

Significance
The activation of nuclear factor κB (NFκB) in the normal inflammatory response is rapidly down regulated, whereas constitutive NFκB activation is a hallmark of cancer. We now reveal cross signaling between EGF receptor (EGFR) and Toll-like receptor 4 (TLR4). NFκB activation in response to EGF requires, in addition to EGFR, TLR4 and two downstream proteins. Conversely, EGFR is required for TLR4-mediated activation of NFκB in response to lipopolysaccharide (LPS). The LYN proto-oncogene (LYN) is required for NFκB activation in response to either ligand. In mice, the EGFR inhibitor erlotinib greatly reduces both cytokine expression and endotoxicity in response to LPS, suggesting that EGFR inhibitors may find use in treating septic shock.
Keywords: EGFR, TLR4, erlotinib, LPS, NFκB
Abstract
Several components of the canonical pathway of response to lipopolysaccharide (LPS) are required for the EGF-dependent activation of NFκB. Conversely, the ability of Toll-like Receptor 4 (TLR4) to activate NFκB in response to LPS is impaired by down regulating EGF receptor (EGFR) expression or by using the EGFR inhibitor erlotinib. The LYN proto-oncogene (LYN) is required for signaling in both directions. LYN binds to the EGFR upon LPS stimulation, and erlotinib impairs this association. In mice, erlotinib blocks the LPS-induced expression of tumor necrosis factor α (TNFα) and interleukin-6 (IL-6) and ameliorates LPS-induced endotoxity, revealing that EGFR is essential for LPS-induced signaling in vivo.
The NFκB family of signal-activated transcription factors plays a pivotal role in regulating inflammation, survival, and growth. The family consists of five members, p65 (RelA), Rel B, c-Rel, p105/p50, and p100/p52 (1). In unstimulated cells, NFκB is present in the cytoplasm as inactive hetero- and homodimers through its interaction with inhibitory IκB proteins. NFκB is activated in response to a wide variety of stimuli, including tumor necrosis factor-α (TNFα), interleukin-1β (IL-1β), or pathogen-derived components such as lipopolysaccharide (LPS). Growth factors and nonreceptor tyrosine kinases can also activate NFκB (1–4). Upon activation, IκBα, which inhibits RelA, is phosphorylated on S32 and S36 by IκB kinase (IKK), leading to its degradation and to the translocation of released NFκB p65/p50 heterodimers and p65/p65 homodimers to the nucleus, where they activate the transcription of target genes (5). In the normal inflammatory response, the activation of NFκB is rapidly down-regulated, mainly through the resynthesis of IκB (6, 7).
The EGF receptor (EGFR) is a transmembrane protein consisting of an extracellular domain to which ligands bind, a transmembrane domain, and an intracellular domain that includes a tyrosine kinase. Upon activation, EGFR is phosphorylated on about 20 tyrosine residues (8), leading to the activation of several downstream signaling pathways. EGFR is highly expressed in a variety of solid tumors, and consitutive or ligand-induced EGFR-dependent signaling in tumor cells has been linked to increased cell survival, proliferation, and metastasis (9). We recently showed that EGFR plays a key role in the constitutive activation of NFκB in several cancer cell lines (4).
Both receptor and nonreceptor protein tyrosine kinases are essential for many cellular signaling pathways that regulate growth, differentiation, apoptosis, and immune responses (10), and members of the SRC family of tyrosine kinases are vital signaling intermediates (11). LYN, a member of this family, is a key regulator of several intracellular signaling cascades (12).
Toll-like receptors (TLRs), a family of type 1 membrane glycoproteins, are expressed on immune cells, such as macrophages, dendritic cells (DCs), B cells, and neutrophils, as well as on nonimmune cells, including epithelial cells, fibroblasts, and keratinocytes. They enable the innate immune system to recognize pathogen-associated molecular patterns (PAMPs) by activating signal transduction pathways (13). TLRs have an extracellular domain containing leucine-rich repeats, which are responsible for ligand binding, a transmembrane domain, and a cytoplasmic Toll/IL-1 receptor (TIR) domain, which is required for signaling (13). Upon activation, TLRs recruit a set of adaptor proteins that also have TIR domains, resulting in downstream signaling cascades that lead to the activation of NFκB and members of the IFN-regulatory factor (IRF) family, which in turn direct the induction of proinflammatory cytokines and chemokines (13, 14). In humans, the 10 functional TLRs can be subdivided according to their subcellular locations. TLRs 1, 2, 4, 5, 6, and 10 are expressed on cell surfaces and recognize lipid and protein ligands, whereas TLRs 3, 7, 8, and 9 are expressed on intracellular organelles, principally endosomes and the endoplasmic reticulum (15–18). Several TLRs participate in innate immune responses by activating EGFR in airway epithelial cells (19), and recent work has shown that EGFR is required for dsRNA-mediated TLR3-dependent signaling (20).
TLR4 is crucial for effective host cell responses to LPS from Gram-negative bacteria (21, 22). Upon LPS binding, TLR4 oligomerizes and recruits adaptors to its intracellular TIR domains, triggering downstream signaling (23). TLR4 is the only family member that can signal through both MYD88 (myeloid differentiation primary response gene 88)-dependent and MYD88-independent, TRIF-dependent pathways (23). Signaling via MYD88 involves the rapid activation of NFκB, which leads to the production of proinflammatory cytokines (24). TRIF-dependent signaling involves a slower activation of NFκB and also activation of IFN regulatory factor 3 (IRF3), leading to the production of type I IFN (IFN α/β), IFN-inducible gene products, and the full innate immune response (25). It is well-known that chronic inflammation can facilitate the development of cancer (26–29) and TLR4 plays a key role in carcinogenesis. It has been reported that the TLR4 ligand LPS activates EGFR in several different cell lines and in vivo models (30–34).
Sepsis is a severe inflammatory response to infection, leading to an imbalance between pro- and anti-inflammatory responses (35). LPS induces a systemic inflammation that mimics many of the initial clinical features of sepsis, including increases in proinflammatory cytokines (36). During sepsis the majority of cytokines have multiple intrinsic effects, mediating not only immune defenses but also pathological manifestations. Treatment of LPS-injected animals with neutralizing antibodies against proinflammatory cytokines resulted in improved outcomes (37, 38). However, several clinical trials of antiinflammatory cytokines, including TNFα and anti-IL-1 therapy failed to improve the survival of septic patients (39, 40).
In this study we elucidate the role of TLR4 in the EGF-induced activation of NFκB, which requires a functional interaction between EGFR and TLR4. The EGFR inhibitor erlotinib blocks TLR4-mediated NFκB activation, indicating that the kinase activity of EGFR is necessary. Down-regulating the expression of the SRC family member LYN impairs EGF-mediated NFκB activation. Furthermore, EGFR is required for TLR4 to activate NFκB. LYN binds to both EGFR and TLR4 in response to LPS, and this binding is blocked by erlotinib. Importantly, erlotinib also inhibits LPS-induced NFκB-dependent cytokine production in mice and protects mice from LPS-induced lethality. These in vivo findings reveal a potential therapeutic role for erlotinib in protection against septic shock.
Results
EGFR-Mediated NFκB Activation Requires MYD88 and TAK1.
Our recent study elucidated the important role of EGF in mediating NFκB activation (4). Signaling to NFκB might depend solely on EGFR, or might also involve another receptor. TLRs activate NFκB (13), and MYD88, a universal adaptor protein, is crucial for the ability of all TLR/IL-1R family members, except TLR3, to induce NFκB activation (41). Therefore, it was logical to determine whether MYD88 is required for EGF-dependent NFκB activation. Before we could study MYD88 expression in human mammary epithelial (HME) cells, we needed to prevent apoptosis by expressing a high level of the antiapoptotic protein BCL2. Increased IKK phosphorylation and IκB phosphorylation, degradation, and resynthesis were observed in control HME-BCL2 cells treated with EGF but not in MYD88 knockdown cells (Fig. S1A). Knockdown of MYD88 diminished EGF-induced ERK phosphorylation as well (Fig. S1A). TGF-β-activated kinase 1 (TAK1) phosphorylates and activates IKK in TLR/IL-1 pathways, leading to the phosphorylation of IκB and activation of NFκB (42). To test the involvement of TAK1 in EGFR-dependent NFκB activation, stable pools of HME cells expressing shRNAs against TAK1 or scrambled shRNA were generated. Down-regulation of TAK1 impaired EGF- or IL-1-stimulated phosphorylation of IKK, IκB, and ERK, and also impaired the degradation and resynthesis of IκB (Fig. S1B). We conclude that MYD88 and TAK1, which are essential for TLR/IL-1-mediated NFκB activation, are also required for NFκB activation in response to EGF.
Fig. S1.

MYD88 and TAK1 are essential for the EGF-induced activation of NFκB. (A, Upper) HME-BCL2 cells were infected with a vector encoding nontargeted shRNA (NTshRNA) or an shRNA against MYD88 and immunoblotted for MYD88 and β-actin expression. (Lower) The cells were serum starved for 24 h and then stimulated with 100 ng/mL EGF. Phosphorylated EGFR, IKK, IκB, ERK, and AKT were detected with phospho-specific antibodies. An anti-IκB antibody detected the degradation and resynthesis of this protein. (B, Upper) HME cells were infected with a vector encoding a scrambled shRNA (Con sh) or TAK1 shRNA. TAK1 was detected by the Western method. (Lower) EGF-starved cells were stimulated with EGF (100 ng/mL) for the indicated times or with IL-1β for 5 min.
TLR4 Silencing Impairs EGF-Induced NFκB Activation.
Next we investigated the role of individual TLRs in this pathway. Because we observed rapid activation of NFκB in response to EGF, we reasoned that a cell surface TLR was most likely to be involved. We began by focusing on TLR4. In HME cells, decreasing the expression of TLR4 inhibited the EGF-dependent phosphorylation of IKK and IκB (Fig. 1A). TLR4 down-regulation in nonsmall cell lung carcinoma (NSCLC) A549 cells also inhibited EGF-dependent phosphorylation of IKK and IκB, as well as the subsequent degradation and resynthesis of IκB (Fig. S2A). Because A549 cells already have high constitutive levels of activated NFκB, the ability of EGF to drive a further increase in IκB phosphorylation is limited. The EGF-induced phosphorylation of EGFR was similar in control and TLR4-deficient cells (Fig. 1A and Fig. S2A). These results indicate that TLR4 is necessary for EGF-dependent NFκB activation in both nonmalignant and malignant human cells.
Fig. 1.

TLR4 is required for EGF-induced NFkB activation and EGF-stimulated phosphorylation of TLR4 is inhibited by erlotinib. (A, Left) HME cells were transfected with nontargeted siRNA (NTsiRNA) or with an siRNA against TLR4. After 48 h, the TLR4 mRNA expression level was determined by RT-PCR. (Right) EGF-starved cells were treated with EGF for 5 min and the levels of phosphorylated EGFR, IKK, and IkB were analyzed by the Western method. β-actin was used as a loading control. (B) EGF-starved HME cells were stimulated with EGF, and phosphorylated and total TLR4 levels were detected by the Western method. (C) EGF-starved HME cells were pretreated with erlotinib (50 µM) for 1 h or left untreated. The cells were then stimulated with EGF and immunoblotted for phosphorylated and total TLR4. Each experiment in A–C was carried out two or three times independently, with results similar to the representative examples that are shown. (D) A diagram showing that, upon EGF stimulation, activated EGFR phosphorylates TLR4, leading to NFkB activation through MYD88 and TAK1. Erlotinib inhibits the kinase activity of EGFR and suppresses TLR4 phosphorylation.
Fig. S2.

TLR4 is required for EGFR dependent activation of NFκB in A549 cells. (A, Upper) A549 cells were infected with a vector encoding NTshRNA or an shRNA against TLR4, and then selected with puromycin. TLR4 mRNA expression was determined by RT-PCR. (Lower) Serum-starved cells were treated with EGF and the levels of phosphorylated and total proteins were analyzed by the Western method. (B) Serum-starved A549 cells were stimulated with EGF and immunoblotted for phosphorylated and total TLR4. Each of these experiments was carried out two or three independent times, with results similar to the representative examples that are shown.
TLR4 Is Phosphorylated in Response to EGF.
Tyrosine phosphorylation of the cytosolic Toll/interleukin-1 receptor (TIR) domain of TLR4 is required for NFκB activation in response to LPS (43, 44). Because TLR4 is essential for EGF-induced NFκB activation, we investigated whether EGF causes TLR4 phosphorylation. Using an antibody that recognizes phosphorylated tyrosine residue 674 (44), we observed a substantial increase in TLR4 phosphorylation in HME cells and A549 cells stimulated with EGF (Fig. 1B and Fig. S2B). Pretreatment with erlotinib for 1 h blocked the EGF-dependent phosphorylation of TLR4 (Fig. 1C), indicating that the kinase activity of EGFR is required for TLR4 phosphorylation in response to EGF. Our mechanistic findings are summarized in Fig. 1D.
EGFR Is Essential for LPS-Induced Activation of NFκB.
Because HME cells die following knockdown of EGFR, we used HME-BCL2 cells to study the role of EGFR in the response to LPS. The substantial increases in the phosphorylation of EGFR, IKK, and IκB and the degradation and resynthesis of IκB in response to LPS were impaired when EGFR was down-regulated (Fig. 2A). The phosphorylation of v-akt murine thymoma viral oncogene homolog (AKT) and ERK was increased upon LPS stimulation in control cells, but not in EGFR-knockdown cells (Fig. 2A), indicating that EGFR is required for LPS-mediated AKT and ERK phosphorylation. EGFR also plays a role in TLR4-dependent signaling in cancer cells, because the ability of LPS to activate NFκB was impaired when EGFR expression was down-regulated in A549 and OVCAR3 cells (Fig. 2B and Fig. S3A). To determine whether the kinase activity of EGFR is required for LPS-dependent signaling to NFκB, we treated HME cells with erlotinib for 1 h before stimulating them with LPS. Erlotinib blocked the LPS-dependent phosphorylation of IKK and IκB, and the degradation and resynthesis of IκB (Fig. 2C). Inhibition of EGFR kinase activity by erlotinib also diminished LPS-induced TLR4 phosphorylation in A549 cells (Fig. 2D), impaired NFκB activation in A549 and OVCAR3 cells, and blocked ERK and AKT phosphorylation (Figs. 2E and S3B).
Fig. 2.

EGFR is essential for LPS-induced activation of NFκB. (A) HME-BCL2 cells were transfected with NTshRNA or with an shRNA against EGFR and then selected with puromycin. The cells were then stimulated with LPS (10 µg/mL), and phosphorylated and total EGFR, as well as phosphorylated IKK, IκB, ERK, and AKT, were detected by the Western method. An anti-IκB antibody detected the degradation and resynthesis of this protein. (B) A549 cells were treated with LPS (10 µg/mL) and the phosphorylated and total protein levels were determined by the Western method. (C–E) The kinase activity of EGFR is required for NFκB activation. (C) HME cells were pretreated with 50 μM erlotinib for 1 h, or left untreated, and then stimulated with LPS. Phosphorylated EGFR, IKK, and IκB were detected by the Western method. An anti-IκB antibody detected the degradation and resynthesis of this protein. (D) LPS-mediated TLR4 phosphorylation is inhibited by erlotinib. A549 cells were pretreated with 10 μM of erlotinib for 1 h or left untreated, then stimulated with LPS for 5 or 15 min. Phosphorylated TLR4 was analyzed by the Western method. (E) A549 cells were pretreated with 10 μM of erlotinib for 1 h or left untreated, and then stimulated with LPS. Phosphorylated and total protein levels were detected by the Western method. The experiments above were repeated thrice, with very similar results.
Fig. S3.

EGFR is required for TLR4-dependent activation of NFκB in OVCAR3 cells. (A) OVCAR3 cells were treated with LPS (10 µg/mL) for 5 min and phosphorylated IKK, IκB, and total EGFR were determined by the Western method. (B) OVCAR3 cells were pretreated with 10 μM of erlotinib for 1 h or left untreated, and then stimulated with LPS. Phosphorylated and total protein levels were detected by the Western method. The experiments above were repeated thrice, with very similar results.
Kinases in the SRC Family Are Involved in EGFR-TLR4 Signaling to NFκB.
Surprisingly, we were not able to observe binding of EGFR and TLR4 to each other in response to EGF or LPS using confocal microscopy or coimmunoprecipitation (Fig. S4 A and B). These negative results make it unlikely that EGFR phosphorylates TLR4 directly but do not rule it out completely. This finding is distinct from the results of Yamashita et al. (20), with a different TLR family member. These workers showed that TLR3 binds to EGFR in response to dsRNA. For EGFR-TLR4 signaling to NFκB we assumed that one or more additional kinases are required. We began by investigating the SRC family of kinases, because SRC is well known to mediate EGFR phosphorylation (45, 46) and a SRC family member is known to be involved in LPS-dependent NFκB activation (43), and also in TLR3-dependent signaling (20). In response to LPS TLR4 can be activated by SRC family members through the phosphorylation of Y674 (44). The EGF-dependent phosphorylation of IKK and IκB was substantially inhibited by prior exposure of HME or A549 cells to the SRC family inhibitor PP2, which also greatly diminished EGF-induced EGFR phosphorylation and eliminated downstream AKT and ERK phosphorylation (Figs. S4 and S5). This result suggests an important role for one or more SRC family members in EGF-dependent NFκB activation. LPS-induced NFκB activation in A549 cells was also inhibited by pretreatment with PP2 (Fig. S5C), suggesting that a SRC family kinase is also involved in TLR4 signaling to NFκB, consistent with the previous finding of Medvedev et al. (43).
Fig. S4.

Association of EGFR and TLR4 was not observed in response to EGF or LPS. (A) Confocal microscopy of HME cells stimulated with EGF (100 ng/mL) or LPS (10 µg/mL) and stained for EGFR (red) and TLR4 (green). No overlap of red and green was seen. (B) Attempts to coimmunoprecipitate (Co-IP) EGFR and TLR4. HME cells over expressing full-length, flag-tagged TLR4 were stimulated with EGF or LPS for 5 min (Upper) or with EGF for the indicated times (Lower). Whole cell lysates were used for Co-IP with normal rabbit IgG or rabbit polyclonal antibodies against EGFR. Rabbit polyclonal antibodies against EGFR or mouse monoclonal antibodies against Flag were used for Western analysis.
Fig. S5.

Kinases in the SRC family are involved in EGFR-TLR4 signaling to NFκB. (A) EGF-starved HME cells were pretreated with 50 μM of the SRC family inhibitor PP2 for 2 h or were left untreated, and the cells were then stimulated with EGF. Phosphorylation of EGFR, IKK, IκB, SRC, AKT, and ERK was analyzed by the Western method. (B) Serum-starved A549 cells were pretreated with 50 μM of PP2 for 2 h or left untreated, and then and stimulated with EGF. (C) A549 cells were pretreated with 50 μM of PP2 for 2 h or left untreated, and then stimulated with LPS. Phosphorylated and total proteins in B and C were detected by the Western method. Each experiment was carried out independently twice, with results similar to the representative examples that are shown.
LYN Is Required for EGFR-TLR4 Activation of NFκB.
The involvement of LYN in LPS-mediated TLR4 signaling had been reported earlier (43, 47). To elucidate whether LYN is also involved in the activation of NFκB in response to EGF, we knocked its expression down in HME cells. Reduction of LYN expression attenuated the EGF-dependent phosphorylation of IKK and the degradation and resynthesis of IκB (Fig. 3A). Down regulation of LYN also impaired EGF-mediated IKK and IκB phosphorylations in A549 cells (Fig. S6). An association of LYN with constitutively activated EGFR in lung adenocarcinoma cells has been reported by Sutton et al. (46). Coimmunoprecipitation experiments in HME cells demonstrated that, upon stimulation with LPS, LYN is recruited to both EGFR and TLR4 (Fig. 3B), consistent with the previous finding of Medvedev et al. (43) for TLR4. The LPS-stimulated increase in the association of LYN with EGFR or TLR4 in A549 cells was blocked by erlotinib (Fig. S7), showing that the kinase activity of EGFR is required. Our current understanding of how EGFR participates in LPS-mediated NFκB activation is illustrated in Fig. 3C.
Fig. 3.

LYN is required for EGFR-TLR4 signaling to NFκB. (A, Left) HME cells were infected with a vector encoding NTshRNA or an shRNA against LYN, selected with puromycin, and immunoblotted for LYN and β-actin expression. (Right) EGF-starved cells were treated with EGF and the levels of phosphorylated and total proteins were analyzed by the Western method. This experiment was done twice, with results similar to the representative example that is shown. (B) LPS stimulates the recruitment of LYN to EGFR and TLR4. HME cells stimulated with LPS were analyzed by coimmunoprecipitation (Co-IP). Total cell lysates were assayed with normal rabbit IgG or rabbit polyclonal antibodies against LYN. Rabbit polyclonal antibodies against EGFR or mouse monoclonal antibodies against LYN or TLR4 were used for Western analysis. The experiments above were repeated thrice, with very similar results. (C) A diagram showing the importance of the SRC family member LYN as a key kinase in mediating the cross talk between EGFR and TLR4 that leads to NFκB activation. In response to LPS, LYN associates with EGFR and also with TLR4, leading to downstream signaling. Erlotinib inhibits the kinase activity of EGFR, and blocks the LPS stimulated interaction of LYN with EGFR and TLR4.
Fig. S6.

LYN is involved in EGFR-TLR4 signaling to NFκB in A549 cells. (Left) The cells were transfected with NTsiRNA or an siRNA against LYN, and immunoblotted for LYN and β-actin expression. (Right) Serum-starved cells were treated with EGF and the levels of phosphorylated proteins were analyzed by the Western method. β-actin was used as a loading control. This experiment was done twice, with results similar to the representative example.
Fig. S7.
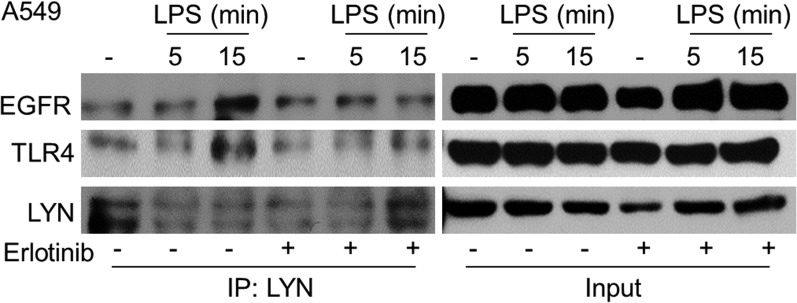
The kinase activity of EGFR is required for the LPS-stimulated recruitment of LYN to EGFR and TLR4 in A549 cells. The cells were treated with 10 μM erlotinib for 1 h or left untreated, and then stimulated with LPS. Co-IP was performed using rabbit polyclonal antibodies against LYN. Rabbit polyclonal antibodies against EGFR or mouse monoclonal antibodies against LYN or TLR4 were used for Western analysis. The experiments above were repeated thrice, with very similar results.
Erlotinib Blocks LPS-Induced Cytokine Expression in Vivo and Protects Mice from LPS-Mediated Lethality.
Administration of LPS to mice triggered the appearance of IL-6 and TNFα in plasma after 6 h, and this induction was decreased substantially by pretreatment with erlotinib (Fig. 4 A and B). Additionally, erlotinib pretreatment significantly inhibited the induction of mRNAs encoding IL-6, TNFα, and CXCL1 in splenocytes 6 h after administration of LPS (Fig. 4C). These results demonstrate that a kinase that is inhibited by erlotinib, almost certainly EGFR, is required for LPS-induced NFκB activation in vivo. We next investigated a potential therapeutic role for erlotinib in LPS-induced endotoxicity in mice. As shown in Fig. 4D, 80% of mice treated with erlotinib (100 mg/kg) by oral gavage survived 48 h after LPS administration (10 mg/kg), much longer than control mice injected with LPS alone. Erlotinib alone was not toxic. These results reveal that erlotinib, and probably other EGFR inhibitors, have the potential to prevent or treat inflammatory diseases that involve functional interactions between EGFR and one or more TLRs.
Fig. 4.

Erlotinib treatment blocks LPS-induced cytokine expression in vivo. (A and B) Mice were pretreated with an erlotinib suspension at a dosage of 100 mg/kg body weight, or with vehicle, by oral gavage 16 h before LPS injection. The mice were injected intraperitoneally with LPS at 10 mg/kg body weight. A second dose of erlotinib (100 mg/kg) or vehicle was administered at the same time as the LPS. Another set of mice (controls) were injected with medium without LPS or erlotinib. Plasma were collected 6 h after LPS injection, and the concentrations of IL-6 (A) and TNFα (B) were measured by ELISA. Plasma was also collected from control mice 6 h after the injection of medium. IL-6 and TNFα were not detected in these samples. The data reflect the means ± SD for two sets of experiments (n = 5 and n = 3). The ELISAs were repeated thrice with very similar results. (C) Mouse splenocytes were isolated 6 h after LPS injection. Total RNAs from these cells were analyzed by real-time PCR for IL-6, TNFα, and CXCL-1 mRNAs. The experiments were repeated twice and each measurement was performed in triplicate. Data are expressed as means ± SD (n = 5). (D) Erlotinib protects mice from LPS-induced endotoxicity. C57BL/6 mice were pretreated with erlotinib (100 mg/kg) (n = 15) or vehicle control PEG (n = 15) 16 h before LPS (i.p., 10 mg/kg) injection. A second dose of erlotinib (100 mg/kg) or vehicle was administered at the same time as LPS. Mice were treated again with erlotinib or vehicle once daily for 3 more days. The control group of mice (n = 10) received erlotinib alone (100 mg/kg) in the same way as the treatment group. Survival was monitored after LPS injection. Survival data were analyzed by the Kaplan–Meier method and log-rank test, *P < 0.0018 versus the vehicle pretreated group.
Discussion
EGF-Induced NFκB Activation Requires EGFR, TLR4, MYD88, TAK1, and LYN.
We showed previously that NFκB is activated by EGF in nonmalignant human epithelial cells and that the EGF/EGFR pathway is responsible for the constitutive activation of NFκB in cells derived from several different types of tumors (4). We now show that down-regulation of TLR4 impairs EGF-induced NFκB activation in nonmalignant and malignant human cells, and we conclude that there is an important connection between TLR4 and EGFR in NFκB activation in response to EGF. Down regulation of MYD88 in HME cells impaired the NFκB activation that was observed within 5 min of EGF stimulation (Fig. S1A), showing that this protein is essential for the EGFR-NFκB pathway. MYD88 activates NFκB through TAK1 (48) and kinase-inactive TAK1 impairs NFκB activation in response to LPS (49). Consistently, TAK1 is also necessary for EGFR-dependent NFκB activation (Fig. S1B). To the best of our knowledge, this is the first report showing that EGF-induced NFκB activation requires both TLR4 and two downstream components in the canonical TLR4-dependent pathway. Tyrosine phosphorylation of TLR4 is essential for signaling, and the TIR domain TLR4 mutants Y674A and Y680A are defective in the LPS-dependent activation of NFκB (43). Although a number of protein tyrosine kinases have been implicated in TLR4-dependent signaling, it is not clear how these key residues of TLR4 become phosphorylated. However, it has been shown recently that TLR3 activation requires two tyrosine kinases, EGFR and SRC (20). We observed that treatment of HME and A549 cells with EGF leads to the phosphorylation of TLR4 at Y674, and that the kinase activity of EGFR is required for this activation. This result indicates that the EGF-dependent phosphorylation of TLR4 is essential for NFκB activation in response to EGF, as depicted in Fig. 1D. It is well known that SRC phosphorylates EGFR on multiple tyrosine residues, including some located in or near the kinase domain (50, 51), and that these phosphorylations are critical for different aspects of EGF-dependent signaling (52). We now show that the EGF-induced phosphorylation of EGFR at tyrosine 1068 was impaired by the SRC family kinase inhibitor PP2 and that PP2 blocks EGFR-mediated NFκB activation in both nonmalignant and malignant cells, indicating that the kinase activity of SRC or a family member is required for EGFR-mediated NFκB activation. We show that down-regulation of the SRC family member LYN impairs EGF-mediated NFκB activation indicating that LYN is essential for EGF-mediated NFκB activation.
LPS-Mediated NFκB Activation Requires EGFR and LYN.
Activation of TLR4-dependent signaling by LPS is a critical upstream event in response to infections by Gram-negative bacteria. In response to LPS, TLR4 activates NFκB through both MYD88-dependent and MYD88-independent pathways. TLR activation initiates a complex and integrated signaling cascade that activates EGFR in airway epithelial cells (19). LPS induces the expression of vascular cell adhesion molecule-1 (VCAM-1), a systemic inflammation marker, through EGFR-dependent activation of AKT (53, 54).
The involvement of TLR4 in EGFR-dependent NFκB activation prompted us to focus on understanding whether cross talk between TLR4 and EGFR might trigger NFκB activation. Accordingly, we show that knockdown of EGFR or inhibition of EGFR kinase activity by erlotinib impaired LPS-stimulated NFκB activation in nontumorigenic HME cells as well as in cancer cells. Earlier studies have shown that the transactivation of EGFR is required for LPS-induced COX-2 activation (32, 33, 55) or NRAS activation (34). LPS-induced increase in human beta-defensin-3 expression requires EGFR activation in oral squamous cell carcinoma cells (31). However, it has not been established that EGFR is essential for the LPS-dependent activation of NFκB. We now show in addition that EGFR activation is required for LPS-induced NFκB activation.
Basu et al. (56) reported that EGFR binds to TLR4 in response to the Helicobacter pylori secretory protein HP0175 in human gastric epithelial cells. However, the interaction was observed only after 60 min, and we have found that EGF- or LPS-mediated NFκB activation is strongly induced within 5–10 min. Furthermore, we were not able to observe any interaction between EGFR and TLR4 in response to EGF or LPS (Fig. S4), suggesting that additional kinases are likely to mediate an indirect interaction between these two receptors. The SRC family inhibitor PP2 blocks LPS-mediated NFκB activation (43) and, consistent with this report, we now show that PP2 blocks NFκB activity upon LPS stimulation in A549 cells.
LPS stimulation leads to the recruitment of LYN, a SRC family member, to TLR4 in HEK293TLR4/MD-2 stable transfectants (43). Consistent with this earlier report, we now show that LPS stimulation leads to the recruitment of LYN to TLR4 in HME and A549 cells. We also demonstrate that stimulation with LPS leads to the recruitment of LYN to EGFR, and this association is blocked by erlotinib, indicating that the kinase activity of EGFR is required. Erlotinib also blocks the LPS-stimulated recruitment of LYN to TLR4, revealing that the kinase activity of EGFR is also necessary for this association. The involvement of LYN in an LPS mediated pathway was reported earlier (43, 47, 57). In this study, we observed the involvement of LYN specifically in the activation of EGFR in response to LPS and also in the activation of TLR4 in response to EGF, and conclude that LYN is a key kinase in establishing cross talk between EGFR and TLR4, leading to downstream signaling (Fig. 3C). Additional studies are necessary to explore further details of how LYN functions in response to LPS in mediating TLR4-dependent signaling. In particular, because EGFR and TLR4 do not bind to each other in response to LPS, and because LYN can be activated by oligomerization of TLR4 alone, it is not clear why EGFR is needed in order for LPS to activate NFκB.
LPS, a potent immunostimulatory component of Gram-negative bacteria, can induce systemic inflammation and sepsis (58) by triggering the release of many cytokines, including TNFα, IL-1β, and IL-6 (59). We now demonstrate that treatment of mice with erlotinib inhibits the production of inflammatory cytokines following LPS administration. It is noteworthy that erlotinib, a well known drug used extensively in cancer treatment, is also beneficial in suppressing the inflammatory signal triggered by LPS. Importantly, we also show that erlotinib protects mice from LPS-mediated lethality. Because too much activation of LPS/TLR4 signaling can lead to acute endotoxicity and chronic inflammatory disorders, our findings highlight the potential utility of erlotinib in inhibiting these devastating responses to infection. Septic shock is a complex disease for which preventive and therapeutic strategies are unfortunately lacking. Developing a better understanding of its pathophysiology underpins the development of more efficacious management regimes. Therefore, further investigation of the use of erlotinib, or other EGFR inhibitors, to modulate LPS-mediated endotoxicity may contribute to the development of a novel strategy for therapeutic intervention to ameliorate septic shock in the future.
Materials and Methods
The human mammary epithelial cell line hTERT-HME1, from Clontech, and the human cancer cell line A549, from American Tissue Culture Collection, were used to show that knockdown of TLR4 prevented activation of NFκB in response to EGF and that TLR4 was phosphorylated by EGFR. HME1 cells expressing BCL2 were used to show that activation of NFκB by LPS-TLR4 requires the kinase activity of EGFR. Inhibitors and knockdown experiments showed that LYN is required for signaling in both directions. C57BL/6J mice from the Jackson Laboratory were used to show that inhibiting EGFR blocked IL-6 and TNFα expression in response to LPS, using ELISA assays, and that pretreatment with an EGFR inhibitor protected the mice from LPS-mediated endotoxicity. Experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the Cleveland Clinic. Detailed materials and methods are provided in SI Materials and Methods.
SI Materials and Methods
Cells and Reagents.
Human mammary epithelial cells hTERT-HME (Clontech) were grown in Medium 171 with mammary epithelial growth supplement (Invitrogen), 50 U/mL penicillin, and 50 μg/mL streptomycin. The cancer cell line A549 was obtained from ATCC, and OVCAR3 cells were obtained from Daniel Lindner, Taussig Cancer Center, Cleveland Clinic, Cleveland, OH. A549 cells were maintained in DMEM supplemented with 5% (vol/vol) heat-inactivated FBS, 50 U/mL penicillin, and 50 μg/mL streptomycin. OVCAR3 cells were maintained in RPMI medium supplemented with 10% (vol/vol) FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. All cultures were kept in 5% (vol/vol) CO2 at 37 °C. Antibodies against phospho-EGFR (Tyr1068), phospho-ERK (Thr202/Tyr204), ERK, phospho-AKT (Ser473), AKT, phospho-IKKα/β (Ser176/180), phospho-IκB (Ser32/36), Phospho-SRC (Tyr416), and MYD88 were from Cell Signaling. The antibody against phosphorylated TLR4 (Tyr674) was from Imgenex. Antibodies against p65, IκB, IKKα/β, TLR4, TAK1, LYN, and normal rabbit IgG were from Santa Cruz Biotechnology. The antibody against EGFR was from Bethyl Laboratories. Anti-Flag M2 antibody and anti β-actin antibody were from Sigma Aldrich. Human recombinant EGF was from Millipore and recombinant human IL-1β was from Peprotech. Lipopolysaccharide (Escherichia coli 055:B5) was from Sigma Aldrich. Erlotinib was from Santa Cruz Biotechnology and PP2 was from EMD Biosciences.
Constructs and Transfections.
For the knockdown of TLR4 in HME cells, and LYN in A549 cells, we used ON-TARGET plus SMART pool human TLR4 siRNAs or LYN siRNAs and nontargeted control siRNAs (Thermo Scientific). Cells at about 50% confluence were transfected with siRNA pools using the DharmaFECT1 reagent (Thermo Scientific) following the manufacturer’s instructions. After 72 h, immunoblotting experiments were performed. In shRNAs in the lentiviral vector pLKO-puro targeting TLR4 (NM_003266.2–1056s1c1, CCGGCGTTTG GTTCTGGGAG AATTTCTCGA GAAATTCTCC CAGAACCAAA CGTTTTTG) MYD88 (NM_002468.2–723s1c1, CCGGGCAGAG CAAGGAATGT GACTTCTCGA GAAGTCACAT TCCTTGCTCT GCTTTTT) EGFR (NM_005228.3–4682s1c1, CCGGGCTGAG AATGTGGAAT ACCTACTCGA GTAGGTATTC CACATTCTCA GCTTTTTG) LYN (NM_002350.2–318s21c1 CCGGGAGTGA CGATGGAGTA GATTTCTCGA GAAATCTACT CCATCGTCAC TCTTTTTG) were obtained from Sigma-Aldrich. The shRNA construct against TAK1 and the scrambled shRNA control were kind gifts from Paul J. Chiao, The University of Texas M. D. Anderson Cancer Center, Houston. The BCL-2 construct was a kind gift from Scott K. Durum, National Cancer Institute, Frederick, MD, whom we also thank for the suggestion to use BCL2 expression to prevent apoptosis. We obtained the full-length flag-tagged human TLR4 plasmid as a kind gift from Catherine Greene, Beaumont Hospital, Dublin, and recloned it in the lentiviral vector pLCMV-puro, a gift from Peter Chumakov, Cleveland Clinic, at the SnaB1 and BamH1 sites. To produce infectious virus, 293T cells were transfected transiently with pCMVDR8.2 and pVSV-G helper plasmids (gifts from D. Trono, University of Geneva), as well as the plasmid of interest, by using Lipofectamine Plus (Invitrogen). The virus produced was collected 24 and 48 h after infection, supplemented with 4 μg/mL polybrene, and used to infect cells.
mRNA Expression, Immunoblotting, and Immunoprecipitation.
To determine mRNA expression levels, total cellular RNA was extracted using a Qiagen kit. The RT reaction was performed using the SuperScript III First-Strand Synthesis System, and PCR was done using human TLR4-specific primers.
Immunoblotting was performed as described (4). For immunoprecipitations, cells were lysed in buffer containing 20 mM Hepes (pH 7.4), 150 mM NaCl, 1.5 mM MgCl2, 2 mM DTT, 2 mM EGTA, 1% Nonidet p-40, and a mixture of protease and phosphatase inhibitors (Roche Applied Science). After incubation on ice for 20 min, cell debris was removed by centrifugation. Equivalent amounts of cell lysates were precleared with rabbit IgG (IgG) agarose (Sigma) for 1 h, and then mixed with an antibody against LYN, or IgG as a control, at 4 °C overnight, with rotation. In another set of experiments, the precleared lysates were incubated with an antibody against EGFR or IgG as a control. Immunocomplexes were precipitated with protein A/G agarose beads (Santa Cruz). The beads were washed four times with IP buffer, and the immunoprecipitates were analyzed by the Western method.
IL-6 and TNFα ELISA.
The concentrations of IL6 and TNFα were measured in mouse plasma by using the Mouse IL-6 ELISA Ready-SET-Go (eBioscience) or Mouse TNF-alpha ELISA Ready-SET-Go (eBioscience) ELISA kit, following the instructions provided by the manufacturer.
Quantitative Real-Time PCR.
cDNA was synthesized from total RNA, using a random hexamer and SuperScript III (Invitrogen). Quantitative real-time PCR was performed in the AB 7300 Real Time PCR System, and the expression of mouse IL-6, TNFα, and CXCL1 (KC) mRNAs was examined by using the SYBR GREEN PCR Master Mix (Applied Biosystems). PCR amplifications were performed in triplicate. The reaction protocol included preincubation at 95 °C to activate FastStart DNA polymerase for 10 min, 40 cycles of amplification for 15 s each at 95 °C, and annealing for 60 s each at 60 °C. Levels of IL-6, TNFα and CXCL1 (KC) mRNAs were normalized to the level of actin mRNA. Primer sequences were designed using AlleleID 6.0. The following primers were used: mouse IL-6 forward, GGACCAAGACCATCCAATTC; mouse IL-6 reverse, ACCACAGTGAGGAATGTCCA; mouse TNFα forward, CAAAGGGAGAGTGGTCAGGT; mouse TNFα reverse, ATTGCACCTCAGGGAAGAGT; mouse CXCL1 forward, TAGGGTGAGGACATGTGTGG; mouse CXCL1 reverse, AAATGTCCAAGGGAAGCGT; mouse actin forward, GGTCATCACTATTGGCAACG; mouse actin reverse, ACGGATGTCAACGTCACACT.
Confocal Microscopy.
Cells were grown on glass coverslips. After fixation and permeabilization with 4% paraformaldehyde and 0.2% Triton X-100 (15 min each), the samples were blocked with 5% normal goat serum. To detect EGFR, the cells were labeled with rabbit monoclonal anti-EGFR (D38B1, Cell Signaling) and goat anti-rabbit–Alexa Fluor 568 (Life technologies). For TLR4 detection, the cells were stained with mouse monoclonal anti-TLR4 (76B357.1, Abcam), followed by biotin-XX–labeled goat anti-mouse and Streptavidin-conjugated Alexa Fluor 488 (Life technologies). Coverslips were mounted on glass slides in VECTASHIELD/DAPI (4′,6-diamidino-2-phenylindole), and images were taken using a Leica TCS-SP5 confocal microscope (Leica Microsystems).
Erlotinib Treatment and LPS Injection.
C57BL/6J mice (6–8 wk old) were purchased from the Jackson Laboratory and kept under standard laboratory conditions. Mice were housed in a temperature-controlled room with a 12-h light/dark cycle and allowed food and water ad libitum.
Erlotinib HCl (OSI-744), purchased from Selleckchem was suspended in polyethylene glycol (PEG) 400. Mice were pretreated with an erlotinib suspension at a dosage of 100 mg/kg body weight, or vehicle PEG400, by oral administration 16 h before LPS injection. LPS (Escherichia coli 055:B5; Sigma) was suspended in DMEM before use, and injected intraperitoneally (i.p.) into mice (10 mg/kg body weight; single doses). Control mice received an equivalent volume of medium i.p. A second dose of erlotinib (100 mg/kg) or vehicle was administered at the same time as LPS administration. At 6 h after administration of LPS or medium, mice were anesthetized, and blood and spleens were collected. Blood samples treated with EDTA, an anticoagulant, were immediately placed on ice and processed within 1 h. After centrifugation at 3,000 × g for 10 min, plasma was collected, and aliquots were stored at −80 °C until assayed for cytokine concentrations. Survival studies in mice were done in the Case Western Reserve University Mouse Metabolic Phenotyping Center. Mice were placed into three groups with similar mean body weights and were treated with erlotinib by oral gavage (100 mg/kg) or vehicle PEG. After 16 h, LPS (10 mg/kg) was administered intraperitoneally. The mice were treated with erlotinib once daily for 3 more days. Control mice received only erlotinib.
Splenocyte Isolation.
Mouse spleens were collected under sterile conditions and splenocytes were gently smashed through a nylon cell strainer into RPMI medium to create single-cell suspensions. Cells were centrifuged at 1,000 × g for 5 min, and the part of pellet was resuspended in 1 mL per spleen of ACK lysing buffer (Lonza) for 5 min at room temperature. The remaining white blood cells were used to analyze cytokine mRNAs by Quantitative real-time PCR.
Statistical Analysis.
Values are expressed as means ± SD. Data were analyzed using Student's t test, and P values of <0.05 were considered statistically significant. Survival data were analyzed by the Kaplan–Meier method and a log-rank test, using GraphPad Prism software.
Acknowledgments
We thank Maojing Yang for excellent technical assistance, Yuxin Wang for help with the manuscript and Judy Drazba of the Imaging Core of the Lerner Research Institute, Cleveland Clinic for help with confocal microscopy. This work was funded by National Institutes of Health Grant PO1 CA062220 (to G.R.S. and X.L.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1511794112/-/DCSupplemental.
References
- 1.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 2.Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006;210:171–186. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 3.Brown M, Cohen J, Arun P, Chen Z, Van Waes C. NF-kappaB in carcinoma therapy and prevention. Expert Opin Ther Targets. 2008;12(9):1109–1122. doi: 10.1517/14728222.12.9.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De S, Dermawan JK, Stark GR. EGF receptor uses SOS1 to drive constitutive activation of NFκB in cancer cells. Proc Natl Acad Sci USA. 2014;111(32):11721–11726. doi: 10.1073/pnas.1412390111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18(18):2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 6.Gilmore TD. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene. 2006;25(51):6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 7.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4):a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morandell S, Stasyk T, Skvortsov S, Ascher S, Huber LA. Quantitative proteomics and phosphoproteomics reveal novel insights into complexity and dynamics of the EGFR signaling network. Proteomics. 2008;8(21):4383–4401. doi: 10.1002/pmic.200800204. [DOI] [PubMed] [Google Scholar]
- 9.Okamoto I. Epidermal growth factor receptor in relation to tumor development: EGFR-targeted anticancer therapy. FEBS J. 2010;277(2):309–315. doi: 10.1111/j.1742-4658.2009.07449.x. [DOI] [PubMed] [Google Scholar]
- 10.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 11.Ingley E. Src family kinases: Regulation of their activities, levels and identification of new pathways. Biochim Biophys Acta. 2008;1784(1):56–65. doi: 10.1016/j.bbapap.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 12.Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23(48):7906–7909. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]
- 13.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13(11):460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 14.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7(5):353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 15.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 16.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 17.Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003;85(2):85–95. doi: 10.1016/s0165-2478(02)00228-6. [DOI] [PubMed] [Google Scholar]
- 18.Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. 2009;9(1):57–63. doi: 10.1038/nrc2541. [DOI] [PubMed] [Google Scholar]
- 19.Koff JL, Shao MX, Ueki IF, Nadel JA. Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1068–L1075. doi: 10.1152/ajplung.00025.2008. [DOI] [PubMed] [Google Scholar]
- 20.Yamashita M, et al. Epidermal growth factor receptor is essential for Toll-like receptor 3 signaling. Sci Signal. 2012;5(233):ra50. doi: 10.1126/scisignal.2002581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Medzhitov R, Janeway C., Jr Innate immunity. N Engl J Med. 2000;343(5):338–344. doi: 10.1056/NEJM200008033430506. [DOI] [PubMed] [Google Scholar]
- 22.Stoll LL, Denning GM, Weintraub NL. Endotoxin, TLR4 signaling and vascular inflammation: Potential therapeutic targets in cardiovascular disease. Curr Pharm Des. 2006;12(32):4229–4245. doi: 10.2174/138161206778743501. [DOI] [PubMed] [Google Scholar]
- 23.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42(2):145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 24.Akira S. Toll-like receptor signaling. J Biol Chem. 2003;278(40):38105–38108. doi: 10.1074/jbc.R300028200. [DOI] [PubMed] [Google Scholar]
- 25.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 26.Ferrantini M, Capone I, Belardelli F. Dendritic cells and cytokines in immune rejection of cancer. Cytokine Growth Factor Rev. 2008;19(1):93–107. doi: 10.1016/j.cytogfr.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 27.Khatami M. Developmental phases of inflammation-induced massive lymphoid hyperplasia and extensive changes in epithelium in an experimental model of allergy: Implications for a direct link between inflammation and carcinogenesis. Am J Ther. 2005;12(2):117–126. doi: 10.1097/01.mjt.0000143699.91156.21. [DOI] [PubMed] [Google Scholar]
- 28.Khatami M. ‘Yin and Yang’ in inflammation: Duality in innate immune cell function and tumorigenesis. Expert Opin Biol Ther. 2008;8(10):1461–1472. doi: 10.1517/14712598.8.10.1461. [DOI] [PubMed] [Google Scholar]
- 29.Smyth MJ, Godfrey DI, Trapani JA. A fresh look at tumor immunosurveillance and immunotherapy. Nat Immunol. 2001;2(4):293–299. doi: 10.1038/86297. [DOI] [PubMed] [Google Scholar]
- 30.Hsu D, et al. Toll-like receptor 4 differentially regulates epidermal growth factor-related growth factors in response to intestinal mucosal injury. Lab Invest. 2010;90(9):1295–1305. doi: 10.1038/labinvest.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shuyi Y, et al. Human beta-defensin-3 (hBD-3) upregulated by LPS via epidermal growth factor receptor (EGFR) signaling pathways to enhance lymphatic invasion of oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;112(5):616–625. doi: 10.1016/j.tripleo.2011.02.053. [DOI] [PubMed] [Google Scholar]
- 32.McElroy SJ, et al. Transactivation of EGFR by LPS induces COX-2 expression in enterocytes. PLoS One. 2012;7(5):e38373. doi: 10.1371/journal.pone.0038373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hattar K, et al. Endotoxin induces proliferation of NSCLC in vitro and in vivo: Role of COX-2 and EGFR activation. Cancer Immunol Immunother. 2013;62(2):309–320. doi: 10.1007/s00262-012-1341-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trussoni CE, Tabibian JH, Splinter PL, O’Hara SP. Lipopolysaccharide (LPS)-Induced Biliary Epithelial Cell NRas Activation Requires Epidermal Growth Factor Receptor (EGFR) PLoS One. 2015;10(4):e0125793. doi: 10.1371/journal.pone.0125793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420(6917):885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 36.Doi K, Leelahavanichkul A, Yuen PS, Star RA. Animal models of sepsis and sepsis-induced kidney injury. J Clin Invest. 2009;119(10):2868–2878. doi: 10.1172/JCI39421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McNamara MJ, Norton JA, Nauta RJ, Alexander HR. Interleukin-1 receptor antibody (IL-1rab) protection and treatment against lethal endotoxemia in mice. J Surg Res. 1993;54(4):316–321. doi: 10.1006/jsre.1993.1050. [DOI] [PubMed] [Google Scholar]
- 38.Tracey KJ, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. 1987;330(6149):662–664. doi: 10.1038/330662a0. [DOI] [PubMed] [Google Scholar]
- 39.Fisher CJ, Jr, et al. The Soluble TNF Receptor Sepsis Study Group Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. N Engl J Med. 1996;334(26):1697–1702. doi: 10.1056/NEJM199606273342603. [DOI] [PubMed] [Google Scholar]
- 40.Fisher CJ, Jr, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA. 1994;271(23):1836–1843. [PubMed] [Google Scholar]
- 41.Medzhitov R, Janeway CA., Jr Innate immune recognition and control of adaptive immune responses. Semin Immunol. 1998;10(5):351–353. doi: 10.1006/smim.1998.0136. [DOI] [PubMed] [Google Scholar]
- 42.Takaesu G, et al. TAK1 is critical for IkappaB kinase-mediated activation of the NF-kappaB pathway. J Mol Biol. 2003;326(1):105–115. doi: 10.1016/s0022-2836(02)01404-3. [DOI] [PubMed] [Google Scholar]
- 43.Medvedev AE, et al. Role of TLR4 tyrosine phosphorylation in signal transduction and endotoxin tolerance. J Biol Chem. 2007;282(22):16042–16053. doi: 10.1074/jbc.M606781200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fiorotto R, et al. Loss of CFTR affects biliary epithelium innate immunity and causes TLR4-NF-kappaB-mediated inflammatory response in mice. Gastroenterology. 2011;141(4):1498–1508. doi: 10.1053/j.gastro.2011.06.052. e1491–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu W, Graves LM, Gill GN, Parsons SJ, Samet JM. Src-dependent phosphorylation of the epidermal growth factor receptor on tyrosine 845 is required for zinc-induced Ras activation. J Biol Chem. 2002;277(27):24252–24257. doi: 10.1074/jbc.M200437200. [DOI] [PubMed] [Google Scholar]
- 46.Sutton P, Borgia JA, Bonomi P, Plate JM. Lyn, a Src family kinase, regulates activation of epidermal growth factor receptors in lung adenocarcinoma cells. Mol Cancer. 2013;12:76. doi: 10.1186/1476-4598-12-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Avila M, Martinez-Juarez A, Ibarra-Sanchez A, Gonzalez-Espinosa C. Lyn kinase controls TLR4-dependent IKK and MAPK activation modulating the activity of TRAF-6/TAK-1 protein complex in mast cells. Innate Immun. 2012;18(4):648–660. doi: 10.1177/1753425911435265. [DOI] [PubMed] [Google Scholar]
- 48.Janssens S, Beyaert R. A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem Sci. 2002;27(9):474–482. doi: 10.1016/s0968-0004(02)02145-x. [DOI] [PubMed] [Google Scholar]
- 49.Irie T, Muta T, Takeshige K. TAK1 mediates an activation signal from toll-like receptor(s) to nuclear factor-kappaB in lipopolysaccharide-stimulated macrophages. FEBS Lett. 2000;467(2-3):160–164. doi: 10.1016/s0014-5793(00)01146-7. [DOI] [PubMed] [Google Scholar]
- 50.Stover DR, Becker M, Liebetanz J, Lydon NB. Src phosphorylation of the epidermal growth factor receptor at novel sites mediates receptor interaction with Src and P85 alpha. J Biol Chem. 1995;270(26):15591–15597. doi: 10.1074/jbc.270.26.15591. [DOI] [PubMed] [Google Scholar]
- 51.Biscardi JS, et al. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem. 1999;274(12):8335–8343. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- 52.Biscardi JS, Ishizawar RC, Silva CM, Parsons SJ. Tyrosine kinase signalling in breast cancer: Epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Res. 2000;2(3):203–210. doi: 10.1186/bcr55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lin WN, et al. Involvement of MAPKs and NF-kappaB in LPS-induced VCAM-1 expression in human tracheal smooth muscle cells. Cell Signal. 2007;19(6):1258–1267. doi: 10.1016/j.cellsig.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 54.Lin WN, Luo SF, Wu CB, Lin CC, Yang CM. Lipopolysaccharide induces VCAM-1 expression and neutrophil adhesion to human tracheal smooth muscle cells: Involvement of Src/EGFR/PI3-K/Akt pathway. Toxicol Appl Pharmacol. 2008;228(2):256–268. doi: 10.1016/j.taap.2007.11.026. [DOI] [PubMed] [Google Scholar]
- 55.Finzi L, Shao MX, Paye F, Housset C, Nadel JA. Lipopolysaccharide initiates a positive feedback of epidermal growth factor receptor signaling by prostaglandin E2 in human biliary carcinoma cells. J Immunol. 2009;182(4):2269–2276. doi: 10.4049/jimmunol.0801768. [DOI] [PubMed] [Google Scholar]
- 56.Basu S, et al. Helicobacter pylori protein HP0175 transactivates epidermal growth factor receptor through TLR4 in gastric epithelial cells. J Biol Chem. 2008;283(47):32369–32376. doi: 10.1074/jbc.M805053200. [DOI] [PubMed] [Google Scholar]
- 57.Henricson BE, Carboni JM, Burkhardt AL, Vogel SN. LPS and Taxol activate Lyn kinase autophosphorylation in Lps(n), but not in Lpsd), macrophages. Mol Med. 1995;1(4):428–435. [PMC free article] [PubMed] [Google Scholar]
- 58.Miyake K. Innate recognition of lipopolysaccharide by Toll-like receptor 4-MD-2. Trends Microbiol. 2004;12(4):186–192. doi: 10.1016/j.tim.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 59.Shimizu K, et al. Hepatocyte growth factor inhibits lipopolysaccharide-induced oxidative stress via epithelial growth factor receptor degradation. Arterioscler Thromb Vasc Biol. 2012;32(11):2687–2693. doi: 10.1161/ATVBAHA.112.300041. [DOI] [PubMed] [Google Scholar]