

Abstract
Remyelination is a potent regenerative process in demyelinating diseases, such as multiple sclerosis, the effective therapeutic promotion of which will fill an unmet clinical need. The development of pro-regenerative therapies requires the identification of key regulatory targets that are likely to be involved in the integration of multiple signaling mechanisms. Fibroblast growth factor (FGF) signaling system, which comprises multiple ligands and receptors, potentially provides one such target. Since the FGF/FGF receptor (FGFR) interactions are complex and regulate multiple diverse functions of oligodendrocyte lineage cells, it is difficult to predict their overall therapeutic potential in the regeneration of oligodendrocytes and myelin. Therefore, to assess the integrated effects of FGFR signaling on this process, we simultaneously inactivated both FGFR1 and FGFR2 in oligodendrocytes and their precursors using two Cre-driver mouse lines. Acute and chronic cuprizone-induced or lysolecithin-induced demyelination was established in Fgfr1/Fgfr2 double knockout mice (dKO). We found that in the acute cuprizone model, there was normal differentiation of oligodendrocytes and recovery of myelin in the corpus callosum of both control and dKO mice. Similarly, in the spinal cord, lysolecithin-induced demyelinated lesions regenerated similarly in the dKO and control mice. In contrast, in the chronic cuprizone model, fewer differentiated oligodendrocytes and less efficient myelin recovery were observed in the dKO compared to control mice. These data suggest that while cell-autonomous FGF signaling is redundant during recovery of acute demyelinated lesions, it facilitates regenerative processes in chronic demyelination. Thus, FGF-based therapies have potential value in stimulating oligodendrocyte and myelin regeneration in late-stage disease.
Keywords: oligodendrocyte, myelin
INTRODUCTION
Multiple sclerosis (MS) is a demyelinating disease of the central nervous system. In early stages of MS, remyelination can occur relatively robustly but becomes less efficient as the disease progresses, contributing to the later chronic progressive phase of the disease. Given the potential for endogenous repair, it is important to uncover the molecular mechanisms that contribute to successful repopulation of the lesions by oligodendrocytes and myelin recovery in order to identify therapeutic targets. This is especially important for chronically demyelinated lesions, which characterize the currently untreatable later progressive phase of the disease (Franklin, 2002; Franklin and Gallo, 2014).
FGFs are a family of growth factors that serve diverse functions in multiple cell types. We have shown that oligodendrocyte lineage cells express three of the four FGFRs, which regulate different aspects of oligodendrocyte development and myelination in a developmentally regulated manner (Bansal et al., 1996; Oh et al., 2003; Fortin et al., 2005; Kaga et al., 2006; Furusho et al, 2011; 2012). FGF signaling is also likely to play an important role during the regeneration of oligodendrocytes and myelin, since FGF2 and/or FGFR is upregulated in MS patients and in mouse models of demyelination (Hinks and Franklin, 1999; Messersmith et al., 2000; Clemente et al., 2011; Dutta and Trapp, 2011; Gudi et al., 2011). However, it remains unclear whether the presence of FGF2 and its receptors in and around demyelinated lesions is indicative of increased or decreased repair capacity, since both beneficial and detrimental effects of FGF2 perturbation have been reported (Ruffini et al., 2001; Armstrong et al, 2002; 2006; Rottlaender et al., 2011; Dehghan et al., 2012). Further, since, in addition to oligodendrocytes and their progenitors, other FGFR-expressing cell types, including reactive astrocytes and microglia, are also likely to be affected by modulation of FGF2 in the lesions, it is difficult to assess whether the outcome of FGF perturbation on oligodendrocyte differentiation and subsequent myelin recovery is due to direct or indirect effects on oligodendrocyte lineage cells.
Recently, it has been reported that conditional loss of FGFR1 signaling in oligodendrocyte progenitor cells (OPCs) and oligodendrocytes results in enhanced OPC differentiation and remyelination in the chronic cuprizone model of demyelination (Zhou et al., 2012). However, while FGFR1 is expressed in both OPCs and oligodendrocytes, FGFR2 is the most abundant FGF receptor that is specifically upregulated in mature oligodendrocytes, concomitant with their differentiation (Bansal et al., 1996; Fortin et al., 2005). Thus, deletion of a single FGFR gene is unlikely to reflect the overall impact of FGF signaling on oligodendrocyte differentiation, especially because FGFR1 negatively regulates FGFR2 and because the two receptors are known to produce opposing cellular responses in oligodendrocyte lineage cells (Fortin et al., 2005). We therefore investigated the overall effect of FGFR1 and FGFR2 signaling, focusing largely on the regeneration of oligodendrocytes, in the acute and chronic cuprizone-induced mouse models of demyelination, established in mice lacking both Fgfr1 and Fgfr2 in OPCs and oligodendrocytes. The data suggest that in the acute model, the overall effect of FGFR1/FGFR2 signaling is neither beneficial nor detrimental for oligodendrocyte differentiation. In contrast, in the chronic model, it is beneficial in promoting the endogenous repair process. These studies also emphasize the importance of integrated signaling via both FGFR1 and FGFR2 in elucidating the overall cell-autonomous impact of FGF signaling during the recovery of oligodendrocytes following demyelination.
MATERIALS AND METHODS
Generation of Fgfr1/Fgfr2 double conditional knockout mice
We generated conditional double knockout mice by mating Fgfr1flox/flox;Fgfr2flox/flox (Pirvola et al., 2002; Yu et al., 2003) with CNPcre/+ (2’,3’-cyclic nucleotide 3’-phosphohydrolase; Lappe-Siefke et al., 2003) mice to produce progeny with disrupted Fgfr1 and Fgfr2 genes in CNP-expressing oligodendrocyte-lineage cells and Schwann cells as described previously (Kaga et al., 2006; Furusho et al., 2009; Wang et al., 2009; Furusho et al., 2012). To confirm and extend findings from the Fgfr1flox/flox;Fgfr2flox/flox;CNPcre/+ line, we also generated a second line of conditional Fgfr1/Fgfr2 conditional double knockout mice by mating Fgfr1flox/flox;Fgfr2flox/flox with Olig1cre/+ mice (Lu et al., 2002), to produce progeny in which disruption of Fgfr1 and Fgfr2 genes occurs in Olig1-expressing OPCs even earlier in the lineage than in the CNP-Cre line. The genetic backgrounds of Fgfr1flox/flox;Fgfr2flox/flox lines is 129/Sv, Olig1cre line is CD1 and the CNPcre line is C57BL/6. The mutant mice of genotypes, Fgfr1flox/flox;Fgfr2flox/flox;CNPcre/+ and Fgfr1flox/flox;Fgfr2flox/flox;Olig1cre/+ will be referred to as Fgfr1/Fgfr2-CNPCre and Fgfr1/Fgfr2-Olig1Cre dKO, respectively. Littermates of these mutants lacking Cre will be referred to as “controls”. The conditional loss of FGFR1 and FGFR2 in Fgfr1/Fgfr2 dKO mice was confirmed by PCR, immunoblotting, in situ hybridization and analysis of reporter mice as described previously (Kaga et al., 2006; Furusho et al., 2009; Wang et al., 2009; Furusho et al., 2012).
Demyelination Models
Cuprizone-mediated demyelination model has been extensively described previously (Matsushima and Morell, 2001; Mason et al., 2001; Armstrong et al., 2002; 2006; Skripuletz et al., 2011). Briefly, finely powdered cuprizone (Sigma, St. Louis, MO) was mixed into milled chow and fed daily to 8 week old mutant male mice and their littermate controls for 4 weeks to assess the extent of demyelination. The dose of cuprizone needed to induce reproducible demyelination throughout the corpus callosum without toxicity was first determined for both the mutant lines and was found to be 0.2% for Fgfr1/Fgfr2-CNPCre and 0.25% for Fgfr1/Fgfr2-Olig1Cre line. To assess oligodendrocyte and myelin recovery, mice were fed cuprizone for either 6 weeks (acute demyelination model) or for 12 weeks (chronic demyelination model) and then restored to normal chow for 3 weeks before analysis as described previously for the FGF2-null mice (Armstrong et al., 2002; 2006). Matched coronal sections of the corpus callosum from control and dKO mice were analyzed in the region between the cingulum apexes overlying the fimbria-fornix.
Lysolecithin-mediated focal demyelination was induced as described previously (Pavelko et al., 1998; Shields et al., 1999; O'Leary et al., 2002). Briefly, five-month-old Fgfr1/Fgfr2-CNPCre dKO and littermate control mice of either sex were anesthetized and injected with 2ul of 1% solution of lysolecithin (Sigma, St.Louis, MO) in sterile 0.9% NaCl into the ventral-lateral column of the spinal cord around the T12-T13 thoracic region. Immediately after injection, the wounds were closed using suture clips. The day of lysolecithin injection was designated as day 0. Mice were perfused with 4% glutaraldehyde at 14 days post-injection. The spinal cords were embedded in resin, and 1 micrometer sections were stained with toluidine blue. Remyelinated axons within the lesions are readily distinguished from normally myelinated axons outside of the lesions by the relative thinness of their myelin sheaths. Using this criteria, extent of remyelination was assessed in the lesions by blind ranking of each semi-thin section obtained from the center of each lesion, the highest rank representing most remyelination. Statistical analysis was performed using a Mann Whitney U-test.
In situ Hybridization
Mice were anesthetized and perfused with 4% paraformaldehyde in PBS (PFA/PBS) and brains were post fixed in 4% PFA/PBS overnight at 4ºC. Following overnight cryoprotection in 20% sucrose in PBS, the tissues were embedded in OCT compound (Sakura Finetechnical Co. Ltd., Tokyo, Japan). Cryosections (15μm) were thaw-mounted onto slide glasses (Fisher scientific, NA, USA), and stored at −30ºC until use.
In situ hybridization was carried out with slight modification of the procedure previously described (Kaga et al., 2006). Riboprobes used were specific for proteolipid protein (PLP) mRNA (Dr. W.B. Macklin, Univ. of Colorado Sch. of Med., Aurora, CO), myelin basic protein (MBP) mRNA (Dr. M. Qiu, Univ. of Louisville, Kentucky) and platelet-derived growth factor receptor-α (Pdgfra) mRNA (Dr. W.D. Richardson, Univ. Collage London, UK). Sections were post-fixed with 4% PFA for 15 min, washed in PBS and incubated in 1 ug/ml proteinase K at 37°C for 30 min. Sections were fixed with 4% PFA and washed with PBS again. Hybridization for each mRNAs was performed overnight at 65 °C or 70°C by using digoxigenin-labeled antisense riboprobes in a solution containing 50% formamide, 5×SSC (750 mM NaCl, 75 mM Sodium Citrate) and 1% SDS. After hybridization, the sections were washed in 50% formamide, 2×SSC and 1% SDS at 65 °C followed by rinses in maleic acid buffer at room temperature. After blocking for nonspecific binding in blocking buffer [phosphate buffered saline, pH7.4, with 0.1% Triton X-100 (TX100), 0.2% bovine serum albumin and 10% normal goat serum (NGS)] for 1 hr, sections were incubated overnight in alkaline phosphatase-conjugated-antidigoxigenin antibody (1:2000; Roche Diagnostics, Penzberg, Germany). Color development in the presence of 4-nitroblue tetrazolium chloride, 5-bromo-4-chloro-3-indolylphosphate was performed in the dark at room temperature. The sections were washed in 1 mM EDTA pH8 and mounted with mount media.
Coronal sections of brain from control and mutant mice were matched using anatomical landmarks and comparisons were made within the same litters. The total numbers of PLP mRNA+ or Pdgfra mRNA+ cells were counted in the corpus callosum region between the cingulum apexes overlying the fimbria-fornix from three sections each from 3–6 control and mutant mice. Statistical significance was determined using unpaired Student’s t-test.
Immunohistochemistry and Histology
Cryosections were prepared as described above. Following antigen retrieval by heat treatment (95ºC, 5 min), sections were blocked in 10% normal goat serum, 0.2% TX100 for 1 hr and then incubated overnight (4ºC) in mouse CC1 antibody (1:40; Calbiochem, Germany). Sections were then incubated in goat anti-mouse Cy3 (1:500; Jackson Immuno Research, West Grove, PA) for 1 hr. For immuno-labeling for MBP, sections were immersed in 100% ethanol, washed in PBS, blocked for 1hr in 10% normal goat serum, 5% bovine serum albumin, and 0.1% gelatin in PBS and incubated overnight (4°C) in polyclonal anti-rabbit MBP (1:3,000; R. Bansal, Farmington, CT). After three washes in PBS, the sections were incubated for 1hr in goat anti-rabbit IgG conjugated to Alexa 488 (1:500; Molecular Probes, Eugene, OR). For immuno-labeling the myelin protein oligodendrocyte glycoprotein (MOG) the sections were incubated in 100% ethanol for 10 min, then immersed in 0.1% H2O2 in PBS for 30 min to inactivate endogenous peroxidase. Next the sections were blocked and incubated overnight (4ºC) in mouse anti-MOG antibody (1:1000; gift from Dr. C.Linington, Aberdeen, UK). Sections were then incubated in biotinylated anti-rabbit IgG (1:200; Vector Laboratories, Burlingame, CA) for 1 hr and in the ABC reagents (VECTASTAIN Elite ABC kit: Vector Laboratories) for 40 min at room temperature before color development with 0.05% 3-3-diaminobenzidine / 0.015% H2O2 (DAB; Research Genetics, Huntsville, AL). Parallel sections processed identically but without the incubation in the primary antibody served as negative controls. Sections were stained with Luxol Fast Blue (LFB) according to the standard Klüver-Barrera protocol (Klüver and Barrera, 1953). Briefly, the frozen sections were dehydrated, de-fated with methanol/chloroform (1:1) overnight, hydrated back in 95% ethanol and then incubated in LFB staining solution (0.1% in 95% ethanol with 0.5% acetic acid) at 56ºC for 4h. The sections were differentiated in lithium carbonate solution (0.05%), rinsed in 70% ethanol, and then incubated in 0.5% periodic acid for periodic acid-Schiff staining (pink color). The sections were then incubated in Schiff reagent (SIGMA) and rinsed in lukewarm tap water, dehydrated through 100% ethanol, cleared in Xylene and mounted with Permount (Fisher scientific).
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA) from corpus callosum tissue micro-dissected from the brains. 1ug total RNA was reverse transcribed to cDNA using the iScriptTM Synthesis Kit (BioRad, Hercules, CA) according to the manufacture’s instructions. qRT-PCR was performed using an Eppendorf Mastercycler ep realplex Thermal Cycler (Eppendorf, Hamburg, Germany) and the iQTM SYBR Green Supermix (BioRad, Hercules, CA) according to the manufacture’s instructions. The following primers were used: MBP forward primer, 5’-TACCTGGCCACAGCAAGTAC -3’; MBP reverse primer, 5’-GTCACAATGTTCTTGAAG -3’; Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward primer, 5’-TGTGTCCGTCGTGGATCTG -3’; GAPDH reverse primer, 5’-CATGTAGGCCATGAGGTCCACCAC -3’. qRT-PCR conditions were as follows: denaturation at 95ºC, 30 s; primer annealing at 55.5ºC, 30 s; elongation at 72ºC, 40 s. Quantification of PCR products was performed using the 2- ΔΔCt method. Quantities of mRNA were normalized to the housekeeping gene GAPDH.
Cell Culture and Immuno-labeling
Dissociated cultures of forebrains were prepared individually from mutant and littermate control pups at P2. After removing meninges, the forebrains were chopped with a blade, trypsinized (0.025%) for 30 min at 37ºC, gently triturated and plated at a density of 50,000/cm2 on polyl-D-lysine coated four-well dishes (50 mg/ml; Sigma-Aldrich) in 4% fetal calf serum (FCS)/DMEM. After 17h, the medium was changed to defined media [DMEM with100 ug/ml transferrin, 5.2 ng/ml sodium selenium, 5 ug/ml insulin, 8.8 ug/ml putrescine, 6 ng/ml progesterone, 10,000 units/ml penicillin and 10,000 ug/ml streptomycin (all ingredients from Sigma)] plus 0.5% FCS and cells were grown with half medium changes every 2–3 days.
Forebrain cultures were immunolabeled, as described previously (Furusho et al., 2011; Guardiola-Diez et al., 2012) with marker antibodies of total oligodendrocyte-lineage cells, O4 (1:25; Bansal et al., 1992); immature oligodendrocyte, O1 (1:25; anti-galactocerebroside) or HPC7 (1:25; Baas and Barnstable, 1998; HPC7 antibody labels immature oligodendrocytes similar to O1, Guardiola-Diaz et al., 2012); and mature oligodendrocyte, anti-MBP (1:100; Sternberger Monoclonal Inc., Lutherville, MD).
Immunoblotting
Immunoblotting was performed as described previously (Fortin et al., 2005). Briefly, equal amounts of total proteins from forebrain homogenates were loaded on SDS-PAGE, transferred to PVDF membrane, and immunolabeled for MBP (1:10,000, E. Barbarese, Farmington, CT) and beta-actin (1:5000, Sigma, MO) as a loading control. The MBP isoforms and beta-actin identified on immunoblots were quantified by densitometry and expressed as MBP/beta-actin for each sample.
RESULTS
Disruption of FGFR1/FGFR2 signaling does not affect oligodendrocyte differentiation in the untreated postnatal brain or in cultures initiated from mutant brains
We have previously shown that differentiation of OPCs in the postnatal spinal cord is not affected in either Fgfr1/Fgfr2-CNPCre or Fgfr1/Fgfr2-Olig1Cre double knockout mice (Furusho et al, 2012). In view of the raising concept that oligodendrocytes may constitute a heterogeneous population, it is plausible that there may be regional differences in their response to the loss of FGFR1/FGFR2 signaling between the spinal cord and forebrain. Therefore, prior to examining the effect of Fgfr1/Fgfr2 ablation on oligodendrocyte differentiation during remyelination, we examined the effects of their ablation on oligodendrocyte differentiation during normal developmental myelination in the forebrain. For this, we analyzed the forebrains of control and mutant mice from the untreated CNP-Cre line and also from the Olig1-Cre driver line in which Fgfr1/Fgfr2 were ablated from OPCs even more efficiently and at an earlier stage than in the CNP-Cre line. The complete ablation of Fgfr1 in OPCs was previously confirmed by qRT-PCR analysis of isolated OPCs from neonatal forebrains of Fgfr1/Fgfr2 dKOs (Furusho et al., 2012), and ablation of FGFR2 was demonstrated by immunoblotting, showing reduced expression of FGFR2 protein in Fgfr1/Fgfr2 dKOs and Fgfr2 KO (Kaga et al., 2006; Furusho et al., 2009; Wang et al., 2009; Furusho et al., 2012). To examine the differentiation of OPCs into oligodendrocytes, we used PLP in situ hybridization as markers of differentiated oligodendrocytes in the corpus callosum of Fgfr1/Fgfr2-CNPCre and Fgfr1/Fgfr2-Olig1Cre mutants and littermate controls on postnatal Day 8 (P8), when active oligodendrocyte differentiation is going on in the forebrain. We also compared the numbers of oligodendrocytes in the 4-month old control and mutant mice (Fig. 1A, B). Quantification of the numbers of oligodendrocytes showed that there were no significant differences in the numbers of differentiated oligodendrocytes in the corpus callosum at these ages in either of the two lines of mutant mice (Fig. 1C).
Figure 1. Disruption of FGFR1/FGFR2 signaling does not affect oligodendrocyte differentiation in the untreated postnatal brain.

Coronal brain sections of postnatal Day 8 (P8) and 4-month (4M)-old Fgfr1/Fgfr2-CNPCre (A) or Fgfr1/Fgfr2-Olig1Cre (B) dKO mice and their littermate controls were analyzed for the expression of PLP mRNA by in situ hybridization to identify differentiated oligodendrocytes. C. Quantification of the numbers of PLP mRNA+ oligodendrocytes at P8 and 4M in matched regions of the corpus callosum from control and dKO show that at both ages, the numbers of oligodendrocytes were similar between controls and dKOs in both the CNP-Cre and Olig1-Cre lines of mice. D. Coronal sections of Fgfr1/Fgfr2-Olig1Cre and control mice were analyzed for the expression of Pdgfra mRNA by in situ hybridization to identify OPCs. Quantification of the numbers in the white matter near the cingulum area shows no difference between the control and dKO mice. E. Dissociated cultures initiated from forebrains of newborn mutant and control mice were immuno-labeled at 7 or 15 days in vitro (DIV) with HCP7 or O1 to identify immature oligodendrocytes or anti-MBP to identify mature oligodendrocytes and O4 to identify total oligodendrocyte lineage cells. Quantification of the numbers of double immuno-labeled cells showed that the percentage of total oligodendrocyte lineage cells that were immature oligodendrocytes (%HCP7+/O4+) and the percentage of immature oligodendrocyte that were mature oligodendrocytes (%MBP+/O1+) did not differ between the control and mutant mice. N=6–9 cultures from individual brains of each genotype. Scale bar, 20 um. F. Coronal sections of control and mutant mice immunolabeled with anti-MBP at P8 show no obvious difference in the patterns of staining. Three sections per animal were analyzed from 3–4 mice per group. G. Immunoblot analysis and quantification of MBP levels in the forebrains of adult control and Fgfr1/Fgfr2-CNPCre mice show no difference between the two. Beta-actin is shown as a protein loading control. Error bars represent S.E.M. N=3. Scale bars, 200 um. cc, corpus callosum; cg, cingulum; ctx, cortex; Hipp, hippocampus.
To examine OPC numbers at P8, we performed in situ hybridization on coronal sections of the brain of Fgfr1/Fgfr2-Olig1Cre dKO and control mice for Pdgfra mRNA expression, a recognized marker of OPCs (Fig. 1D). Quantification of the numbers of Pdgfra mRNA+ cells showed that the numbers of OPCs were not affected in the mutants compared to controls. We also evaluated the numbers of OPCs in the corpus callosum of untreated adult control and Fgfr1/Fgfr2-CNPCre mice at ages corresponding roughly to the ages when cuprizone treated mice were evaluated. We found no significant differences between the control and mutant mice (Pdgfra+ cells at 3-3.5 months of age: Control: 204+/−26; Mutants: 190+/−13. N=3 each, p=0.666).
The effect of Fgfr1/Fgfr2 ablation on oligodendrocyte differentiation was also examined in vitro (Fig. 1E). Dissociated cultures initiated from forebrains of newborn Fgfr1/Fgfr2-Olig1Cre and control mice were immuno-labeled with antibodies that mark different stages of oligodendrocyte differentiation (HCP7 and O1 antibodies identify immature oligodendrocytes as they first enter terminal differentiation, and MBP identifies the next stage in the lineage when they become mature oligodendrocytes). Quantification of the numbers of immuno-labeled cells showed that the percentage of total oligodendrocyte lineage cells (marked by O4 antibody) that were at the immature oligodendrocyte stage (%HCP7+/O4+ cells) or the percentage of immature oligodendrocytes (O1+ cells) that were at the mature oligodendrocyte stage (%MBP+/O1+ cells) did not differ between the control and mutants suggesting that the differentiation and/or survival of oligodendrocytes at these stages were not affected in vitro.
Finally, we examined the effect of Fgfr1/Fgfr2 ablation on the initiation of myelination in the forebrains of Fgfr1/Fgfr2-CNPCre and control mice at P8. Coronal sections immunolabeled for the myelin protein MBP showed no obvious differences in the pattern of MBP expression between control and mutant mice (Fig. 1F). Further, evaluation of the levels of MBP by immunoblotting in the forebrains of untreated adult control and Fgfr1/Fgfr2-CNPCre mice at an age corresponding roughly to the age when cuprizone treated mice were evaluated also showed no difference between the control and mutant mice (Fig. 1G).
Taken together, we conclude from these in vivo and in vitro studies carried out in two independent lines of mice that, similar to the spinal cord, the loss of FGFR1/FGFR2 signaling in the forebrain does not adversely affect OPC numbers or the extent and timing of their differentiation into oligodendrocytes during developmental myelination.
Repopulation of acutely demyelinated lesions by oligodendrocytes remains unaffected in the forebrains of Fgfr1/Fgfr2-CNPCre conditional knockout mice
We next asked if loss of FGFR1/FGFR2 signaling in the forebrain had an effect on the repopulation of acutely demyelinated lesions by oligodendrocytes. We employed the well-established acute cuprizone model of CNS demyelination with spontaneous remyelination (Matsushima and Morell, 2001; Mason et al., 2001). In this model, cuprizone ingestion results in oligodendrocyte and myelin loss in the mouse forebrain. Demyelinated lesions develop primarily in the corpus callosum following cuprizone ingestion, which is followed by spontaneous recovery of oligodendrocytes and myelin upon its removal from the diet. The new oligodendrocytes are generated by differentiation of OPCs that are not adversely affected by cuprizone feeding. Using this model, we first evaluated the extent of oligodendrocyte loss (PLP mRNA+ cells) and demyelination (LFB myelin staining) in the corpus callosum of control and Fgfr1/Fgfr2-CNPCre mice that were kept on a daily diet of chow containing 0.2% cuprizone for 4 weeks. We found that this dose consistently led to similar losses of oligodendrocytes and myelin in the corpus callosum of control and dKO mice (Fig. 2A, B, C; 4 wks Cupz), indicating that the mutant mice demyelinated similar to controls.
Figure 2. Repopulation of acutely demyelinated lesions by oligodendrocytes remain unaffected in Fgfr1/Fgfr2-CNPCre conditional knockout mice.

Control and Fgfr1/Fgfr2- CNPCre mice were fed 0.2% cuprizone (Cupz) for 4 weeks (wks) or 6 wks followed by normal chow for 3 wks. A. Coronal sections of the brains analyzed by PLP mRNA in situ hybridization or CC1 immuno-labeling to identify oligodendrocytes show that the loss of oligodendrocytes during demyelination (4 wks of Cup) or their recovery during remyelination (6 wks Cupz+3 wks no Cupz) was similar in controls and mutant mice. B. Quantification of PLP mRNA+ and CC1+ oligodendrocytes in the corpus callosum shows no significant differences in the numbers of oligodendrocytes between the controls and Fgfr1/Fgfr2-CNPCre dKO mice. Three sections per animal were counted from 3–5 mice per group. C. Analysis of coronal sections of the corpus callosum shows that myelin loss after 4 wks of cuprizone feeding (LFB staining, blue) and recovery of myelin protein (MOG immuno-labeling) and mRNA (MBP in situ hybridization) after 3 wks of recovery were also similar in controls and dKO mice. D. Quantification by qRT-PCR of the relative levels of MBP mRNA shows no difference between the control and dKO mice. Scale bars, 200 μm. Error bars represent S.E.M.; N=3–5.
We next assessed the ability of mutant OPCs to generate new oligodendrocytes and repopulate the acutely demyelinated lesions in the acute model of demyelination. Following a similar experimental paradigm that was used in a previous study for the evaluation of oligodendrocyte differentiation during the early stages of remyelination in FGF2-null mice (Armstrong et al., 2002), we fed Fgfr1/Fgfr2-CNPCre mice cuprizone for 6 weeks and analyzed them 3 weeks later after resuming a cuprizone-free diet. Oligodendrocytes in control and mutant mice were identified by PLP mRNA in situ hybridization and by CC1 immunolabeling (Fig. 2A), and their numbers were quantified (Fig. 2B) from matched corpus callosum regions between the cingulum apexes overlying the fimbria-fornix. We found that similar numbers of oligodendrocytes (PLP mRNA+ and CC1+ cells) were generated in the control and mutant mice, suggesting that oligodendrocyte differentiation was unaffected in the Fgfr1/Fgfr2-CNPCre dKO mice.
To assess the extent of myelin recovery in the acute model of demyelination, we immuno-labeled the coronal sections of the brain for the expression of myelin protein MOG (Fig. 2C, middle panel). Several previous studies have analyzed the expression level of different myelin protein in matched corpus callosum regions between the cingulum apexes overlying the fimbria-fornix as a gross measure of relative myelin recovery between control and mutant mice (Armstrong et al., 2002; 2006). Since this region is almost completely demyelinated by 4–6 weeks of cuprizone feeding, the appearance of MOG immunostaining in this region at 3 weeks of recovery indicates regenerated, rather than original, myelin. As an additional measure of myelin recovery, we compared the expression pattern of MBP mRNA (present in myelinated fibers) in the corpus callosum of control and mutant mice (Fig. 2C lower panel). No obvious differences were observed in MOG or MBP mRNA expression patterns between the control and Fgfr1/Fgfr2-CNPCre mice. Further, since these are not quantitative methods, we compared MBP mRNA expression in the corpus callosum of control and mutant mice by qRT-PCR (Fig. 2D). Once again, no obvious differences were observed between the control and Fgfr1/Fgfr2-CNPCre mice, suggesting that myelin recovery in the dKO mice occurred similarly as in the control mice.
To further confirm the results obtained from the analysis of Fgfr1/Fgfr2-CNPCre dKO mice, we examined oligodendrocyte differentiation during remyelination in another independent line of mice—i.e., the Olig1-Cre driver mice, in which Fgfr1/Fgfr2 were ablated in all Olig1-expressing oligodendrocyte lineage cells (Fig. 3).
Figure 3. Repopulation of acutely demyelinated lesions by oligodendrocytes remain unaffected in Fgfr1/Fgfr2-Olig1Cre conditional knockout mice.

Control and Fgfr1/Fgfr2- Olig1Cre mice were fed 0.25% cuprizone (Cupz) for 4 weeks (wks) or 6 wks followed by normal chow for 3 wks. A. Coronal sections of the brains, analyzed by PLP mRNA in situ hybridization to identify oligodendrocytes, show that the loss of oligodendrocytes during demyelination (4 wks of Cupz) or their recovery during remyelination (6 wks Cupz+3 wks no Cupz), respectively, was similar in controls and Fgfr1/Fgfr2-Olig1Cre dKO mice. B. Quantification of PLP mRNA+ oligodendrocytes in the corpus callosum shows no significant differences in the numbers of oligodendrocytes between the controls and Fgfr1/Fgfr2-Olig1Cre dKO mice. Three sections per animal were counted from 3–5 mice per group. C. Analysis of coronal sections of the corpus callosum by LFB staining shows that myelin loss after 4 wks of cuprizone feeding and recovery after 3 wks of normal diet were also similar in controls and dKO mice. Scale bars, 200 μm. Error bars represent S.E.M.’ N=3–6.
After 4 weeks of cuprizone feeding, both control and Fgfr1/Fgfr2-Olig1Cre mice showed similar losses of oligodendrocytes (Fig. 3A, B, PLP mRNA+ cells; 4 wks Cupz) and myelin (Fig. 3C, LFB staining, 4 wks Cupz). As was seen for the Fgfr1/Fgfr2-CNPCre mice, we found that after 3 weeks of cuprizone removal from the diet, the numbers of oligodendrocytes that were generated in Fgfr1/Fgfr2-Olig1Cre mice were similar to those in controls (Fig. 3A, B). Further, the recovery of myelin, examined at 3 weeks following 6 weeks of cuprizone ingestion, appeared comparable in the control and Fgfr1/Fgfr2-Olig1Cre mice (Fig. 3C, lower panels).
These data confirm the findings in the Fgfr1/Fgfr2-CNPCre dKO mice and together indicate that the combined loss of FGFR1/FGFR2 signaling in OPCs and oligodendrocytes does not positively or negatively affect the differentiation of oligodendrocytes during the recovery phase of acutely demyelinated lesions, as was observed during normal developmental myelination in the forebrain (Fig. 1).
Lysolecithin-induced demyelinated lesions remyelinate similarly in the spinal cords of Fgfr1/Fgfr2-CNPCre dKO and control mice
The acute cuprizone model (above) showed no perturbation of oligodendrocyte differentiation and myelin recovery in the corpus callosum of Fgfr1/Fgfr2-CNPCre dKO mice. Since the spinal cord is not demyelinated in this model, we next examined the lysolecithin-induced demyelination model to not only assess the role of FGFR1/FGFR2 signaling on remyelination in the spinal cord but also to use another independent model of demyelination. The lysolecithin-induced demyelination model is extensively used to reliably assess the onset and extent of remyelination by the examination of semi-thin sections at the light microscopic level (Pavelko et al., 1998; Shields et al., 1999; O'Leary et al., 2002). Remyelinated axons within the lesions are readily distinguished from normally myelinated axons outside of the lesions by the relative thinness of their myelin sheaths. Using these criteria, the extent of remyelination can be assessed semi-quantitatively in the lesions by blind ranking of each semi-thin section obtained from the center of each lesion, with the highest rank representing the most remyelination. We therefore induced focal demyelination in control and Fgfr1/Fgfr2-CNPCre mice by injection of lysolecithin into the lateral-ventral columns of the spinal cords. At 14 days post-injection, lesions in semi-thin sections of spinal cords were examined for the extent of remyelination, as described previously (Pavelko et al., 1998; Shields et al., 1999; O'Leary et al., 2002). The ranking analysis of semi-thin sections showed no statistically significant differences between the control and dKO mice (Fig. 4A, B), suggesting that similar to the forebrain, regeneration of myelin in the spinal cords of Fgfr1/Fgfr2-CNPCre dKO mice was comparable to that in the controls.
Figure 4. Lysolecithin-induced demyelinated lesions remyelinate similarly in the spinal cords of Fgfr1/Fgfr2-CNPCre dKO and control mice.
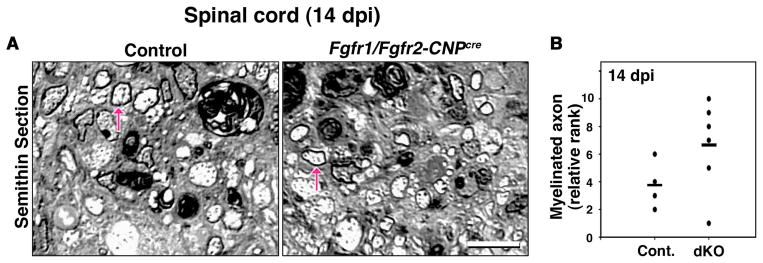
Focal demyelination was induced in control and Fgfr1/Fgfr2-CNPCre mice by the injection of lysolecithin into the lateral-ventral columns of the spinal cords. A. Representative images of toluidine blue-stained semi-thin cross-sections at 14 days postinjection (dpi) show that axons (arrows) which are similarly remyelinated in the lesioned area of the control and dKO mice. B. Quantification of the extent of remyelination by blind relative ranking analysis confirms that there is no statistically significant difference between the control and dKO mice. Mann-Whitney U-test p>0.1 (P=0.1356); N=4–6. The highest rank represents the most remyelination. Scale bars, 10 um.
Repopulation of chronically demyelinated lesions by oligodendrocytes is impaired in both CNP-Cre and Olig1-Cre lines of Fgfr1/Fgfr2 conditional knockout mice
We next investigated the effects of Fgfr1/Fgfr2 ablation on the regeneration of oligodendrocytes in chronically demyelinated lesions. Once again, following a similar experimental paradigm as was used for the evaluation of oligodendrocyte and myelin recovery in the FGF2-null mice (Armstrong et al., 2002), we fed cuprizone to Fgfr1/Fgfr2-CNPCre mice for 12 weeks and analyzed them 3 weeks later after resuming a cuprizone-free diet (Fig. 5). It has been shown previously that recovery of new oligodendrocytes after 12 weeks of cuprizone feeding (chronic demyelination model) was much less robust than after 6 weeks of feeding (acute demyelination model) and that the number of oligodendrocytes did not return to normal levels after 6 weeks of no cuprizone feeding in mixed strains of mice (Matsushima and Morell, 2001; Armstrong et al., 2006; Vana et al., 2007; Zhou et al., 2012), although complete remyelination was noted after 12 weeks of recovery in C57BL/6 mice following chronic demyelination (Lindner et al., 2009). The recovery of oligodendrocytes in our cuprizone fed control mice, which are of mixed strain, was much less in the chronic than in the acute model (compare Fig. 2B and 5B). Interestingly, comparison of the Fgfr1/Fgfr2-CNPCre and control mice in the chronic model showed that even fewer oligodendrocytes were present in the corpus callosum of dKO compared to control mice (Fig. 5A). This was confirmed by quantification of the numbers of PLP mRNA+ and CCI+ oligodendrocytes showing a ~70% reduction in the numbers of oligodendrocytes in the mutants compared to controls (Fig. 5B). Consistent with the reduced numbers of oligodendrocytes, the immuno-labeling of coronal sections of brains from control and dKO mice for the myelin protein MOG showed reduced expression in the brains of dKO compared to control mice (Fig. 5C). These data show a dramatic impairment of recovery by 3 weeks in the mutant mice compared to controls. However, the possibility remains that if the controls regained normal numbers of oligodendrocytes over time the relative recovery in the mutants could also improve.
Figure 5. Repopulation of chronically demyelinated lesions by oligodendrocytes is impaired in Fgfr1/Fgfr2-CNPCre conditional knockout mice.

Control and Fgfr1/Fgfr2-CNPCre dKO mice were fed 0.2% cuprizone (Cupz) for 12 weeks (wks), followed by normal chow for 3 wks. A. Coronal sections of the brains analyzed by PLP mRNA in situ hybridization or CC1 immunolabeling to identify oligodendrocytes show that after 3 wks of normal chow feeding, fewer oligodendrocytes appear in the Fgfr1/Fgfr2-CNPCre dKO mice compared to controls. B. Quantification of the numbers of PLP mRNA+ or CC1+ oligodendrocytes in the corpus callosum confirms the significant reduction in the dKOs compared to controls. Three sections per animal were counted from 3–5 mice per group. C. Coronal sections of the brain analyzed by MOG immunolabeling show reduced signals in the dKO, indicative of impaired recovery of myelin. Scale bars, 200 μm. Error bars represent S.E.M.; N=3–5. *p<0.05, **p<0.01.
We also investigated the effects of Fgfr1/Fgfr2 ablation on the regeneration of oligodendrocytes in chronically demyelinated lesions in the Fgfr1/Fgfr2-Olig1Cre dKO mice. Similar to the Fgfr1/Fgfr2-CNPCre dKOs, reduced oligodendrocyte numbers (Fig. 6A, B; PLP mRNA+ cells) and impaired myelin recovery (Fig. 6C; MOG, LFB) was observed in the corpus callosum after 3 weeks on a normal diet following 12 weeks of cuprizone feeding.
Figure 6. Repopulation of chronically demyelinated lesions by oligodendrocytes is impaired in Fgfr1/Fgfr2-Olig1Cre conditional knockout mice.

Control and Fgfr1/Fgfr2- Olig1Cre dKO mice were fed 0.25% cuprizone (Cupz) for 12 weeks (wks), followed by normal chow for 3 wks. A. Coronal sections of the brains, analyzed by PLP mRNA in situ hybridization to identify oligodendrocytes, show that after 3 wks of normal chow feeding, fewer oligodendrocytes appear in the Fgfr1/Fgfr2-Olig1Cre dKO mice compared to controls. B. Quantification of the numbers of PLP mRNA+ oligodendrocytes in the corpus callosum confirms the significant reduction in the dKOs compared to controls. Three sections per animal were counted from 3–5 mice per group. C. Coronal sections of the brain, analyzed by MOG immunolabeling and LFB staining, show reduced signals in the dKO, indicative of impaired recovery of myelin. Scale bars, 200 μm. Error bars represent S.E.M.; N=3. **p<0.01.
We conclude that in contrast to acute lesions, recovery following chronically cuprizone-induced demyelination is impaired by the combined loss of FGFR1/FGFR2 signaling in oligodendrocyte lineage cells.
OPC response remains unaltered during the cuprizone-induced demyelination or recovery phase in Fgfr1/Fgfr2-CNPCre and Fgfr1/Fgfr2-Olig1Cre conditional knockout mice
We next asked whether the reduced numbers of oligodendrocytes observed during remyelination of chronically demyelinated lesions in the Fgfr1/Fgfr2 dKO mice were due to a reduction in the numbers of OPCs that repopulate the demyelinated regions to generate new oligodendrocytes for remyelination. We identified OPCs and examined their response by in situ hybridization for the expression of Pdgfra mRNA in the corpus callosum after 4 weeks of cuprizone feeding. No differences were observed between the controls and Fgfr1/Fgfr2 dKO mice in either the CNP-Cre (Fig. 7A, C; 4 wks) or the Olig1-Cre line (Fig. 7B, C; 4 wks) of mice, indicating that the initial OPC response to cuprizone-induced demyelination was not altered by the loss of FGFR1/FGFR2 signaling. Similarly, when we examined the OPC numbers in acutely or chronically demyelinated lesions at 3 weeks after acute (6 wks Cupz) or chronic (12 wks Cupz) demyelination, no differences were observed in the corpus callosum of either Fgfr1/Fgfr2-CNPCre (Fig. 7A,C) or Fgfr1/Fgfr2-Olig1Cre (Fig. 7B,C) dKO mice compared to their respective controls.
Figure 7. OPC response remains unaltered during the cuprizone-induced demyelination and recovery phases in Fgfr1/Fgfr2-CNPCre and Fgfr1/Fgfr2-Olig1Cre conditional knockout mice.

Fgfr1/Fgfr2-CNPCre (A) or Fgfr1/Fgfr2-Olig1Cre (B) dKO mice and their littermate controls were fed 0.2% or 0.25% cuprizone (Cupz), respectively, for 4 weeks (wks) or for 6 wks and 12 wks, followed by a normal chow diet for 3 wks. Coronal sections of the brains, analyzed by Pdgfra mRNA in situ hybridization as a marker for OPCs, show no difference between controls and either line of dKO. C. Quantification of the numbers of Pdgfra mRNA+ OPC in the corpus callosum of Fgfr1/Fgfr2-CNPCre or Fgfr1/Fgfr2-Olig1Cre mice confirms that the OPC numbers were not significantly different between controls and dKOs at any of the time points analyzed. Two to three sections per animal were counted from 3–5 mice per genotype. Scale bars, 200 um. Error bars represent S.E.M.; N=3–5.
We conclude that since the OPC response was not altered in Fgfr1/Fgf2 dKO mice either after initial demyelination or after chronic demyelination, the reduction in oligodendrocytes observed after chronic demyelination in the dKO mice is not caused by limiting OPC numbers in the lesions but could reflect an impairment of oligodendrocyte differentiation and/or survival.
DISCUSSION
Promoting endogenous remyelination is an ideal approach to enhance remyelination in MS (Franklin and ffrench-Constant 2008; Franklin and Gallo 2014). However, this requires a better understanding of the molecular mechanisms that control oligodendrocyte and myelin regeneration in order to identify therapeutic targets. A role of FGF2 signaling in promoting the recovery of MS lesions has been suggested from the findings that FGF2 expression and FGFR1-expressing OPCs are increased in MS tissue in active plaques and in chronic inactive peri-plaques, where remyelination normally occurs (Clemente et al., 2011). Here, we show that in an animal model of demyelination, simultaneous loss of FGFR1/FGFR2 signaling from oligodendrocytes and their progenitors results in impaired recovery of oligodendrocytes and myelin of chronically demyelinated lesions, suggesting that cell-autonomous FGF receptor signaling provides an important molecular mechanism for promoting endogenous repair of chronic lesions. In contrast, following acute demyelination, oligodendrocyte differentiation and myelin recovery remained unaltered in these mice, suggesting an overall dispensable role of FGF signaling during the early phases of regeneration. Further, in contrast to the general belief that FGF signaling is inhibitory to OPC differentiation (McKinnon et al., 1990; Bansal 2002; Fortin et al., 2005; Armstrong et al., 2002; 2006; Zhou et al., 2012), simultaneous loss of Fgfr1 and Fgfr2 function has revealed that the overall cell-autonomous role of FGF signaling in the CNS is not inhibitory to OPC differentiation either during normal development or during remyelination.
Our findings that suggest a beneficial role of FGFR1/FGFR2 signaling during regeneration of chronically demyelinated lesions are highly significant, since very few regulatory molecules are known to have a positive impact during this phase. Thyroid hormone is one of the few known factors that promote remyelination in chronic lesions (Harsan et al., 2008). Further, since we observed this positive effect only during regeneration of chronic but not acute lesions, it is possible that the difference in the micro-environment of the two types of lesions may play a role in governing the outcome of FGFR1/FGFR2 loss. For example, microglia are strongly activated in acute lesions but not in chronic lesions (Hiremath et al., 1998; Mason et al., 2004; Lindner et al., 2009; Hibbits et al., 2012). Therefore, it is plausible that FGF signaling in oligodendrocyte lineage cells may become redundant in acute lesions, which may already be primed by several other microglia-derived growth and differentiation factors (Gudi et al., 2011; Voss et al., 2012), while in the chronic phase, astrocyte-derived FGF2, which persists in these lesions (Armstrong et al., 2006; Furusho et al., unpublished observations), may play a more prominent role, thus unmasking the full potential of cell-autonomous FGF signaling in promoting recovery.
Numerous studies in different experimental paradigms have proposed a plethora of often contradictory roles of FGF2 signaling in remyelination and clinical recovery. For example, on the positive side, administration of a viral vector expressing FGF2 to an animal model of MS ameliorated pathological signs of chronic experimental autoimmune encephalomyelitis (EAE) and led to an increase in the number of OPCs and myelin-forming oligodendrocytes in the demyelinated areas (Ruffini et al., 2001); mice lacking FGF2 manifested more severe symptoms of EAE, decreased remyelination, and increased nerve fiber degeneration (Rottlaender et al., 2011). Also, intra-peritoneal injection of FGF2 following lysolecithin-induced demyelination in the optic chiasm and nerve increased MBP gene expression and restored visual evoked potential, indicating FGF2-mediated potentiation of regeneration and functional recovery (Dehghan et al., 2012), and intracranial injection of FGF2 in a growth factor cocktail enhanced remyelination in the cuprizone mouse model of demyelination (Kumar et al., 2007). A beneficial role of FGF signaling in remyelination is also inferred by correlating the spatial and temporal upregulated expression patterns of FGF and FGF receptors with remyelination in MS tissue and in animal models of MS. Specifically, FGF1 and/or FGF2 was upregulated during remyelination of lysolecithin-induced lesions in rodent spinal cords (Tourbah et al., 1992; Hinks and Franklin, 1999) and of cuprizone-induced lesions in the corpus callosum (Gudi et al., 2011); FGF2 transcripts peaked during the initial stages of remyelination, and multiple FGF receptors were expressed in oligodendrocyte lineage cells and astrocytes in the murine hepatitis virus-induced model of demyelination (Messersmith et al., 2000). FGF2 and FGFR1 expression in activated microglia is upregulated in the spinal cord in EAE (Liu et al., 1998), and an increase in the expression of FGF2 mRNA was induced by antibody-mediated demyelination during the early stages of recovery in brain aggregate cultures (Copelman et al., 2000) and importantly, FGF2 expression and FGFR1-expressing OPCs were increased in MS tissue in active plaques and in chronic inactive peri-plaques, where remyelination normally occurs (Clemente et al., 2011). While these studies collectively suggest that FGF2 might be a favorable factor for remyelination, others found increased numbers of oligodendrocytes and improved remyelination of cuprizone-induced lesions in mice lacking FGF2 or FGFR1, proposing an inhibitory role of FGF signaling in oligodendrocyte differentiation and remyelination (Armstrong et al, 2002; Armstrong et al., 2006; Zhou et al., 2012).
These conflicting reports could be due in part to the fact that FGF2 is a pleotropic growth factor that elicits a multitude of responses in multiple cell types, including reactive astrocytes, microglia, neurons, and hematopoietic cells. These cells in turn produce numerous regulatory factors that can positively or negatively influence oligodendrocyte lineage cells and, thus, their potential for remyelination (Moore et al., 2011). Therefore, it is plausible that the effects on oligodendrocytes and OPCs in response to modulation of FGF2 in the lesion environment, as in FGF2-null mice, could potentially be indirect. A further confounding factor is that FGF2 is only one of the 22 FGF family members, many of which are expressed in lesions during the course of demyelination and remyelination (Zhou et al., 2012), presumably by astrocytes and/or microglia, like FGF2 (Liu et al., 1998; Gehrmann et al., 1996; Madiai et al., 2003; Gudi et al., 2011). Eighteen of these FGFs signal through four cell surface FGF receptors, providing a changing repertoire of complex ligand-receptor interactions (Ornitz et al., 1996; Itoh and Ornitz, 2004). Both FGFR-1 and -2 are prominently expressed in oligodendrocytes in a developmentally regulated manner (Bansal et al. 1996; Miyake et al., 1996; Fortin et al., 2005). Therefore, deletion of a single FGF family member (e.g., FGF2) or FGF receptor type (e.g., FGFR1) is unlikely to provide a complete understanding of the overall role of FGF signaling within oligodendrocyte lineage cells during remyelination. This is reflected by our results, which reveal that in contrast to the conclusions drawn from the ablation of FGF2 or Fgfr1 alone (Armstrong et al, 2002; Armstrong et al., 2006; Zhou et al., 2012), simultaneous ablation of both Fgfr1 and Fgfr2, specifically from both OPCs and oligodendrocytes, favors the interpretation that the overall role of cell-autonomous FGF signaling is not detrimental to oligodendrocyte differentiation in either the acute or the chronic cuprizone models of demyelination; on the contrary, it facilitates the recovery of chronically demyelinated lesions.
This seemingly contradictory finding indicates that the two FGFRs may be regulated differently by FGF2 and perhaps play opposing roles in the regulation of oligodendrocyte function. Consistent with this notion, our earliest studies have shown that exposure of isolated oligodendrocytes to FGF2 results in the upregulation of FGFR1 and downregulation of FGFR2 transcripts (Bansal et al., 1996). Subsequently, using receptor type-specific blocking antibodies and activating ligands, we showed that upregulated FGFR1 signaling was responsible for downregulating FGFR2 expression in oligodendrocytes (Fortin et al, 2005). We also showed that signaling via FGFR1 leads to apparently deleterious outcomes—i.e., downregulation of myelin proteins, loss of myelin-like membranes, and abnormal cell cycle reentry of mature oligodendrocytes—eventually resulting in their death (Bansal and Pfeiffer, 1997; reviewed in Bansal, 2002 and Fortin et al., 2005). In contrast, signaling via FGFR2 leads to positive outcomes—i.e., enhanced oligodendrocyte process growth (Fortin et al., 2005), a function further supported by our in vivo gene knockout studies, which indicate a requirement of FGFR2 signaling for upregulating myelin gene expression and promoting myelin growth during developmental myelination in the spinal cord (Furusho et al., unpublished observations). In addition to oligodendrocytes, opposing effects of FGF2, mediated by different FGFRs, have also been observed in chondrocytes (Yan et al., 2011). These studies together present additional layers of complexity in understanding the integrated functions of the FGF signaling system and indicate that a fine balance between FGFR1 and FGFR2 signaling seems to be essential for maintaining normal function. Therefore, dysregulation of this exquisite balance may result in diverse and sometimes opposing outcomes.
In conclusion, we have identified FGF receptor signaling, specifically within oligodendrocyte lineage cells, as an indispensable contributor for promoting the endogenous repair of chronically demyelinated lesions in an animal model. Since there is an urgent unmet need to identify targets for promoting the repair of chronic MS lesions, these studies are clearly a significant advance towards that goal. However, given the broad-spectrum nature of the FGF signaling system and the complexity of interactions between FGF receptor types, manipulation of this system for therapeutic intervention in demyelinating diseases should be considered with caution.
Acknowledgments
We would like to thank Dr. D.M. Ornitz (Washington Univ. Sch. of Med., St. Louis, MI) for providing the Fgfr1 and Fgfr2 floxed mice, Dr. K-A Nave (Max Planck Institute of Experimental Medicine, Goettingen, Germany) for the CNP-Cre mice, and Dr. D.H. Rowitch (Univ. of California at San Francisco, CA) for Olig1-Cre mice. We would also like to thank Dr. Akihiro Ishii for performing the immunoblots. This work was supported by NIH Grant NS38878, a Wellcome Trust Research Training Fellowship (AR), and in part by grants from the National Multiple Sclerosis Society, RG 4087-A-3 and NIH, NS41078 to RB and the UK Multiple Sclerosis Society to RF.
References
- Armstrong RC, Le TQ, Frost EE, Borke RC, Vana AC. Absence of fibroblast growth factor 2 promotes oligodendroglial repopulation of demyelinated white matter. J Neurosci. 2002;22:8574–8585. doi: 10.1523/JNEUROSCI.22-19-08574.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RC, Le TQ, Flint NC, Vana AC, Zhou YX. Endogenous cell repair of chronic demyelination. J neuropathol Exp neurol. 2006;65:245–256. doi: 10.1097/01.jnen.0000205142.08716.7e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas D, Barnstable CJ. HPC-7: A novel oligodendrocyte lineage protein, which appears prior to galactcerebroside. Glia. 1998;23:169–179. [PubMed] [Google Scholar]
- Bansal R, Stefansson K, Pfeiffer SE. Proligodendroblast antigen (POA), a developmental antigen expressed by A007/O4-positive oligodendrocyte progenitors prior to the appearance of sulfatide and galactocerebroside. J Neurochem. 1992;58:2221–2229. doi: 10.1111/j.1471-4159.1992.tb10967.x. [DOI] [PubMed] [Google Scholar]
- Bansal R, Kumar M, Murray K, Morrison RS, Pfeiffer SE. Regulation of FGF receptors in the oligodendrocyte lineage. Mol Cell Neurosci. 1996;7:263–275. doi: 10.1006/mcne.1996.0020. [DOI] [PubMed] [Google Scholar]
- Bansal R, Pfeiffer SE. FGF-2 converts mature oligodendrocytes to a novel phenotype. J Neurosci Res. 1997;50:215–228. doi: 10.1002/(SICI)1097-4547(19971015)50:2<215::AID-JNR10>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Bansal R. Fibroblast growth factors and their receptors in oligodendrocyte development: Implications for demyelination and remyelination. Dev Neurosci. 2002;24:35–46. doi: 10.1159/000064944. [DOI] [PubMed] [Google Scholar]
- Clemente D, Ortega MC, Arenzana FJ, de Castro F. FGF-2 and Anosmin-1 are selectively expressed in different types of multiple sclerosis lesions. J Neurosci. 2011;31:14899–14909. doi: 10.1523/JNEUROSCI.1158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copelman CA, Cuzner ML, Groome N, Diemel LT. Temporal analysis of growth factor mRNA expression in myelinating rat brain aggregate cultures: increments in CNTF, FGF-2, IGF-I, and PDGF-AA mRNA are induced by antibody-mediated demyelination. Glia. 2000;30:342–351. doi: 10.1002/(sici)1098-1136(200006)30:4<342::aid-glia30>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Dehghan S, Javan M, Pourabdolhossein F, Mirnajafi-Zadeh J, Baharvand H. Basic fibroblast growth factor potentiates myelin repair following induction of experimental demyelination in adult mouse optic chiasm and nerves. J Mol Neurosci. 2012;48:77–85. doi: 10.1007/s12031-012-9777-6. [DOI] [PubMed] [Google Scholar]
- Dutta R, Trapp BD. Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog Neurobiol. 2011;93:1–12. doi: 10.1016/j.pneurobio.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin D, Rom E, Sun H, Yayon A, Bansal R. Distinct fibroblast growth factor (FGF)/FGF receptor signaling pairs initiate diverse cellular responses in the oligodendrocyte lineage. J Neurosci. 2005;25:7470–7479. doi: 10.1523/JNEUROSCI.2120-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJM. Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci. 2002;3:705–714. doi: 10.1038/nrn917. [DOI] [PubMed] [Google Scholar]
- Franklin RJM, ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- Franklin RJM, Gallo V. The Translational Biology of Remyelination: Past, Present, and Future. GLIA. 2014 doi: 10.1002/glia.22622. in press. [DOI] [PubMed] [Google Scholar]
- Furusho M, Dupree JL, Bryant M, Bansal R. Disruption of fibroblast growth factor receptor signaling in nonmyelinating Schwann cells causes sensory axonal neuropathy and impairment of thermal pain sensitivity. J Neurosci. 2009;29:1608–14. doi: 10.1523/JNEUROSCI.5615-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusho M, Kaga Y, Ishii A, Hébert JM, Bansal R. Fibroblast growth factor signaling is required for the generation of oligodendrocyte progenitors from the embryonic forebrain. J Neurosci. 2011;31:5055–66. doi: 10.1523/JNEUROSCI.4800-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusho M, Dupree JL, Nave KA, Bansal R. Fibroblast growth factor receptor signaling in oligodendrocytes regulates myelin sheath thickness. J Neurosci. 2012;32:6631–6641. doi: 10.1523/JNEUROSCI.6005-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrmann J, Lannes-Vieira J, Wekerle H. Differential expression of fibroblast growth factor-2 and receptor by glial cells in experimental autoimmune encephalomyelitis (EAE) Glia. 1996;16:93–100. doi: 10.1002/(SICI)1098-1136(199602)16:2<93::AID-GLIA1>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Guardiola-Diaz HM, Ishii A, Bansal R. Erk1/2 MAPK and mTOR signaling sequentially regulates progression through distinct stages of oligodendrocyte differentiation. Glia. 2012;60:476–486. doi: 10.1002/glia.22281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudi V, Škuljec J, Yildiz Ö, Frichert K, Skripuletz T, Moharregh-Khiabani D, Voss E, Wissel K, Wolter S, Stangel M. Spatial and temporal profiles of growth factor expression during CNS demyelination reveal the dynamics of repair priming. PLoS One. 2011;6:e22623. doi: 10.1371/journal.pone.0022623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harsan LA, Steibel J, Zaremba A, Agin A, Sapin R, Poulet P, Guignard B, Parizel N, Grucker D, Boehm N, Miller RH, Ghandour MS. Recovery from chronic demyelination by thyroid hormone therapy: Myelinogenesis induction and assessment by diffusion tensor magnetic resonance imaging. J Neurosci. 2008;28:14189–14201. doi: 10.1523/JNEUROSCI.4453-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbits N, Yoshino J, Le TQ, Armstrong RC. Astrogliosis during acute and chronic cuprizone demyelination and implications for remyelination. ASN Neuro. 2012;4:e00100. doi: 10.1042/AN20120062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinks GL, Franklin RJM. Distinctive Patterns of PDGF-A, FGF-2, IGF-I, and TGF-b1 Gene Expression during Remyelination of experimentally-Induced spinal cord demyelination. Mol Cell Neurosci. 1999;14:153–168. doi: 10.1006/mcne.1999.0771. [DOI] [PubMed] [Google Scholar]
- Hiremath MM, Saito Y, Knapp GW, Ting JP-Y, Suzuki K, Matsushima GK. Microglia/macrophage accumulation during cuprizone-induced demyelination in C57BL/6 mice. J Neuroimmunol. 1998;92:38–49. doi: 10.1016/s0165-5728(98)00168-4. [DOI] [PubMed] [Google Scholar]
- Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Kaga Y, Shoemaker WJ, Furusho M, Bryant M, Rosenbluth J, Pfeiffer SE, Oh L, Rasband M, Lappe-Siefke C, Yu K, Ornitz DM, Nave KA, Bansal R. Mice with conditional inactivation of fibroblast growth factor receptor-2 signaling in oligodendrocytes have normal myelin but display dramatic hyperactivity when combined with Cnp1 inactivation. J Neurosci. 2006;26:12339–12350. doi: 10.1523/JNEUROSCI.3573-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluver H, Barrera E. A method for the combined staining of cells and fibers in the nervous system. J Nueopathol Exp Neurol. 1953;12:400–403. doi: 10.1097/00005072-195312040-00008. [DOI] [PubMed] [Google Scholar]
- Kumar S, Biancotti JC, Yamaguchi M, de Vellis J. Combination of growth factors enhances remyelination in a cuprizone-induced demyelination mouse model. Neurochem Res. 2007;32:783–797. doi: 10.1007/s11064-006-9208-6. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Lindner M, Fokuhl J, Linsmeier F, Trebst C, Stangel M. Chronic toxic demyelination in the central nervous system leads to axonal damage despite remyelination. Neurosci Lett. 2009;453:120–125. doi: 10.1016/j.neulet.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Liu X, Mashour GA, Webster HF, Kurt A. Basic FGF and FGF receptor 1 are expressed in microglia during experimental autoimmune encephalomyelitis: temporally distinct expression of midkine and pleiotrophin. Glia. 1998;24:390–397. doi: 10.1002/(sici)1098-1136(199812)24:4<390::aid-glia4>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109:75–86. doi: 10.1016/s0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- Madiai F, Hussain SR, Goetti VM, Burry RW, Stephens RL, Jr, Hackshaw KV. Upregulation of FGF-2 in reactive spinal cord astrocytes following unilateral lumbar spinal nerve ligation. Exp Brain Res. 2003;148:366–376. doi: 10.1007/s00221-002-1286-3. [DOI] [PubMed] [Google Scholar]
- Mason JL, Langaman C, Morell P, Suzuki K, Matsushima GK. Episodic demyelination and subsequent remyelination within the murine central nervous system: changes in axonal calibre. Neuropathol Appl Neurobiol. 2001;27:50–58. doi: 10.1046/j.0305-1846.2001.00301.x. [DOI] [PubMed] [Google Scholar]
- Mason JL, Toews A, Hostettler JD, Morell P, Suzuki K, Goldman JE, Matsushima GK. Oligodendrocytes and progenitors become progressively depleted within chronically demyelinated lesions. Am J Pathol. 2004;164:1673–1682. doi: 10.1016/S0002-9440(10)63726-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon RD, Matsui T, Dubois-Dalcq M, Aaronson SA. FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron. 1990;5:603–614. doi: 10.1016/0896-6273(90)90215-2. [DOI] [PubMed] [Google Scholar]
- Messersmith DJ, Murtie JC, Le TQ, Frost EE, Armstrong RC. Fibroblast growth factor 2 (FGF2) and FGF receptor expression in an experimental demyelinating disease with extensive remyelination. J Neurosci Res. 2000;62:24–56. doi: 10.1002/1097-4547(20001015)62:2<241::AID-JNR9>3.0.CO;2-D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake S, Makimura M, Kanegae Y, Harada S, Sato Y, Takamori K, Tokuda C, Saito I. Efficient generation of recombinant adenoviruses using adenovirus DNA-terminal protein complex and a cosmid bearing the full-length virus genome. Proc Natl Acad Sci USA. 1996;93:1320–1324. doi: 10.1073/pnas.93.3.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CS, Abdullah SL, Brown A, Arulpragasam A, Crocker SJ. How factors secreted from astrocytes impact myelin repair. J Neurosci Res. 2011;89:13–21. doi: 10.1002/jnr.22482. [DOI] [PubMed] [Google Scholar]
- Oh LY, Denninger A, Colvin JS, Vyas A, Tole S, Ornitz DM, Bansal R. Fibroblast growth factor receptor 3 signaling regulates the onset of oligodendrocyte terminal differentiation. J Neurosci. 2003;23:883–894. doi: 10.1523/JNEUROSCI.23-03-00883.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary MT, Hinks GL, Charlton HM, Franklin RJ. Increasing local levels of IGF-I mRNA expression using adenoviral vectors does not alter oligodendrocyte remyelination in the CNS of aged rats. Mol Cell Neurosci. 2002;19:32–42. doi: 10.1006/mcne.2001.1062. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M. Receptor specificity of the fibrobrast growth factor family. J Biol Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- Pavelko KD, van Engelen BG, Rodriguez M. Acceleration in the rate of CNS remyelination in lysolecithin-induced demyelination. J Neurosci. 1998;18:2498–2505. doi: 10.1523/JNEUROSCI.18-07-02498.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirvola U, Ylikoski J, Trokovic R, Hébert JM, McConnell SK, Partanen J. FGFR1 is required for the development of the auditory sensory epithelium. Neuron. 2002;35:671–680. doi: 10.1016/s0896-6273(02)00824-3. [DOI] [PubMed] [Google Scholar]
- Rottlaender A, Villwock H, Addicks K, Kuerten S. Neuroprotective role of fibroblast growth factor-2 in experimental autoimmune encephalomyelitis. Immunology. 2011;133:370–378. doi: 10.1111/j.1365-2567.2011.03450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffini F, Furlan R, Poliani PL, Brambilla E, Marconi PC, Bergami A, Desina G, Glorioso JC, Comi G, Martino G. Fibroblast growth factor-II gene therapy reverts the clinical course and the pathological signs of chronic experimental autoimmune encephalomyelitis in C57BL/6 mice. Gene Ther. 2001;8:1207–1213. doi: 10.1038/sj.gt.3301523. [DOI] [PubMed] [Google Scholar]
- Shields SA, Gilson JM, Blakemore WF, Franklin RJ. Remyelination occurs as extensively but more slowly in old rats compared to young rats following gliotoxin-induced CNS demyelination. Glia. 1999;28:77–83. doi: 10.1002/(sici)1098-1136(199910)28:1<77::aid-glia9>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Tourbah A, Baron-Van Evercooren A, Oliver L, Raulais D, Jeanny JC, Gumpel M. Endogenous aFGF expression and cellular changes after a demyelinating lesion in the spinal cord of adult normal mice: immunohistochemical study. J Neurosci Res. 1992;33:47–59. doi: 10.1002/jnr.490330107. [DOI] [PubMed] [Google Scholar]
- Vana AC, Flint NC, Harwood NE, Le TQ, Fruttiger M, Armstrong RC. Platelet-derived growth factor promotes repair of chronically demyelinated white matter. J Neuropathol Exp Neurol. 2007;66:975–88. doi: 10.1097/NEN.0b013e3181587d46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss EV, Škuljec J, Gudi V, Skripuletz T, Pul R, Trebst C, Stangel M. Characterisation of microglia during de- and remyelination: can they create a repair promoting environment? Neurobiol Dis. 2012;45:519–28. doi: 10.1016/j.nbd.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Wang SJ, Furusho M, D’Sa C, Kuwada S, Conti L, Morest DK, Bansal R. Inactivation of Fibroblast Growth Factor Receptor signaling in myelinating glial cells results in significant loss of adult spiral ganglion neurons accompanied by age-related hearing impairment. J Neurosci Res. 2009;87:3428–3437. doi: 10.1002/jnr.22164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Chen D, Cool SM, van Wijnen AJ, Mikecz K, Murphy G, Im HJ. Fibroblast growth factor receptor 1 is principally responsible for fibroblast growth factor 2-induced catabolic activities in human articular chondrocytes. Arthritis Res Ther. 2011;13:R130. doi: 10.1186/ar3441. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- Zhou YX, Pannu R, Le TQ, Armstrong RC. Fibroblast growth factor 1 (FGFR1) modulation regulates repair capacity of oligodendrocyte progenitor cells following chronic demyelination. Neurobiol Dis. 2012;45:196–205. doi: 10.1016/j.nbd.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]