HPLC-MS. These observations suggested that all enzymes for oxidation of SQ to SGL, followed by lactone hydrolysis of SGL to SG, dehydration of SG to KDSG, and cleavage of KDSG to the C3-intermediate SL (Fig. 1B), were active in extracts of SQ-grown cells. Further, these enzymes appeared to be inducibly produced.
Fig. 2.

Complete disappearance of SQ in cell-free extract of SQ-grown P. putida SQ1 cells concomitant with a transient formation of metabolites SGL, SG, and KDSG, and formation of the end-product SL. The reaction in soluble protein extract (soluble protein fraction) was started by addition of NAD+ (not shown) and followed in samples that were taken at intervals and analyzed by HPLC-MS; the time points of sampling are indicated. The total-ion chromatograms (TICs) recorded in the negative-ion mode from the MS/MS fragmentation of the quasi-molecular ions ([M-H]−) of SQ, SGL, SG, and KDSG, as well as SL, are shown. Note that SGL and KDSG were each observed as [M-H]− ions of identical mass (m/z = 241) but that the compounds eluted at different HPLC retention times, as indicated for t = 5 min. Discussion of the MS/MS fragmentation of the metabolites SGL, SG, and KDSG, as well as SL, is provided in SI Materials and Methods.
Fig. S1.

MS/MS fragmentation of SGL, SG, and KDSG. (A) Fragment ions of the [M-H]− ions of SGL. Fragmentation led to a loss of water (−18) and carbon dioxide (−44), and to the formation of HSO3− (81) ions. (B) Fragment ions of the [M-H]− ions of SG. Fragmentation led to a loss of water (−18), CH2O2 (−46), C2H4O3 (−76), and SO3− (−80), concomitant with the formation of HSO3− (81) ions. (C) Fragment ions of the [M-H]− ions of KDSG. Fragmentation led mainly to a loss of water (−18) and carbon dioxide (−44), and to the formation of HSO3− (81) ions.
Fig. S2.

MS/MS fragmentation of SLA and SL. (A) Fragment ions of the [M-H]− ions of SLA. Fragmentation led to a loss of water (−18) and to the formation HSO3− (81) and C3H3O2− (71) ions. (B) Fragment ions of the [M-H]− ions of SL. Fragmentation led to a loss of water (−18) and to the formation HSO3− (81) ions.
Identification of a Gene Cluster for SQ Degradation in P. putida SQ1.
We compared the proteome of P. putida SQ1 during growth with SQ or glucose by 2D-PAGE, and all prominent SQ-specific protein spots were identified by peptide fingerprinting (PF)-MS (Fig. 3A). In addition, non–gel-based, total-proteome analyses of SQ- and glucose-grown cells were performed (Fig. 3B). As described in the following section, the proteomic data strongly suggested that one gene cluster (shown in Fig. 1C) is involved in SQ-degradation in P. putida SQ1. Interestingly, such a gene cluster is not encoded in the genomes of P. putida strains KT2440, F1, and W619, which cannot grow with SQ (12, 21).
Fig. 3.

Results of proteomic and transcriptional analyses indicating the involvement of a single SQ-degradation gene cluster in P. putida SQ1. A schematic representation of the gene cluster is illustrated in Fig. 1C. (A) Soluble proteins in SQ- and glucose-grown cells were separated by 2D-PAGE, and all prominent protein spots on the gels from SQ-grown cells that suggested inducibly produced proteins were identified by peptide PF-MS. The genes identified are indicated next to each protein spot (i.e., their locus tag number in the IMG annotation), with all genes located within the gene cluster labeled in red (0088–0090 and 0100); labeled in black are two prominent spots that identified genes considered unlikely to be involved in SQ degradation directly (0897, a translation elongation factor gene, and 4549, a gene for an ATP-binding cassette (ABC)-transport periplasmic binding protein). (B) Extract of the results of the total proteome (Orbitrap-MS) analyses of SQ-grown cells (blue bars) and glucose-grown cells (gray bars), illustrating the detected strong expression of proteins encoded within the newly identified gene cluster specifically during growth with SQ (0088–0091, 0094, and 0100), but not during growth with glucose. For comparison, the strong expression of enzymes of the Entner–Doudoroff pathway detected specifically during growth with glucose, but not during growth with SQ, is shown (Edd, Glk, Zwf-1, and Eda), as well as the expression level of enzymes for a further conversion of GAP and pyruvate (Gap and AceE) and of two constitutively produced proteins (SucC and AtpD) for each growth condition. (C) Differential transcriptional analysis (RT-PCR) of all genes encoded within the newly identified gene cluster (Fig. 1C), which indicated their strong and inducible transcription specifically during growth with SQ, but not during growth with glucose. A constitutively expressed gene (3160, for citrate synthase) served as a positive control, and the negative control was a PCR assay without RT (RNA) to confirm the absence of DNA contamination in the RNA preparations used. Each result in A–C was replicated with material from an independent growth experiment.
Four of the most prominent protein spots found exclusively on the 2D gels of SQ-grown cells (Fig. 3A) identified four predicted genes in the same gene cluster (on the same contig) in the draft genome sequence of strain SQ1: [Integrated Microbial Genomes (IMG) locus tags] PpSQ1_00088, 00090, 00100, and 00094 (in the following sections, the IMG locus tag prefix PpSQ1_0 is omitted). The total proteome analysis confirmed and expanded on these results, in that two other loci in the gene cluster were also identified by proteins that also appeared to be highly abundant specifically in SQ-grown cells. These additional loci were 0089 and 0091 (Fig. 3B). In contrast, strong expression of the well-known enzyme homologs for the Entner–Doudoroff pathway for glucose (well known in other Pseudomonas species) was detected for glucose-grown cells, but not for SQ-grown cells, of P. putida SQ1 (Fig. 3B).
The newly identified gene cluster (Fig. 1C) encodes five candidate enzymes for the proposed Entner–Doudoroff pathway for SQ (Fig. 1B), and all five candidates were detected in the proteomic approach, specifically in SQ-grown cells (Fig. 3 A and B): two candidates for NAD(P)+-dependent SQ or SLA dehydrogenases, respectively; that is, 0090, a predicted short-chain alcohol dehydrogenase, and 0088, a predicted succinate semialdehyde dehydrogenase. Further, one candidate each (Figs. 1C and 3 A and B) for a lactonase (0091, predicted gluconolactonase), dehydratase (0089, predicted phosphogluconate dehydrogenase), and aldolase (0100, predicted 2,4-dihydroxyhept-2-ene-1,7-dioic acid aldolase) was identified. The other predicted genes in the cluster (Fig. 1C) might encode for transport (uptake of SQ and export of SL), regulation, and funneling of other SQ metabolites into the pathway (Discussion).
Transcriptional analyses (RT-PCR) (Fig. 3C) of the whole 13-gene cluster (Fig. 1C) confirmed that all of these genes were inducibly, and strongly, transcribed during growth with SQ, but not during growth with glucose. In addition, we constructed an insertion mutant via homologous recombination in gene 0090 (for SQ dehydrogenase, as discussed below) in P. putida SQ1, and the mutant strain did grow with glucose but had lost the ability to grow with SQ (Fig. S3).
Fig. S3.

Growth experiment with P. putida SQ1 WT (A) and its insertion mutant (B) in gene 0090 (SQ dehydrogenase). Duplicate growth experiments are shown with glucose (open symbols) and SQ (solid symbols) as growth substrate in liquid cultures (6 mM each). Growth was monitored as optical density (OD580).
Recombinant Expression of the SQ and SLA Dehydrogenase Candidates and Activities of the Purified Enzymes.
The two identified dehydrogenase candidates of the SQ-gene cluster, 0090 and 0088, were cloned and heterologously overexpressed in E. coli, and the proteins were purified (SI Materials and Methods and Fig. S4); for comparison, the GP dehydrogenase of strain SQ1 (3570, Zwf-1; Fig. 3B) was also produced recombinantly and purified (Fig. S4). Only one dehydrogenase candidate, the predicted short-chain alcohol-dehydrogenase 0090, catalyzed an NAD+-dependent reaction with SQ, and this reaction produced SGL from SQ (as discussed below). In the photometrical assay, recombinant SQ dehydrogenase 0090 exhibited a much higher catalytic efficiency (kcat/Km) with NAD+ than with NADP+ as an electron acceptor (Table 1). Further, SQ dehydrogenase 0090 exhibited no activity with GP as a substrate and with both cosubstrates tested, NAD+ and NADP+, under the same assay conditions. For comparison, the recombinant GP dehydrogenase 3570 (Zwf-1) exhibited similar high catalytic efficiency with GP and NADP+, as well as NAD+, as a cosubstrate (Table 1), but no significant activity was detectable with SQ as a substrate and both cosubstrates tested; notably, the additional presence of SQ did not inhibit its activity with GP. The pH optimum for the SQ dehydrogenase reaction was between 8 and 9. Thus, we demonstrated that gene 0090 of the SQ-gene cluster encodes for an NAD+-dependent SQ dehydrogenase, which is poor at catalyzing a reaction with GP. In addition, we confirmed that NAD(P)+-dependent GP dehydrogenase 3570 of the Entner–Doudoroff pathway is poor at catalyzing a reaction with SQ.
Fig. S4.
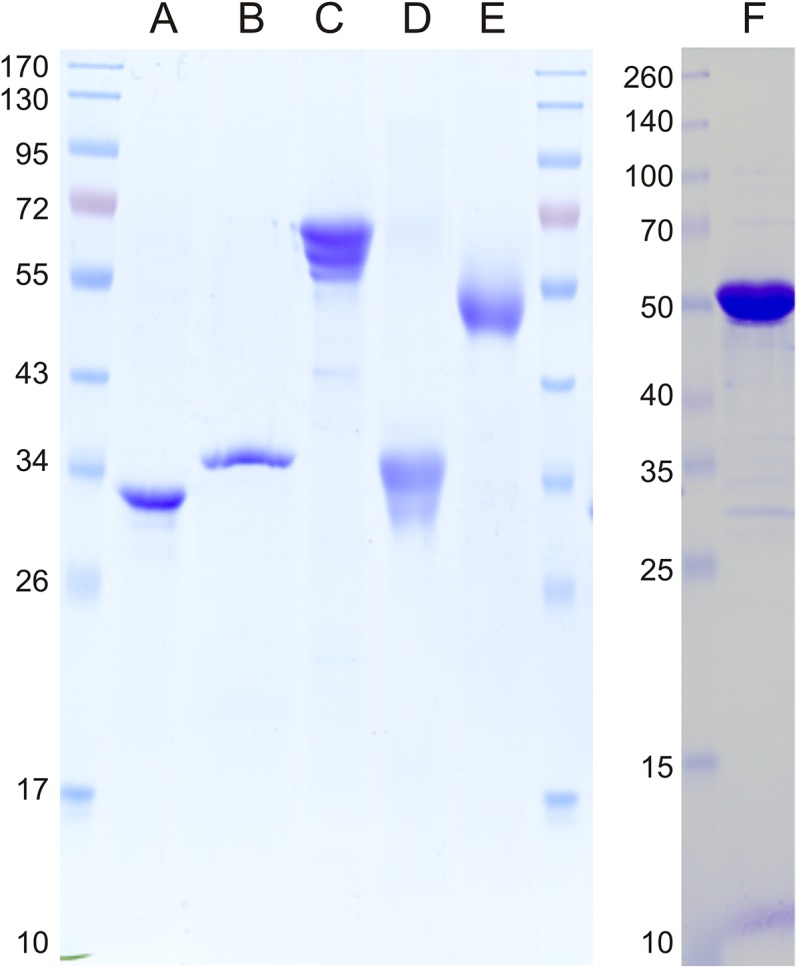
Analysis of the purity of heterologously overproduced enzymes using SDS/PAGE. Marker proteins are indicated (kilodaltons). Lane A, SQ dehydrogenase (PpSQ1_00090); lane B, SGL lactonase (PpSQ1_00091); lane C, SG dehydratase (PpSQ1_00089); lane D, KDSG aldolase (PpSQ1_00100); lane E, SLA dehydrogenase (PpSQ1_00088); and lane F, GP dehydrogenase (Zwf-1, PpSQ1_03570). The protein bands with a lower molecular mass than expected in lane C represent C-terminally truncated versions of the recombinant His-tagged protein, as determined by PF-MS.
Table 1.
Kinetic parameters determined for recombinant dehydrogenases from P. putida SQ1
| Enzyme | Substrate | Km, mM | Vmax, μmol⋅min−1⋅mg−1 | kcat, s−1 | kcat/Km, 103 s−1⋅M−1 |
| SQ dehydrogenase (PpSQ1_00090) | SQ with NAD+ | 0.5 ± 0.1 | 62.8 ± 2.0 | 33.8 | 67.6 |
| SQ with NADP+ | 2.4 ± 0.1 | 2.7 ± 0.1 | 1.4 | 0.6 | |
| GP dehydrogenase (Zwf-1) (PpSQ1_03570) | GP with NAD+ | 1.3 ± 0.6 | 21.4 ± 3.1 | 21.1 | 16.2 |
| GP with NADP+ | 0.6 ± 0.3 | 12.8 ± 1.8 | 12.6 | 21.0 | |
| SLA dehydrogenase (PpSQ1_00088) | SSA* with NAD+ | 0.14 ± 0.02 | 12.6 ± 0.4 | 11.8 | 84.2 |
| SSA* with NADP+ | 0.14 ± 0.02 | 12.2 ± 0.4 | 11.4 | 81.4 |
SSA, succinate semialdehyde.
There was no SLA available as a substrate in the defined concentration (main text).
The second identified dehydrogenase of the SQ gene cluster, the annotated succinate semialdehyde dehydrogenase 0088 (Fig. 1C), turned out to catalyze the NAD(P)+-dependent oxidation of SLA to SL, which was thus the last reaction of the proposed pathway (Fig. 1B): The enzyme exhibited high activity with SLA and both NAD+ and NADP+, but no activity with SQ (or GP) in the photometrical assay, and the product of the SLA reaction was SL, as confirmed by HPLC-MS (as discussed below). Notably, SLA as a substrate had to be generated enzymatically, either through a reaction of recombinant SLA reductase (YihU) of E. coli K-12 in reverse with DHPS and NAD+ (12) or directly from SQ by an in vitro reconstitution of the complete SQ pathway (as discussed below); therefore, we could not determine the kinetic parameters for the SLA dehydrogenase reaction. The enzyme did not catalyze a reverse reaction with SL and NADH when tested, which is a common observation for aldehyde dehydrogenases. Further, the enzyme exhibited high activity with succinate semialdehyde, with both NADP+ and NAD+ (Table 1).
In Vitro Reconstitution of an Entner–Doudoroff Pathway for SQ.
The identified candidate lactonase (0091), dehydratase (0089), and aldolase (0100) genes for SQ degradation were also heterologously expressed in E. coli, and the proteins were purified (Fig. S4). The five proteins were added sequentially to a reaction mixture with SQ, and after each reaction step, samples were taken for HPLC-MS analysis, as illustrated in Fig. 4.
Fig. 4.

In vitro reconstitution of the Entner–Doudoroff pathway for SQ. The transformation of SQ to SGL, SG, KDSG, and SLA, and of SLA to SL, by successive addition of recombinantly produced pathway enzymes was followed by HPLC-MS. The initial substrate concentrations were 2 mM SQ and 3 mM NAD+, and 50 μg⋅mL−1 of each enzyme was added. (A) Sample of SQ in reaction buffer (t = 0 min). (B) Sample taken 45 min after addition of SQ-dehydrogenase (gene PpSQ1_00090) (t = 45 min). (C) Sample taken 45 min after addition of SGL-lactonase (gene 0091) (t = 90 min). (D) Sample taken 45 min after addition of SG-dehydratase (gene 0089) (t = 135 min). (E) Sample taken 45 min after addition of KDSG-aldolase (gene 0100) (t = 180 min). (F) Sample taken 45 min after addition of SLA-dehydrogenase (gene 0088) and 2 mM NAD+ (t = 225 min). The TICs recorded in the negative-ion mode from the MS/MS fragmentation of the quasi-molecular ions ([M-H]−) of SQ, SG, SGL, and KDSG, as well as SLA and SL, are shown; note that SGL and KDSG were each observed as [M-H]− ions of identical mass (m/z = 241) but that the compounds eluted at different HPLC retention times, as indicated in the duplicated panels for TIC MS2 of m/z = 241. Pyruvate, as the second product of the KDSG-aldolase reaction (Fig. 1), could not be detected under the HPLC-MS conditions we used but was confirmed by other analytical methods.
The SQ dehydrogenase 0090 produced SGL from SQ (Fig. 4 A and B), as indicated by the formation of a peak with a matching mass of the molecular ([M-H]−) ion and MS/MS fragmentation (Fig. S1A); the same peak was observed during the reactions with cell-free extracts of SQ-grown P. putida cells (Fig. 2). A second minor peak that also appeared represented SG, as identified by a matching mass of the [M-H]− ion and MS/MS fragmentation (Fig. S1B); hence, the lactone hydrolyzed spontaneously to SG. After addition of the predicted SGL lactonase 0091, the peak for SGL had disappeared completely and the peak for SG had further increased (Fig. 4 B and C). Thus, we confirmed that 0091 catalyzes a conversion of SGL to SG (Fig. 1C). Next, the predicted SG dehydratase 0089 was added, which resulted in an intermediate (Fig. 4 C and D) that was identified as KDSG by a peak with a different retention time but identical molecular mass as SGL (compare Figs. 2 and 4), as well as matching MS/MS fragmentation that was different from the MS/MS fragmentation of SGL (compare Fig. S1 A and C); the same peak was observed during the reactions with cell-free extracts of SQ-grown P. putida cells (Fig. 2). Correspondingly, the peak for SG had decreased (Fig. 4 C and D). Thus, 0089 is a dehydratase that converts SG to KDSG (Fig. 1C). After addition of the predicted aldolase 0100, the KDSG peak had decreased and SLA had formed (Fig. 4 D and E), as identified by comparison with SLA standard that had been generated enzymatically from DHPS (as discussed above). In addition, the existence of pyruvate after the aldolase reaction was confirmed through a positive lactate dehydrogenase reaction and by HPLC cochromatography with authentic pyruvate standard. Thus, we demonstrated that protein 0100 is an aldolase that cleaves KDSG to SLA and pyruvate (Fig. 1C). Finally, with the addition of the last enzyme of the SQ pathway, SLA dehydrogenase 0088, the SLA peak had disappeared completely and SL had formed (Fig. 4 E and F).
We also tested SG dehydratase 0089 and KDSG aldolase 0100 under the same reaction conditions for their potential activity with the analogous substrates of the classical Entner–Doudoroff pathway, PG and KDPG (Fig. 1A), respectively, which are commercially available. No substrate turnover and product formation could be detected in the HPLC-MS assay (not shown); hence, these enzymes of the SQ pathway in P. putida SQ1 also appeared to be highly specific for the sulfonated substrate, as is SQ dehydrogenase.
SI Materials and Methods
Chemicals.
SQ and SL were synthesized chemically and identified by NMR and MS as described elsewhere (11, 15). GP, PG, KDPG, and GAP were from Sigma–Aldrich. Biochemicals (NAD+, NADP+, NADH, and NADPH) were purchased from Biomol. All other chemicals were of the highest purity available from Sigma–Aldrich, Fluka, Roth, or Merck.
Growth Conditions.
P. putida SQ1 (DSM 100120) (11) was grown in phosphate-buffered, mineral salts medium (pH 7.2) (33) and supplemented with SQ (6 or 12 mM), glucose (6 mM), or succinate (9 mM) as the sole carbon and energy source. Cultures on the 2-mL and 5-mL scales were incubated in glass tubes (Corning) in a roller, and cultures on the 30-mL and 200-mL scales were incubated in capped Erlenmeyer flasks on a shaker, each at 30 °C in the dark. The cultures were inoculated (1%) with precultures grown with the same substrate. For cultures in Erlenmeyer flasks, 0.8-mL samples were taken at intervals to monitor growth at OD580.
Preparation of Cell-Free Extracts.
P. putida cells from cultures on the 200-mL scale were harvested at an OD580 of about 0.5 by centrifugation (20,000 × g, 15 min, 4 °C) and were disrupted by three passages through a chilled French pressure cell (140 MPa; Aminco) in the presence of DNase (25 μg⋅mL−1). Whole cells and cell debris were removed by centrifugation (11,000 × g, 10 min, 4 °C), which yielded crude extract, and the membrane fragments were collected by ultracentrifugation (100,000 × g, 60 min, 4 °C); this supernatant was called soluble protein fraction.
Proteomics.
A draft genome sequence of P. putida SQ1 for proteomics was established under contract by GATC Biotech using the Illumina HiSeq2000 platform and a 100-bp paired-end library, which resulted in 23,816,201 reads (1.85 × 109 total bases); trimming, mapping, and de novo assembly of the unmapped raw reads were performed at the Genomics Center of the University of Konstanz (21). The sequences were annotated within the Joint Genome Institute’s analysis system (34), and the annotation (http://img.jgi.doe.gov; IMG Project ID Gp0039102) was used to build a local Mascot database (35) for PF-MS. For 2D isoelectric focusing (IEF)-SDS/PAGE (2D-PAGE) of the soluble protein fraction of P. putida cells, the Bio-Rad ReadyStrip immobilized pH gradient (IPG) system was used for the first-dimension separation (17-cm length; pI range, pH 4–7) and 12% SDS/PAGE gels (17 × 20 cm; no stacking gel) were used for the second-dimension separation; sample preparation and IEF separation conditions were essentially as described in the manufacturer’s instructions (Bio-Rad Ready-Strip IPG strip instruction manual), with the modifications described previously (36). Stained protein spots were cut from the gel and subjected to PF-MS at the Proteomics Facility of the University of Konstanz (www.proteomics-facility.uni-konstanz.de); the parameters for mass spectral analysis and Mascot database searching and scoring were set as previously described (36). For total proteomics of the soluble protein fraction, the sample preparation, gel purification, tryptic digest, high-resolution PF-MS (Orbitrap-MS) analysis, and Mascot database searching were performed according to our previously published protocol (38).
Transcriptional Analysis.
RNA preparation and RT-PCR assays were carried out according to our previously published protocol (37). Briefly, cells were grown in the appropriate selective medium on a 30-mL scale and harvested in the mid-exponential growth phase (OD580 ∼ 0.28) by centrifugation (15,000 × g, 15 min, 4 °C). The cell pellets were stored at −20 °C in RNAlater RNA stabilization solution (Applied Biosystems). Total RNA was prepared using the E.Z.N.A. Bacterial RNA Kit (Omega Bio-Tek) following the manufacturer’s instructions; the RNA preparations were treated with RNase-free DNase (2 units, 30 min, 37 °C; Fermentas). For complementary cDNA synthesis, Maxima reverse transcriptase (Fermentas) was used. For PCR reactions, Taq polymerase (Fermentas) was used with cDNA from RT reactions as a template (2 μL of RT reaction in 20 μL of PCR mixture). The primer pairs used (Microsynth) are listed in Table S1.
Table S1.
Primer pairs used for RT-PCR
| Primer name | Sequence (5′–3′) | Product length, bp |
| 0088_FOR | AGCCGACCGTGCTCACTGAAGTTA | 215 |
| 0088_REV* | GACGCCGACCATGCCGTATTC | |
| 0089_FOR | CACGGCGCTGATGGAGGACTA | 262 |
| 0089_REV* | CTCGGCCGCGGAGGATTT | |
| 0090_FOR | AAGGCCTGCGTGGCAGAAGTG | 269 |
| 0090_REV* | GCTTTGGCGGTGGTGTAGCAGA | |
| 0091_FOR | GGCGGGCGATGTGATGGTATC | 288 |
| 0091_REV* | CGCGTCGGCAGCTGGATTT | |
| 0092_FOR | TGCCCGAGGCCTGGGACTT | 243 |
| 0092_REV* | CCGTGGGCTCAGGGAAGTTGAT | |
| 0093_FOR | GGGCGCCAACTACATGCATACTCA | 254 |
| 0093_REV* | GCGAGAAACCCTGGGTGGAGTG | |
| 0094_FOR | AACCGCCCGGACCAGAACCT | 224 |
| 0094_REV* | AGCAGATCGCGCCCATACAGGT | |
| 0095_FOR | ATGGCCGCCCGAAACCTG | 261 |
| 0095_REV* | CGTGGCCGTGCCATTGATG | |
| 0096_FOR | ACGGCGACGGGCCACAAG | 235 |
| 0096_REV* | AGGGTGCCCGCTTCAATCCA | |
| 0097_FOR | ACTGGCCAACGCGCTGGTTAT | 262 |
| 0097_REV* | CCCGGAAACACTGGCGAAAAA | |
| 0098_FOR | CGTCGCCCGGCAAGTGGT | 242 |
| 0098_REV* | GCTCGGCCAACGTCAGTAACGAC | |
| 0099_FOR | CTGCTCGGCGGCCTCTGTT | 204 |
| 0099_REV* | CATGACCGCCCAGCCTAGTGA | |
| 0100_FOR | CGTCAGCGCGGTCAAGTATCC | 247 |
| 0100_REV* | CGCCGGGTTGCCGATATG | |
| 3160_FOR | CTGGAAACCTGCTACCTGCTGCTAAA | 228 |
| 3160_REV* | TTCGCGGTGTTGCGGGTTATT |
FOR, forward; REV, reverse.
Primer used for RT reaction.
Construction of an Insertion Mutant.
For the generation of an insertion mutant in the SQ-dehydrogenase gene PpSQ1_00090, an internal 500-bp sequence of gene 0090 was amplified with Primer pKO_0090_F (5′-GGGCAAGGGCTGGC-3′) and pKO_0090_R (5′-TGAAGGCCTGGCTGGCC-3′) by PCR assay using Taq polymerase. The PCR fragment was cloned into the vector PCR2.1 using a Topo TA Cloning Kit (Invitrogen), and the correct sequence of the resulting plasmid PCR2.1[KO0090] was confirmed by sequencing (GATC Biotech). One feature of this plasmid is its restricted host range, which makes it suitable as a vector for insertional mutagenesis via homologous recombination in P. putida SQ1. For preparation of electrocompetent cells, strain SQ1 was grown in 100 mL of LB (OD600 = 0.8; shaking at 18 rpm, 30 °C). Cells were harvested and washed three times by centrifugation at 9.500 × g for 5 min and resuspended in 50 mL, 25 mL, and 10 mL of sucrose solution (300 mM), respectively. Finally, the cells were resuspended in 500 μL of sucrose solution (300 mM). Each 100-μL portion of electrocompetent cells was transferred into a 0.2-cm electroporation cuvette (Biorad); mixed with 10 μL (800 ng) of PCR2.1[KO0090] plasmid DNA; and electroporated using an Eppendorf Gene Pulser at 2.5 V, 200 Ω, and 25 μF. After recovery in 2 mL of LB in a 15-mL Falcon tube (shaking at 180 rpm, 30 °C), the insertion mutants were selected on LB-agar containing 20 μg⋅mL−1 kanamycin. After incubation for 24 h at 30 °C, colonies were restreaked onto fresh plates and the correct insertion of PCR2.1 into the target gene 0090 was confirmed by colony PCR assay using the primers 0089_REV (Table S1) and M13mod_FOR (5′-TTGTAAAACGACGGCCAGTGAAT-3′); these primers bind downstream of the target gene 0090 and in the PCR2.1 sequence, respectively. Growth of mutants was tested in 6 mM SQ- or glucose-minimal salts medium (as discussed above) supplemented with μg⋅mL−1 kanamycin.
Heterologous Overproduction and Purification of His-Tagged Proteins.
Heterologous expression of candidate genes and subsequent purification of the recombinant proteins were carried out according to our previously published protocol (39). In brief, chromosomal DNA of P. putida SQ1 was isolated (Illustra bacterial genomic Prep Mini Spin kit; GE Healthcare), the candidate genes were amplified by PCR using Phusion HF DNA Polymerase (Finnzymes), and the primer pairs are listed in Table S2. The PCR conditions (30 cycles) were 15 s of denaturation at 98 °C, 20 s of annealing at 58 °C, and 60 s of elongation at 72 °C for genes PpSQ1_00088 and 0090; 32 s of elongation at 72 °C for gene PpSQ1_00091; 20 s of annealing at 62 °C for gene PpSQ1_00089; and 20 s of annealing at 57 °C and 30 s of elongation at 72 °C for gene PpSQ1_00100. The PCR products were purified (QIAquick MinElute PCR Purification Kit, Qiagen) and ligated into an N-terminal His6-tagged expression vector (Champion pET 100 directional TOPO expression kit; Invitrogen). The constructs were transformed into OneShot TOP10 E. coli cells (Invitrogen) and confirmed by DNA sequencing (GATC Biotech). BL21 Star (DE3) OneShot E. coli cells (Invitrogen) were transformed with the constructs and grown at 37 °C in LB (100 mg/L ampicillin). Cultures were induced (0.5 mM isopropyl-β-d-thiogalactopyranoside) at an OD580 of 0.8 and grown for an additional 5 h at 20 °C. After harvesting by centrifugation (15,000 × g, 15 min, 4 °C), the cells were resuspended in 20 mM Tris⋅HCl (pH 8.0; buffer A) containing 0.03 mg/mL DNase I (Sigma) and then disrupted by four passages through a chilled French pressure cell (140 MPa; Aminco). Whole cells and debris were removed by centrifugation (15,000 × g, 15 min, 4 °C), and the membrane fragments were removed by ultracentrifugation (100,000 × g, 60 min, 4 °C). The soluble protein fractions were loaded onto a Ni2+-chelating Agarose affinity column (Macherey–Nagel). After a washing step (30 mM imidazole in buffer A), the His-tagged proteins were eluted (200 mM imidazole in buffer A), concentrated via a Vivaspin column (Sartorius), and stored at −20 °C in glycerol (30% vol/vol final concentration).
Table S2.
Primer pairs used for directional cloning
| PpSQ1_00088f* | CACCATGCTCGAACTCAAAGACCCTTCT |
| PpSQ1_00088r | GTATCGGACATGGGGCTACTCTATG |
| PpSQ1_00089f* | CACCATGTCCGAAAAGCACAAGAAA |
| PpSQ1_00089r | TCAGTGGATGGGTGGCTC |
| PpSQ1_00090f* | CACCATGAACCGTCATACCGATACGCATTAC |
| PpSQ1_00090r | CTGACGTCAATCAGGGTGGATTACTG |
| PpSQ1_00091f* | CACCATGAATGAAACGCTGAAGTGTGTGG |
| PpSQ1_00091r | GGTTCATGATAGGAAGCTCGTGAATG |
| PpSQ1_00100f* | CACCATGTCCACTGCTGTCACG |
| PpSQ1_00100r | TCAGTAGGCTGTCGCTAC |
| PpSQ1_03570f* | CACC ATGGCTGCGATCAGTGTTGAACCT |
| PpSQ1_03570r | TGATGCGCCTTGACCGATGTC |
Primer with adaptor sequence for directional cloning (underlined).
Enzyme Assays.
Recombinant purified SQ and GP dehydrogenase, as well as SQ and GP dehydrogenase activities in native cell-free extracts of P. putida SQ1 cells, were routinely assayed spectrophotometrically as the formation of the coproduct NADH/NADPH recorded at 365 nm for 1 min; the standard reaction mixture (1 mL) contained 50 mM Tris⋅HCl (pH 8.0), 1 mM SQ or GP, 2 mM NAD+ or NADP+, and 2 μg of recombinant protein or 50 μg of soluble protein fraction, respectively. The kinetic parameters for recombinant SQ and GP dehydrogenase were determined when the substrate concentrations were varied, and the activities were plotted using hyperbolic fit in Origin (Microcal Software, Inc.); the pH optimum was determined when the pH of the reaction buffer was varied (40). SLA as a substrate for the SLA dehydrogenase assay was prepared in reactions of recombinant SLA reductase of E. coli K-12 (YihU; 5 μg⋅mL−1) (12) with DHPS (1 mM) and NAD+ (4 mM) in reaction buffer [50 mM Tris⋅HCl (pH 8.0)]; the reverse reaction of the SLA reductase was monitored spectrophotometrically as formation of the coproduct NADH at 365 nm in the presence and absence of recombinant SLA dehydrogenase. Analysis of substrate disappearance and product formation by HPLC-MS (as discussed below) in reactions with cell-free extract of SQ- or glucose-grown P. putida SQ1 cells was done on the 0.3-mL scale in 50 mM Tris⋅HCl buffer (pH 7.3) at room temperature (∼20–23 °C). The reactions contained a soluble protein fraction (50 μg of protein per milliliter) and SQ or GP (1 mM), and were started by addition of NAD+ (3 mM); at intervals (t = 0, 5, 30, and 90 min), samples (66 μL) were taken, to which acetonitrile (33 μL) was added. Analysis of substrate disappearance and product formation by HPLC-MS in reactions with recombinant enzymes was carried out on a 1-mL scale in 50 mM Tris⋅HCl buffer (pH 7.5) stirred at room temperature. The recombinant enzymes (50 μg⋅mL−1 each) were added one after another to reaction mixtures that contained SQ (2 mM) and NAD+ (3 mM); after 45 min of reaction time, a sample (66 μL) was taken, to which acetonitrile (33 μL) was added, and the next enzyme was added until the reaction sequence had been completed in the following order: SQ dehydrogenase, lactonase, dehydratase, aldolase, and SLA dehydrogenase. Activity of recombinant SG dehydratases and KDSG aldolase with authentic PG and KDPG as substrates, respectively (2 mM), was tested by HPLC-MS/MS analysis under the same reaction and sampling conditions.
Analytical Methods.
For HPLC-electrospray ionization (ESI)-MS/MS, an Agilent 1100 HPLC system fitted with a ZIC-HILIC column (5 μm, 200 Å, 150 × 4.6 mm; Merck) was connected to an LCQ ion trap mass spectrometer (Thermo Fisher Scientific). The HPLC conditions were as follows: from 90% B to 65% B in 25 min, 65% for 10 min, back to 90% B in 0.5 min, and 90% B column equilibration for 9.5 min. Solvent A was 90% 0.1 M ammonium acetate and 10% acetonitrile, and solvent B was acetonitrile. The flow rate was set to 0.75 mL⋅min−1. The mass spectrometer was run in ESI-negative mode. The retention times and ESI-MS/MS fragmentation patterns of SQ and SLA were the same as described and shown previously (12). The retention times and ESI-MS/MS fragmentation patterns of the other analytes were observed as follows: SGL retention time, 20.4 min; SGL ESI-MS m/z (% base-peak) 241 (100); SGL ESI-MS/MS of [M-H]− 241: 241 (26), 223 (4), 197 (99), 179 (100), 161 (30), 167 (6), 153 (8), 137 (2), 97 (14), 83 (1), 81 (6); SG retention time, 28.1 min; SG ESI-MS m/z (% base-peak) 259 (100); SG ESI-MS-MS of [M-H]− 259: 259 (8), 241 (100), 213 (8), 197 (29), 183 (27), 179 (21), 161 (6), 153 (1), 137 (1), 123 (1), 115 (1), 97 (3), 81 (1); KDSG retention time, 25.6 min; KDSG ESI-MS m/z (% base-peak) 241 (100); KDSG ESI-MS/MS of [M-H]− 241: 241 (4), 223 (100), 205 (24), 197 (1), 179 (2), 161 (3), 81 (1); SL retention time, 27.0 min; SL ESI-MS m/z (% base-peak) 169 (100); SL ESI-MS/MS of [M-H]− 169: 169 (100), 151 (53), 141 (1), 103 (9), 97 (2), 81 (1), 71 (3). Pyruvate was analyzed by HPLC with a refraction index detector (RID-10A; Shimadzu) as described previously (41); briefly, an Aminex HPX-87H ion-exchange column (BioRad) at 60 °C was used, and the eluent was 5 mM H2SO4 at a flow rate of 0.6 mL⋅min−1; pyruvate eluted at 9.3 min.
Enzyme Nomenclature.
We suggest that SQ dehydrogenase belongs to the NC-IUBMB (Nomenclature Commission of the International Union of Biochemistry and Molecular Biology) subgroup EC 1.1.1, with the name sulfoquinovose dehydrogenase [NAD+] (systematic name sulfoquinovose:NAD+ 1-oxidoreductase). The SLA dehydrogenase would then belong to subgroup EC 1.2.1, with the name sulfolactaldehyde dehydrogenase [NAD(P)+] (systematic name 3-sulfolactaldehyde: NAD(P)+ oxidoreductase). The SGL lactonase would belong to EC 3.1.1, with the name sulfogluconolactonase (systematic name 6-deoxy-6-sulfoglucono-1,5-lactone lactonohydrolase). The SG dehydratase would belong to EC 4.2.1, with the name sulfogluconate dehydratase (systematic name 6-deoxy-6-sulfogluconate hydro-lyase [2-dehydro-3,6-dideoxy-6-sulfogluconate–forming]). The KDSG aldolase would belong to EC 4.1.2, with the name 2-dehydro-3,6-dideoxy-6-sulfogluconate aldolase [systematic name 2-dehydro-3,6-dideoxy-6-sulfogluconate 3-sulfolactaldehyde-lyase (pyruvate-forming)].
Discussion
SQ is one of the most abundant organosulfur compounds in the biosphere, following glutathione, Cys, and Met, and the global production of SQ is estimated at 10 gigatons per year (2, 22). Although the biosynthesis of SQ and SQDG in plants, algae, cyanobacteria, and archaea has been studied in considerable detail (e.g., 23, 24), much less is known about the degradation reactions and the recycling of the carbon and sulfur bound in SQ and SQDG. The lipid can easily be hydrolyzed by plant acyl-hydrolases, which liberate SQ-glycerol (1), and glucosidases can liberate SQ in the next step (25); however, it is still unclear whether higher plants are capable of splitting the carbon/sulfur bond in SQ at significant rates. Indeed, inorganic sulfate is the predominant source of sulfur for growth of plants and algae, for example, and plant growth can become sulfur/sulfate-limited, for example, in soils that are sulfur-deficient due to intensive agriculture and to the effective measures to reduce sulfur emissions to the atmosphere in recent years (26, 27); phytoplankton growth in freshwater environments can also become sulfate/sulfur-limited (28). The recycling of the sulfur bound in SQ is catalyzed by heterotrophic bacteria, which can easily be enriched from soil (5, 7–9), and involves the degradation intermediates DHPS and SL, but no release of sulfate, if SQ-utilizing bacteria are grown in pure culture (10–12). Later work demonstrated that SQ can be degraded completely by two-member bacterial communities, that is, by a primary degrader with transient release of SL or DHPS and a secondary degrader with release of sulfate (11, 12). Pathways for the degradation of DHPS and SL, including desulfonation, have been described (13, 15, 29) where, briefly, DHPS is oxidized to SL and the SL is mineralized via one of three known pathways and desulfonative enzymes [i.e., SL sulfolyase (SuyAB; EC 4.4.1.24), sulfoacetaldehyde acetyltransferase (Xsc; EC 2.3.3.15), cysteate sulfolyase (CuyA; EC 4.4.1.25)]. However, details on the pathways, enzymes, and genes for the conversion of SQ to DHPS and SL were missing. A first pathway, sulfoglycolysis leading to DHPS, was found in E. coli K-12, and this pathway seems to be widespread in Enterobacteria (12). A second pathway, leading to SL, in a typical soil and freshwater bacterium, an isolate of P. putida, has been revealed in this study.
Strain SQ1 is able to access half of the carbon atoms of SQ for its heterotrophic aerobic growth through abstraction of a nonsulfonated C3-unit in the form of pyruvate, whereas the C3-organosulfonate half of the substrate is not dissimilated but excreted in the form of SL (Fig. 1 A and B). Four organosulfonate intermediates were detected (SGL, SG, KDSG, and SLA), and five corresponding enzymes were identified [NAD+-dependent SQ dehydrogenase, SGL lactonase, SG dehydratase, KDSG aldolase, and NAD(P)+-dependent SLA dehydrogenase]. Hence, this primary degradation pathway for SQ in P. putida SQ1 follows a catabolic route that resembles an Entner–Doudoroff pathway for SQ (Fig. 1 A and B), as was obviously suspected previously (10).
It is striking that the enzymes that catalyze the analogous reactions in either the SQ or GP Entner–Doudoroff pathway in P. putida SQ1 are encoded and regulated in different gene clusters (operons), as is the case with sulfoglycolysis and classical glycolysis in E. coli K-12 (12). It is even more surprising that most of these enzymes belong to different enzyme families. Only SG dehydratase and PG dehydratase belong to the same protein family [dihydroxy-acid/6-phosphogluconate dehydratases; Cluster of Orthologous Groups (COG) 0129]. SQ dehydrogenase (0090) belongs to the NAD(P)+-dependent short-chain alcohol dehydrogenase family (COG1028), and thus to a different family than typical GP 1-dehydrogenases (COG0364). Further, the SGL lactonase (0091) is a sugar lactone hydrolase family protein (COG3386), whereas PGL lactonase is an archetypal glucosamine-6-phosphate isomerase/6-phosphogluconolactonase family protein (COG0363). The KDSG aldolase (0100) is a 2-keto-3-deoxyrhamnonate aldolase family protein (COG3836), and is most similar to 2,4-dihydroxyhept-2-ene-1,7-dioic acid/4-hydroxy-2-oxovalerate aldolases of aromatic-ring cleavage pathways, whereas the KDPG aldolase is an archetypal KDPG/4-hydroxy-2-oxoglutarate aldolase family protein (COG0800). These phylogenetic differences are reflected in the observed substrate specificities of the enzymes of the SQ pathway, as far as tested in this study. It remains unclear which factors are responsible for the enzymes’ specificity for 6-deoxy-6-sulfo- (monoanionic) over 6-phospho-substituted (dianionic) substrate analogs, and thus for their recruitment into the pathway, but these questions can now be addressed.
The newly identified SQ degradation gene cluster of P. putida SQ1 also contains other genes that are likely to be involved in SQ utilization. Gene 0097 is predicted to encode a sulfite/small organosulfonate exporter (TauE; PF01925) (30, 31), and this gene was cotranscribed during growth with SQ (Fig. 3C). Hence, gene 0097 most likely encodes for an inducible SL exporter. Gene 0094 is predicted to encode an alpha-glucosidase (COG1501; family 31 glycosyl hydrolase), and this gene was also cotranscribed (Fig. 3C) and highly coexpressed (Fig. 3 A and B), specifically during growth with SQ. Hence, we predict that gene 0094 encodes for hydrolysis of SQ-glyceride (e.g., for utilization of the whole sulfolipid SQDG as a growth substrate). Further, gene 0092 was cotranscribed (Fig. 3C) and is predicted to encode an aldose epimerase (COG2017), most likely for catalysis of the equilibrium of alpha- and beta-anomers of SQ; such function would imply that the SQ Entner–Doudoroff pathway is anomer-specific. Finally, SQ transport might be encoded by the cotranscribed (Fig. 3C) predicted sodium/solute symporter (SSS) family transporter gene (0094) or by the predicted galactonate major facilitator superfamily (MFS) transporter gene (0098); the outer membrane porin (OMP) gene (0093) may be involved in degradation of SQDG.
Thirty genome sequences of P. putida strains are available for genome-wide comparisons within the IMG search platform (as of April 2015), and in three of these genomes, syntenic SQ gene clusters were found, as illustrated in Fig. 5 (in P. putida strains H8234, OUS82, and SJ3). Putative SQ-degradation gene clusters with homologs for the core genes identified in P. putida SQ1 were found in other gamma-Proteobacteria, and in beta- and alpha-Proteobacteria, but as differently assembled (nonsyntenic) gene clusters, as illustrated in Fig. 5. For some of these clusters, a corresponding SLA dehydrogenase candidate gene (indicated in red in Fig. 5) was not found; however, the predicted SL exporter (TauE; gene 0097 in strain SQ1) and alpha-glucosidase gene (0094) appeared to be conserved in most of these candidate SQ-gene clusters (indicated in gray in Fig. 5). Such gene clusters were found in genomes of species of (gamma-Proteobacteria) Vibrio, Aeromonas, and Enterobacteria (Serratia, Plesiomonas, Hafnia, Leminorella, and Edwardsiella); of (beta-Proteobacteria) Burkholderia, Janthinobacterium, and Herbaspirillum; and of (alpha-Proteobacteria) Rhizobium, Ensifer, Microvirga, Salinarimonas, and Acidocella (Fig. 5). Interestingly, some of these strains are isolated gut symbionts (e.g., the Enterobacteria strains Hafnia alvei ATCC51873, Leminorella grimontii DSM5078, Serratia sp. Ag2) and/or potential pathogens (e.g., Vibrio, Plesiomonas, Halomonas), whereas other strains represent typical marine- or saline-associated bacteria (e.g., Halomonas, Salinarimonas) and typical freshwater-, soil-, and plant rhizosphere-associated bacteria (e.g., Rhizobium, Ensifer, Herbaspirillum, Microvirga). Whether these isolated strains are indeed able to use SQ for growth, and whether utilization of SQ is indeed a relevant trait in the habitats from which these strains originated, needs to be addressed.
Fig. 5.

Illustration of the SQ-degradation gene cluster in P. putida SQ1 with its five identified core genes (A) and of homologous, predicted SQ-degradation gene clusters found in genomes of other P. putida, other gamma-Proteobacteria (B), and in beta- and alpha-Proteobacteria (C and D, respectively). Homologous gene clusters in bacterial genomes were retrieved from the Joint Genome Institute’s IMG database by the Gene Cassette Search tool and the Gene Neighborhood viewer. Shown are gene clusters that contain gene homologs for at least four of the five core enzymes of the SQ Entner–Doudoroff pathway (genes are indicated by color coding). Also, candidate genes for SL export and for a predicted SQ-glyceride (SQG) alpha-glucosidase (main text) were frequently found to be conserved within these homolog clusters (gene symbols in gray). Note that other candidate genes in the clusters are not specified in this figure (gene symbols in white); for example, more variable genes were predicted for outer membrane porins, other transporters, or regulation (Fig. 1C).
The use of either the glycolysis (Embden–Meyerhof) or Entner–Doudoroff pathway by microbes for the utilization of glucose as a growth substrate illustrates a direct connection between an organism’s environment and the thermodynamic and biochemical properties of the metabolic pathway it employs (32). We speculate that the same holds true for the two newly discovered analogous pathways for SQ, i.e., for sulfoglycolysis in E. coli K-12, which excretes SLA in its reduced form (as DHPS), and for the SQ Entner–Doudoroff pathway in P. putida SQ1, which excretes SLA in its oxidized form (as SL). In glycolysis, glucose is phosphorylated twice and cleaved into two molecules of GAP, whereas in the Entner–Doudoroff pathway, glucose is phosphorylated only once and oxidized to KDPG and the KDPG is cleaved to one pyruvate and one GAP. In the further reactions from GAP to pyruvate, two molecules of ATP per molecule of GAP can be conserved (as indicated in Fig. 1A), and, accordingly, microbes seem to prefer either the glycolysis (Embden–Meyerhof) or Entner–Doudoroff pathway for the utilization of glucose as a function of their energetic situation. Although energy-deprived, fermentative anaerobes predominantly depend on the higher ATP yield of the glycolysis pathway, the Entner–Doudoroff pathway is common among obligate aerobes and facultative anaerobes (18, 32). Furthermore, the fermentative organisms need to invest pyruvate-carbon as an electron acceptor (e.g., for excreting lactate as a fermentation product), whereas in respiring organisms, the reducing equivalents (NADH) from GP oxidation in the Entner–Doudoroff pathway and from dissimilation of pyruvate to CO2 can be preserved as (additional) ATP via proton-motive force and ATPase. In contrast, life is ascetic for microbes that use SQ as a growth substrate via the SQ Entner–Doudoroff pathway concomitant with the excretion of SL, compared with utilization of glucose by the classical Entner–Doudoroff pathway. First, there is pyruvate and SLA produced from KDSG, not pyruvate and GAP as with KDPG; hence, no ATP can be produced by substrate-level phosphorylation of GAP (Fig. 1A). Accordingly, the proteomic data for P. putida SQ1 suggested that GAP dehydrogenase was strongly down-regulated in SQ-grown cells but highly produced in glucose-grown cells, whereas pyruvate dehydrogenase was highly produced for both (Fig. 3B). Second, there is only half of the carbon to respire from SQ, in form of one pyruvate. However, additional reducing equivalents (NADH) are gained from the last reaction of the SQ pathway in P. putida SQ1, from the oxidation of SLA to SL as catalyzed by the SLA dehydrogenase (Fig. 1B). Hence, the SQ Entner–Doudoroff pathway, inclusive of the SLA dehydrogenase reaction in P. putida SQ1, is represented by the equation
In the sulfoglycolytic pathway, there is DHAP and SLA produced from cleavage of SFP (12) rather than DHAP and GAP, as with fructose-1,6-bisphosphate in glycolysis, and the substrate-level phosphorylation of the one DHAP (via GAP to pyruvate) allows for conservation of ATP. In addition, the reduction of SLA to DHPS, as catalyzed by the NADH-dependent SLA reductase in E. coli K-12, regenerates the electron acceptor NAD+ that is needed for conversion of the DHAP via GAP to pyruvate (Fig. 1A), so that the overall equation of sulfoglycolysis inclusive of the SLA reductase reaction in E. coli K-12 is
We therefore suspect that the SQ Entner–Doudoroff pathway might be preferred among SQ-utilizing microbes with respiratory energy metabolism, whereas SQ-utilizing fermentative anaerobes might depend on the direct ATP yield of the sulfoglycolytic pathway and on the SLA reductase reaction as an additional electron-accepting step that aids in the overall fermentation.
Materials and Methods
A detailed version of the materials and methods used in this study can be found in SI Materials and Methods. P. putida SQ1 (DSM 100120) (11) was cultivated in mineral salts medium at pH 7.2 (33) at 30 °C in the dark, with SQ, glucose, or succinate as the sole carbon and energy source. SQ was synthesized chemically and identified by NMR and MS (11). Whole-genome shotgun sequencing of strain SQ1 was performed under contract by GATC Biotech using the Illumina HiSeq2000 platform and a 100-bp paired-end library; trimming, mapping, and de novo assembly of the unmapped raw reads were performed at the Genomics Center of the University of Konstanz (21). The genome annotation was performed within the Joint Genome Institute’s IMG analysis system (34), and the annotation (http://img.jgi.doe.gov; IMG Project ID Gp0039102) was used to build a local Mascot database (35) for PF-MS at the Proteomics Facility of the University of Konstanz. Two-dimensional PAGE was performed using Bio-Rad’s ReadyStrip immobilized pH gradient system (36). For RT-PCR assays (primer sequences; Table S1), the E.Z.N.A Bacterial RNA preparation kit (Omega Bio-Tek) was used, and for complementary cDNA synthesis, the Maxima reverse transcriptase kit (Fermentas) was used; Taq polymerase (Fermentas) was used for subsequent PCR assays (37). For insertional mutagenesis via homologous recombination, an internal 500-bp sequence of gene 0090 cloned into vector PCR2.1 (Invitrogen) was used; the construct was introduced by electroporation, and mutants were selected on kanamycin LB-agar plates. For heterologous expression, the complete genes were amplified by PCR (primer sequences; Table S2) and cloned into a pET100 vector containing an N-terminal His-tag with the help of the Champion pET100 directional TOPO expression kit (Invitrogen); the proteins were purified using nickel-nitrilotriacetic acid columns (12). The standard enzyme reaction mixture was 2 mM SQ in 50 mM Tris⋅HCl buffer (pH 7.5), plus 3 mM NAD(P)+ and the corresponding enzymes (10–60 μg⋅mL−1 each). SQ, SGL, SG, KDSG, SLA, SL, GP, PGL, PG, KDPG, and GAP were separated using hydrophilic interaction liquid chromatography and detected by electrospray ionization-MS (12).
Acknowledgments
We thank Jaco Vangronsveld (Hasselt University) for providing P. putida strain W619. We thank Thomas Huhn for synthesizing SQ; Alasdair Cook, Karin Denger, Michael Weiss, Paolo Franchini, Nicolai Müller, Ralf Schlesiger, and Bernhard Schink for their support and helpful discussions; and Laura Rexer for help in the heterologous production of some of the enzymes. This research was funded by the University of Konstanz, the Konstanz Young Scholar Fund, and the Deutsche Forschungsgemeinschaft (DFG). The work of A.-K.F. was funded by the Konstanz Research School Chemical Biology, the work of J.K. was funded by DFG Grant KL2340/2-1, and the work of D. Schleheck was funded by the University of Konstanz and by DFG Grants SCHL1936/1 and SCHL1936/3.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The annotation of the Pseudomonas putida SQ1 genome sequence reported in this paper has been deposited in the US Department of Energy Joint Genome Institute's Integrated Microbial Genomes (IMG) analysis system (img.jgi.doe.gov) under IMG Project ID Gp0039102 (genome name, Pseudomonas putida SQ1; locus tag prefix, PpSQ1).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1507049112/-/DCSupplemental.
References
- 1.Benson AA. The plant sulfolipid. Adv Lipid Res. 1963;1:387–394. doi: 10.1016/b978-1-4831-9937-5.50016-8. [DOI] [PubMed] [Google Scholar]
- 2.Harwood JL, Nicholls RG. The plant sulpholipid—A major component of the sulphur cycle. Biochem Soc Trans. 1979;7(2):440–447. doi: 10.1042/bst0070440. [DOI] [PubMed] [Google Scholar]
- 3.Benning C. Biosynthesis and function of the sulfolipid sulfoquinovosyl diacylglycerol. Annu Rev Plant Physiol Plant Mol Biol. 1998;49:53–75. doi: 10.1146/annurev.arplant.49.1.53. [DOI] [PubMed] [Google Scholar]
- 4.Meyer BH, et al. Sulfoquinovose synthase—An important enzyme in the N-glycosylation pathway of Sulfolobus acidocaldarius. Mol Microbiol. 2011;82(5):1150–1163. doi: 10.1111/j.1365-2958.2011.07875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martelli HL, Benson AA. Sulfocarbohydrate metabolism. I. Bacterial production and utilization of sulfoacetate. Biochim Biophys Acta. 1964;93(1):169–171. doi: 10.1016/0304-4165(64)90272-7. [DOI] [PubMed] [Google Scholar]
- 6.Martelli HL. Oxidation of sulphonic compounds by aquatic bacteria isolated from rivers of the Amazon region. Nature. 1967;216(5121):1238–1239. [Google Scholar]
- 7.Strickland TC, Fitzgerald JW. Mineralization of sulfur in sulfoquinovose by forest soils. Soil Biol Biochem. 1983;15(3):347–349. [Google Scholar]
- 8.Pugh C, Roy AB, White GF, Harwood JL. How is sulfolipid metabolised? In: Williams JP, Khan MU, Lem NW, editors. Physiology, Biochemistry and Molecular Biology of Plants. Kluwer; Dordrecht, The Netherlands: 1997. pp. 104–106. [Google Scholar]
- 9.Roy AB, Ellis AJ, White GF, Harwood JL. Microbial degradation of the plant sulpholipid. Biochem Soc Trans. 2000;28(6):781–783. [PubMed] [Google Scholar]
- 10.Roy AB, Hewlins MJE, Ellis AJ, Harwood JL, White GF. Glycolytic breakdown of sulfoquinovose in bacteria: A missing link in the sulfur cycle. Appl Environ Microbiol. 2003;69(11):6434–6441. doi: 10.1128/AEM.69.11.6434-6441.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denger K, Huhn T, Hollemeyer K, Schleheck D, Cook AM. Sulfoquinovose degraded by pure cultures of bacteria with release of C3-organosulfonates: Complete degradation in two-member communities. FEMS Microbiol Lett. 2012;328(1):39–45. doi: 10.1111/j.1574-6968.2011.02477.x. [DOI] [PubMed] [Google Scholar]
- 12.Denger K, et al. Sulphoglycolysis in Escherichia coli K-12 closes a gap in the biogeochemical sulphur cycle. Nature. 2014;507(7490):114–117. doi: 10.1038/nature12947. [DOI] [PubMed] [Google Scholar]
- 13.Denger K, et al. Bifurcated degradative pathway of 3-sulfolactate in Roseovarius nubinhibens ISM via sulfoacetaldehyde acetyltransferase and (S)-cysteate sulfolyase. J Bacteriol. 2009;191(18):5648–5656. doi: 10.1128/JB.00569-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denger K, Cook AM. Racemase activity effected by two dehydrogenases in sulfolactate degradation by Chromohalobacter salexigens: Purification of (S)-sulfolactate dehydrogenase. Microbiology. 2010;156(Pt 3):967–974. doi: 10.1099/mic.0.034736-0. [DOI] [PubMed] [Google Scholar]
- 15.Mayer J, et al. 2,3-Dihydroxypropane-1-sulfonate degraded by Cupriavidus pinatubonensis JMP134: Purification of dihydroxypropanesulfonate 3-dehydrogenase. Microbiology. 2010;156(Pt 5):1556–1564. doi: 10.1099/mic.0.037580-0. [DOI] [PubMed] [Google Scholar]
- 16.Entner N, Doudoroff M. Glucose and gluconic acid oxidation of Pseudomonas saccharophila. J Biol Chem. 1952;196(2):853–862. [PubMed] [Google Scholar]
- 17.Wackett LP. Pseudomonas putida—A versatile biocatalyst. Nat Biotechnol. 2003;21(2):136–138. doi: 10.1038/nbt0203-136. [DOI] [PubMed] [Google Scholar]
- 18.Fuhrer T, Fischer E, Sauer U. Experimental identification and quantification of glucose metabolism in seven bacterial species. J Bacteriol. 2005;187(5):1581–1590. doi: 10.1128/JB.187.5.1581-1590.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.del Castillo T, et al. Convergent peripheral pathways catalyze initial glucose catabolism in Pseudomonas putida: Genomic and flux analysis. J Bacteriol. 2007;189(14):5142–5152. doi: 10.1128/JB.00203-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daddaoua A, Krell T, Ramos JL. Regulation of glucose metabolism in Pseudomonas: The phosphorylative branch and entner-doudoroff enzymes are regulated by a repressor containing a sugar isomerase domain. J Biol Chem. 2009;284(32):21360–21368. doi: 10.1074/jbc.M109.014555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Felux AK, Franchini P, Schleheck D. 2015. Draft genome sequence of the sulfoquinovose-degrading Pseudomonas putida strain SQ1. SIGS, 10.1186/s40793-015-0033-x. [DOI] [PMC free article] [PubMed]
- 22.Heinz E. Recent investigations on the biosynthesis of the plant sulfolipid. In: De Kok L, editor. Sulfur Nutrition and Assimilation in Higher Plants. SPB Academic Publishing; The Hague, The Netherlands: 1993. pp. 163–178. [Google Scholar]
- 23.Benning C. Questions remaining in sulfolipid biosynthesis: A historical perspective. Photosynth Res. 2007;92(2):199–203. doi: 10.1007/s11120-007-9144-6. [DOI] [PubMed] [Google Scholar]
- 24.Zolghadr B, et al. UDP-sulfoquinovose formation by Sulfolobus acidocaldarius. Extremophiles. 2015;19(2):451–467. doi: 10.1007/s00792-015-0730-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shibuya I, Benson AA. Hydrolysis of alpha-sulfo-quinovosides by alpha-galactosidase. Nature. 1961;192(4808):1186–1187. [Google Scholar]
- 26.Hanekamp S, Bloem E, Schnug E. History of sulfur deficiency in crops. In: Jez J, editor. Sulfur: A Missing Link Between Soils, Crops and Nutrition. American Society of Agronomy; Madison, WI: 2008. [Google Scholar]
- 27.Kertesz MA, Mirleau P. The role of soil microbes in plant sulphur nutrition. J Exp Bot. 2004;55(404):1939–1945. doi: 10.1093/jxb/erh176. [DOI] [PubMed] [Google Scholar]
- 28.Giordano M, Norici A, Hell R. Sulfur and phytoplankton: Acquisition, metabolism and impact on the environment. New Phytol. 2005;166(2):371–382. doi: 10.1111/j.1469-8137.2005.01335.x. [DOI] [PubMed] [Google Scholar]
- 29.Denger K, Smits TH, Cook AM. L-cysteate sulpho-lyase, a widespread pyridoxal 5′-phosphate-coupled desulphonative enzyme purified from Silicibacter pomeroyi DSS-3(T) Biochem J. 2006;394(Pt 3):657–664. doi: 10.1042/BJ20051311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weinitschke S, Denger K, Cook AM, Smits TH. The DUF81 protein TauE in Cupriavidus necator H16, a sulfite exporter in the metabolism of C2 sulfonates. Microbiology. 2007;153(Pt 9):3055–3060. doi: 10.1099/mic.0.2007/009845-0. [DOI] [PubMed] [Google Scholar]
- 31.Mayer J, Cook AM. Homotaurine metabolized to 3-sulfopropanoate in Cupriavidus necator H16: Enzymes and genes in a patchwork pathway. J Bacteriol. 2009;191(19):6052–6058. doi: 10.1128/JB.00678-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flamholz A, Noor E, Bar-Even A, Liebermeister W, Milo R. Glycolytic strategy as a tradeoff between energy yield and protein cost. Proc Natl Acad Sci USA. 2013;110(24):10039–10044. doi: 10.1073/pnas.1215283110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thurnheer T, Köhler T, Cook AM, Leisinger T. Orthanilic acid and analogues as carbon sources for bacteria: Growth physiology and enzymic desulphonation. J Gen Microbiol. 1986;132(5):1215–1220. [Google Scholar]
- 34.Markowitz VM, et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014;42(Database issue):D560–D567. doi: 10.1093/nar/gkt963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt A, Müller N, Schink B, Schleheck D. A proteomic view at the biochemistry of syntrophic butyrate oxidation in Syntrophomonas wolfei. PLoS One. 2013;8(2):e56905. doi: 10.1371/journal.pone.0056905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weiss M, Denger K, Huhn T, Schleheck D. Two enzymes of a complete degradation pathway for linear alkylbenzenesulfonate (LAS) surfactants: 4-sulfoacetophenone Baeyer-Villiger monooxygenase and 4-sulfophenylacetate esterase in Comamonas testosteroni KF-1. Appl Environ Microbiol. 2012;78(23):8254–8263. doi: 10.1128/AEM.02412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gutiérrez Acosta OB, Schleheck D, Schink B. Acetone utilization by sulfate-reducing bacteria: Draft genome sequence of Desulfococcus biacutus and a proteomic survey of acetone-inducible proteins. BMC Genomics. 2014;15(1):584. doi: 10.1186/1471-2164-15-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Felux AK, Denger K, Weiss M, Cook AM, Schleheck D. Paracoccus denitrificans PD1222 utilizes hypotaurine via transamination followed by spontaneous desulfination to yield acetaldehyde and, finally, acetate for growth. J Bacteriol. 2013;195(12):2921–2930. doi: 10.1128/JB.00307-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cleland WW. The use of pH studies to determine chemical mechanisms of enzyme-catalyzed reactions. Methods Enzymol. 1982;87:390–405. doi: 10.1016/s0076-6879(82)87024-9. [DOI] [PubMed] [Google Scholar]
- 41.Müller N, Griffin BM, Stingl U, Schink B. Dominant sugar utilizers in sediment of Lake Constance depend on syntrophic cooperation with methanogenic partner organisms. Environ Microbiol. 2008;10(6):1501–1511. doi: 10.1111/j.1462-2920.2007.01565.x. [DOI] [PubMed] [Google Scholar]