

Abstract
Altered metabolism is increasingly acknowledged as an important aspect of cancer, and thus serves as a potentially fertile area for the identification of therapeutic targets or leads. Our recent work using transcriptional data to predict metabolite levels in cancer cells led to preliminary evidence of the antiproliferative role of menaquinone (Vitamin K2) in the Jurkat cell line model of acute lymphoblastic leukemia. However, nothing is known about the direct metabolic impacts of menaquinone in cancer, which could provide insights into its mechanism of action. Here, we used metabolomics to investigate the process by which menaquinone exerts antiproliferative activity on Jurkat cells. We first validated the dose-dependent, semi-selective, pro-apoptotic activity of menaquinone treatment on Jurkat cells relative to non-cancerous lymphoblasts. We then used mass spectrometry-based metabolomics to identify systems-scale changes in metabolic dynamics that are distinct from changes induced in non-cancerous cells or by other chemotherapeutics. One of the most significantly affected metabolites was phosphoethanolamine, which exhibited a two-fold increase in menaquinone-treated Jurkat cells compared to vehicle-treated cells at 24 h, growing to a five-fold increase at 72 h. Phosphoethanolamine elevation was observed prior to the induction of apoptosis, and was not observed in menaquinone-treated lymphoblasts or chemotherapeutic-treated Jurkat cells. We also validated the link between menaquinone and phosphoethanolamine in an ovarian cancer cell line, suggesting potentially broad applicability of their relationship. This metabolomics-based work is the first detailed characterization of the metabolic impacts of menaquinone treatment and the first identified link between phosphoethanolamine and menaquinone-induced apoptosis.
Introduction
An important aspect of cancer now receiving increased attention is its altered metabolism, which provides a continuous and abundant supply of metabolites that is necessary for rapid proliferation. Altered metabolism was recently acknowledged as one of the key hallmarks of cancer1. As a result, metabolism is becoming a promising target for cancer drug development2. Drug development is often oriented towards finding (often synthetic) small molecule inhibitors of proteins; however, metabolites and closely related derivatives have important potential for serving as therapeutic candidates since they play key signaling, regulatory, and biosynthetic roles in cells. The use of metabolites as therapeutics is particularly exciting based on the hypothesis that molecules endogenously produced by cells may be less likely to have broad, off-target toxicity effects than synthetic or exotic compounds. As such, there is significant interest in identifying and characterizing metabolites with anticancer-specific potential that may lead to the development of novel chemotherapeutics.
For metabolites with anti-cancer activity, the processes underlying their activity are typically poorly characterized, limiting the ability to leverage them and their downstream targets in development of therapeutics. However, systems-scale approaches to characterizing metabolism (metabolomics) have the potential to yield significant insight into those processes for metabolites.
This work uses a metabolomics-based approach to characterize the effects and potential mechanism of a putative anti-leukemic metabolite, menaquinone. Acute lymphoblastic leukemia (ALL) is a cancer of the white blood cells in the bone marrow that accounts for 75% of children’s leukemia3, 4. About 52,380 new leukemia cases (all types) were projected to be diagnosed in 2014, with 24,090 people projected to die from leukemia3. Involvement of altered metabolism is well-known in ALL, with one carbon metabolism actually being one of the earliest metabolic pathways targeted for ALL therapeutics5. In fact, derivatives of those compounds are still used as frontline treatment to this day6. Other metabolites, including ceramides and metabolites in lipid signaling pathways, are also being studied as additional metabolism-focused cancer therapeutic candidates7.
In our previous work, we developed a computational metabolomics model based only on transcriptional analysis that predicted the levels of menaquinone (MQ, vitamin K2) to be lowered in the Jurkat cell line model of ALL compared to noncancerous lymphoblast cells8. Initial validation experiments using microwell fluorescence assays suggested significant and semi-selective antiproliferative activity of MQ on Jurkat cells relative to noncancerous lymphoblasts8. MQ’s primary physiological role and mechanism in blood coagulation is well-studied and well-understood9–11. Previous studies have shown that MQ can in fact have antiproliferative effects in certain cancer cell types: it can trigger both apoptotic and autophagic programmed cell death, as well as both intrinsic and extrinsic pathways of apoptosis, in a variety of cell types including colon cancer12, acute myeloid leukemia13, hepatocellular carcinoma14, 15, prostate cancer16, and cervical carcinoma17.
However, the mechanisms underlying MQ’s anticancer activity in ALL are not known and are not necessarily expected to be the same as those previously found for myeloid leukemia. In addition, the metabolic impacts of MQ in cancer cells are (to our knowledge) almost entirely unknown. Metabolomics – the systems-scale analysis of small molecule biochemical intermediates in metabolism – is an ideal approach to begin to address both of these questions. As downstream products of many cellular processes, metabolites provide unique insight into the state of biological systems. Metabolomic analysis of cancer cell cultures has previously enabled noteworthy discoveries18, and has even been used to help elucidate the mechanisms of drugs19.
Here, we applied a metabolomics-based approach to move towards understanding the mechanisms and metabolic impacts of MQ in triggering the dose-dependent, semi-selective cell death of leukemic Jurkat cells. Using two-dimensional gas chromatography coupled to mass spectrometry (GCxGC-MS) to measure the metabolomic dynamics of the Jurkat model of ALL and non-cancerous lymphoblasts in response to treatment with MQ, we identified systems-scale changes in metabolism in MQ-treated cells that were unique compared to treatments with other common chemotherapeutics. One of the most prominent of these changes was in the phospholipid biosynthesis metabolite phosphoethanolamine (PE), which was significantly higher in MQ-treated cancer cells but not in MQ-treated non-cancerous lymphoblast cells or in chemotherapeutic-treated cancer cells. We also found PE to itself display some pro-apoptotic activity. Combined with the specificity of PE upregulation upon MQ-induced cancer cell death, this suggests that these two axes of programmed cell death are linked. Finally, we demonstrated that this phenomenon is likely not limited just to leukemic cell lines based on the observed PE elevation upon MQ treatment of an unrelated cancer cell line.
Experimental Methods
Chemicals and Reagents
RPMI 1640 with L-glutamine and antibiotic-antimycotic solution were purchased from Cellgro, Mediatech Inc. (Manassas, VA, USA). FBS was purchased from Atlanta Biologicals (Lawrenceville, GA, USA). MQ and PE were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Expansion cell culture protocols
The human acute T cell lymphoblastic leukemia cell line Jurkat (ATCC CRL-2570) was purchased from the American Type Culture Collection (ATCC) and cultured in RPMI-1640 with 2mM L-glutamine supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic and antimycotic (AA) solution in T25 flasks. When cell density reached 2 × 106 cells/mL, they were subcultured to 2 × 105 cells/mL. A noncancerous, human B-lymphocyte cell line was purchased from Coriell Institute (Camden, NJ, USA; catalogue #GM 15851) and cultured in RPMI 1640 with L-glutamine medium supplemented with 15% FBS and 1% AA. Lymphoblast cells were subcultured to 2 × 105 cells/mL when they reached a confluence of 1 × 106 cells/mL. All cell lines were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Toxicity and antiproliferative assay
Jurkat and lymphoblast cells at a density of 1.5 × 104 cells per 100 μL in phenol red-free RPMI 1640 with 15% FBS and 1% AA were seeded in 96-microwell plates in triplicate. A 40mM stock solution of MQ in DMSO was diluted with the same medium to prepare working concentrations and 100 μL was added to the cells at 24h after seeding. A DMSO-vehicle control and media control were prepared simultaneously. At 72h of metabolite treatment, cells were treated with 500 nM C12-resazurin and 10 nM SYTOX green dye to stain metabolically active live cells and loss of membrane integrity dead cells, respectively (LIVE/DEAD cell vitality assay kit, Life Technologies, Grand Island, NY, USA; catalog #L34951). Live and dead cells were counted using a flow cytometer (Accuri C6, BD Biosciences, Franklin Lakes, NJ, USA) with excitation at 488 nm and emission at 530 nm (SYTOX green) and 575 nm (C12-resorufin). Percentages of live and dead cells were calculated relative to the vehicle control. Assays were repeated at least twice.
Annexin assay for apoptosis
Jurkat cells were seeded at 6 × 104 cells per 300 μL in 24-well plates, in triplicate, in phenol red-free RPMI with 10% FBS and 1% AA. MQ, PE, chemotherapeutic positive controls (docetaxel and doxorubicin) and vehicle controls (DMSO and water) were administered to the cells at 24h after seeding. Apoptotic cells containing externalized phosphatidylserine were assayed by measuring annexin binding to the phosphatidylserine using the Alexa Fluor 488 conjugate annexin V apoptosis detection reagent (Life Technologies, Grand Island, NY, USA; catalog #A13201) and flow cytometry. Briefly, cells were collected by centrifugation and washed consecutively with ice-cold phosphate-buffered saline (PBS) and annexin binding buffer prior to staining. Annexin V positive staining indicated apoptosis, while cells staining double-positive with SYTOX AADvanced dead cell stain (Life Technologies, Grand Island, NY, USA; catalog #S10349) and annexin indicated late-apoptotic cells or dead cells via apoptosis.
Caspase 3/7 activity assay for apoptosis
Jurkat cells were suspended in phenol red-free RPMI with 10% FBS and 1% AA and seeded at a density of 1.5 × 104 cells per 100 μL in 96-well plates. MQ, PE, chemotherapeutic positive controls (docetaxel and doxorubicin), and vehicle control (DMSO and water) were administered at 24 h after seeding, with each treatment diluted in 100 μL of medium. Caspase 3/7 activity was assayed using a caspase-3/7 flow cytometry assay kit (Life Technologies, Grand Island, NY, USA; catalog #C10427). Briefly, caspase 3/7 reagent was added to the cell suspension at 500 nM concentration and incubated at 37°C for 25 min. SYTOX AADvanced stain was added to stain the membrane compromised dead cells and incubated for 5 additional minutes at 37°C. Caspase 3/7 positive staining indicated apoptosis, while double-positive SYTOX and caspase 3/7 cells indicated dead cells via apoptosis.
MQ treatment and metabolomic extraction
The Jurkat cells and normal lymphoblasts were seeded at a density of 2 × 105cells/ml in T75 tissue culture flasks. Cells were treated with 25 & 35 μM MQ, 0.02 μM docetaxel, 0.4 μM doxorubicin and DMSO-vehicle control in new media at 24h after seeding. All treatments were performed in biological triplicate. Cell suspensions for metabolite extractions were collected from the same flasks at 0, 24, 48 & 72 h after the treatments and subjected to fast filtration and quenching. Briefly, vacuum assisted-fast filtration20, 21 on a vacuum manifold (Pall, Port Washington, NY, USA) was used to quickly remove media, followed by a 10 ml PBS wash. Filters (47 mm, 0.45 μM, PES; Millipore, Billerica, MA, US; catalog #HPWP04700) capturing the cells were immediately transferred to 1.5ml of −80°C methanol in a petri dish (60 mm × 15 mm) on dry ice and stored at −80°C for 24 h for thorough extraction. The filters were rinsed with the methanol cell suspension, and the suspension was transferred to microcentrifuge tubes, vigorously mixed for 30 sec and centrifuged at 5000g for 5 min at −5°C. Supernatants were collected and pellets were re-suspended in 250 μl of cold 100% methanol and centrifuged at the same conditions. Supernatants were pooled, and pellets were re-suspended in 250 μL ice cold ultrapure water and centrifuged at 5000g for 5 min at 4°C. Pooled supernatants were stored at −80°C until analysis. At each time point, a small sample was used to count the live and dead cells using flow cytometry as described above, for use in normalizing injection volumes for metabolomics. An extraction blank was prepared in parallel by following the same protocols to quench and extract a filter with no cells, for use in later data processing.
GC x GC-MS analysis
Metabolite extracts corresponding to 5×105 live cells were dried using a centrifugal vacuum concentrator at 40°C. (Thus, no further normalization of measured analyte peak areas to indicators of cell number was necessary; normalization in all figures is only the result of post-processing and variable scaling.) Residues were derivatized as previously described22–24. Briefly, 5 μL of 40 mg/mL O-methylhydroxylamine hydrochloride (MP Biomedicals, Santa Ana, CA) in pyridine was added to the dried sample and shaken at 1400 rpm for 90 min at 30°C. 45 μL of N-methyl-N-(trimethylsilyl) trifluoroacetamide (MSTFA) + 1% trimethylchlorosilane (TMCS) (Thermo Scientific, Lafayette, CO, USA) was then added to the samples which were then shaken at 1400 rpm for 30 min at 37°C. Samples were centrifuged at 21,100g for 3 minutes and 25 μL of the supernatant was added to an autosampler vial. Samples were spiked with 0.125 μL of a retention time standard solution consisting of fatty acid methyl esters (FAMEs) and an injection standard of nonadecanoic acid methyl ester dissolved in dimethylformamide. In parallel, a master quality control (QC) sample was prepared by pooling 75 μL of extract from each sample; 600 μL of master QC was aliquoted, dried, and derivatized along with every batch of 8–10 samples. Two QC injections were performed at the beginning of the run to condition the system and repeated once every 4–5 sample injections to allow for batch corrections. A derivatization blank was prepared and run with every batch of samples. The extraction blank collected during filtration was derivatized and run as the first sample to use for blank deduction during peak alignment.
A LECO Pegasus 4D instrument with an Aglient 7683B autosampler, Agilent 7890A gas chromatograph and time-of-flight mass spectrometer was used to analyze the samples. The first column was an HP-5, 30 m long × 0.320 mm ID × 0.25 μm film thickness (Agilent, Santa Clara, CA, USA), and the second was an Rtx-200, 2 m long × 0.25 mm ID × 0.25 μm film thickness (Restek, Bellefonte, PA, USA). More detailed gas chromatography, autosampler, and mass spectrometry methods are provided in the ESI.
Data Analysis
Data analysis was performed as previously described23, 24. Briefly, instrument output was first analyzed with ChromaTOF (LECO, St. Joseph, MI) to determine baseline, peak area, and peak identities. Software settings included a baseline offset of 0.5, automatic smoothing, 1st dimension peak width of 24 seconds, 2nd dimension peak width of 0.25 seconds, and a match of 700 was required to combine peaks with a minimum signal-to-noise (S/N) of 10 for all subpeaks. Peaks were required to have a S/N of 100 and a minimum similarity score of 800 before assigning a name. Unique mass was used for peak area calculation.
To align the samples, MetPP (http://metaopen.sourceforge.net/metpp.html) was used25, along with the extraction blank file used for peak deduction. Unknowns were retained during peak alignment. On-the-fly alignment was used with quality control samples manually selected as the peak list for primary alignment. Peak alignment was performed using the default criteria.
After alignment, further processing of the data was done based on a previously described procedure26. Briefly, batch effects were removed from the intracellular data set using LOESS. To remove analytes that were not reproducibly detected, analytes for which more than half of the values were missing in the QC samples or for which the QC samples had a coefficient of variance larger than 0.5 were removed from the data set. Then, missing values were manually corrected using small value correction only if more than half of the values in biological replicates were missing. (However, we note that for the OVCAR-3 validation experiments, since only one metabolite was being tested and there were very few samples, direct results from ChromaTOF were used because the downstream analysis steps were either unnecessary or inappropriate.)
Annotated analytes were then compared to the Kyoto Encyclopedia of Genes and Genomes (KEGG) and the Human Metabolome Database (HMDB); if they were listed in KEGG and HMDB they were considered annotated as metabolites. The metabolites were then verified by a manual confirmation of similarity between the annotated peak spectrum and the library spectrum.
Based on QC injections and manual inspection of chromatograms, we identified that one batch was derivatized poorly, so four samples were removed from further analysis. Additionally, principal component analysis clearly identified one sample, a control Jurkat injection at 24 h, to be a significant outlier from all data, so it was also removed. Finally, one control Jurkat sample at 72 h had over 50% of its metabolite values missing (more than three times more than almost all samples), so it was also removed (though its removal had little impact on overall analyses).
Multivariate and univariate statistical analyses were performed using MetaboAnalyst27 (http://metaboanalyst.ca/). Remaining missing values were k-nearest neighbors (KNN) corrected. Data were log-transformed using generalized logarithm transformation (base 2) and autoscaled.
Results
Menaquinone has a time-dependent, dose-dependent semi-selective antiproliferative effect on Jurkat cells
Our previous work8 suggested that MQ may have selective antiproliferative activity on Jurkat cells compared to noncancerous lymphoblasts, based on a microwell assay. We first sought to confirm and then quantify MQ-based antiproliferative activity on Jurkat cells and control non-cancerous lymphoblasts for varying lengths of treatment (48, 72 and 96 h). Live and dead cells were assayed using flow cytometry after staining with C12 resazurin and SYTOX green. MQ treatment resulted in concentration- and time-dependent anti-proliferative activity on cancer cells (Fig. 1), while normal cells showed weaker anti-proliferative response and even exhibited (insignificant) growth stimulation under certain conditions. 25 μM was identified as an approximate IC50 for MQ in Jurkat cells at 72 h; MQ added to normal lymphoblasts at the same concentration caused statistically insignificant effects at 48 h and 72 h, and still much higher relative proliferation than in Jurkat cells at 96 h (though statistically different from control). The 96 h effect may have been caused in part by environmental stress: 96 h is beyond the normal medium renewal and passaging time for these cells, so they also may be experiencing nutritional stress, which is known to change the toxicity of compounds28. Interestingly, normal lymphoblasts even showed a modest but insignificant growth stimulation at 24 h. In comparison, treatment with the chemotherapeutics docetaxel and doxorubicin at commonly used clinical dosages caused severe toxic effects on normal cells by 72 h (Table S2). Using these data on MQ toxicity, we calculated approximate IC50 values for each cell type for each time point (Table S3). These results indicate that MQ shows a semi-selective anticancer effect without serious side effects to healthy cells for 72 h at 25 μM, though the selectivity decreases as MQ treatment dosage is increased.
Fig. 1. MQ exhibits a time-dependent, dose-dependent semi-selective activity against Jurkat cells.

Panels show ratio of live cells present in menaquinone (MQ) treated A) Jurkat and B) lymphoblast cultures relative to vehicle-treated control at three different treatment times. Error bars represent one standard deviation. * indicates significant difference from the DMSO vehicle control (t-test, p ≤ 0.05).
Menaquinone exhibits a pro-apoptotic effect in Jurkat cells
With the antiproliferative effect of MQ established, we sought to characterize whether its effects were strictly due to decreased growth rate or at least in part due to an increase in apoptosis. To do this, we measured the levels of annexin and caspase 3/7, each of which are known markers for specific mechanisms of apoptosis, in MQ-treated Jurkat cells. Fig. 2 shows that the fraction of apoptotic cells for Jurkat cells treated with MQ are higher for both markers, indicating pro-apoptotic activity of MQ in Jurkat cells.
Fig. 2. Pro-apoptotic activity of MQ on Jurkat cells.

Cells were treated with 35 μM MQ for 72 hours, and apoptosis was assayed using both annexin and caspase 3/7 assays. Plotted is the percentage of early apoptotic or late apoptotic cells, relative to all measured cells. Error bars represent one standard deviation. Asterisks indicate significant differences from control (p < 0.01, two-tailed t-test).
Menaquinone’s antiproliferative activity corresponds with system-wide changes in metabolism
Since the anticancer activity of MQ was originally based on predictions of MQ depletion in Jurkat cells, we hypothesized that there might be significant metabolic changes in these cells when treated with MQ. Thus, we next used two-dimensional gas chromatography coupled to mass spectrometry (GCxGC-MS) to characterize the metabolite profiles of both Jurkat cells and lymphoblast cells in response to two dosage levels of MQ. We chose 25 μM since it is approximately an IC50 for the Jurkat cells at 72 hours (the normal feeding or passage time for Jurkat cells), and 35 μM to potentially capture some effects on the lymphoblasts. We also included two common chemotherapeutics that act via non-metabolic mechanisms, docetaxel and doxorubicin, to capture the metabolic profile that might be more generally associated with cell death and apoptosis but not specifically with MQ treatment.
For the Jurkat cells, we measured 208 reproducible analytes across all samples, with 61 unique metabolite identities annotated by the data processing pipeline and matched to known human metabolites with HMDB and KEGG entries. For the lymphoblast cells, 135 reproducible analytes were measured across all samples, with 36 unique metabolite identities annotated by the data processing pipeline and matched to known human metabolites with HMDB and KEGG entries. A heatmap of the metabolite data is presented in Fig. S1, which includes both annotated and unknown analytes. A dendrogram for these samples is presented in Fig. S2, demonstrating clustering by time point and by treatment within the cell types, with few misclustered samples. Results were similar when only annotated metabolites were considered (Fig. S3).
For each cell type, t-tests and one-way ANOVA were first used to identify significant differences between treatment conditions across all time points (significant annotated metabolites reported in Table 1). For Jurkat cells there were 33 significant analytes between the three groups (MQ-treated, chemotherapeutic-treated, and control) based on a one-way ANOVA across all time points. When considering only the unique annotated metabolites, there were 9 significant based on this one-way ANOVA test. Importantly, in post hoc tests for each significant metabolite where the three classes are all compared pairwise (Fisher’s LSD test), 27 of the 33 analytes were different between the MQ and chemotherapeutic treatment conditions (and 8 of the 9 annotated analytes) – indicating unique changes in metabolism that should not be dismissed as being caused only by cell death. In the noncancerous lymphoblasts, on the other hand, there were no significant changes between MQ treatment and control, underscoring the assertion that little change in metabolism is induced in healthy cells for the MQ levels tested here.
Table 1.
Annotated metabolites significantly different between MQ-treated, chemotherapeutic-treated, and vehicle control-treated Jurkat cells via one-way ANOVA across all time points. All p-values were adjusted for multiple hypothesis testing using a False Discovery Rate (FDR) correction51.
| Metabolite | FDR-corrected p-value |
|---|---|
| Phosphoethanolamine | 1.24E-08 |
| Hypotaurine | 1.89E-06 |
| Beta-Alanine | 2.21E-05 |
| 2-Furoic acid | 0.000809 |
| D-Ribose | 0.00306 |
| Fumaric acid | 0.00636 |
| D-Xylitol | 0.0109 |
| Gamma-aminobutyric acid | 0.0168 |
| Hydroxyproline | 0.0304 |
Multivariate analysis reveals that MQ causes unique metabolic responses compared to other chemotherapeutic agents
Principal component analysis (Fig. 3) recapitulated the results evident in the metabolite heatmaps and univariate statistical analyses, showing that while control-treated Jurkat cells do not display marked and consistent changes over time, MQ-treated Jurkat cells exhibit unique metabolite profiles by 72 hours. In contrast, chemotherapeutic-treated cells separate from controls within 24 hours, suggesting that MQ has unique metabolic impacts and may have a unique mechanism of action compared to other common chemotherapeutics.
Fig. 3. Principal component analysis of (A) Jurkat and (B) lymphoblast metabolomics data.

(A) In Jurkat cells, the chemotherapeutic-treated samples (red) separate from the rest of the samples in the first principal component, while the later MQ-treated samples (when antiproliferative activity is at its highest; blue triangles) separate in the second principal component and are more similar to control samples than to chemotherapeutic samples. Two outlier samples miscluster and increase the confidence interval ellipses. (B) In lymphoblasts, there is little separation between control cells and the two treatment levels of MQ. At early time points when there is modest growth stimulation, the metabolite profiles diverge slightly, but by the end of the time course they are essentially identical. Green markers represent control samples, light blue represents 25 μM MQ treatment, dark blue represents 35 μM MQ treatment, dark red represents docetaxel treatment, and light red represents doxorubicin treatment. Pentagons represent 0 hour samples, circles represent 24 hour samples, squares represent 48 hour samples, and triangles represent 72 hour samples. Dotted lines represent 95% confidence intervals for individual classes.
Importantly, the MQ-treated and chemotherapeutic-treated samples are different from each other, being separated in the first principal component. The two different chemotherapeutic treatments tested have similar metabolic effects based on their similar values in the first principal component. This means that the major axis of variation in the data (PC1) captures the separation of chemotherapeutic-treated Jurkat cells from control and MQ-treated Jurkat cells, rather than separating the dying (chemotherapeutic and MQ-treated cells) from the controls. The metabolic activity associated with the anti-proliferative effects of MQ is captured in the second principal component, which does not correlate with chemotherapeutic treatment. This supports the idea that the metabolic changes observed in MQ-treated cells are not just due to general cell death and apoptosis processes, but are unique to MQ treatment.
By comparison, the MQ-treated lymphoblasts show very little separation from the control cells (Fig. 3B). There is some slight separation at the early time points in the second principal component, when there is a modest growth stimulation at both dosages of MQ. However, by 72 hours, the metabolite profiles are again identical, suggesting little metabolic impact on lymphoblasts. Again, this is consistent with univariate analyses.
Finally, enrichment analysis based on the relative levels of analytes under different treatment conditions across all time points identified multiple pathways that were statistically significantly enriched in the MQ-treated Jurkat cells. Enrichment relative to chemotherapeutic-treated cells is presented in Table 2; other enrichment comparisons are presented in Table S4, and details on the metabolites measured for each pathway are included in Table S5. All p-values are FDR-corrected.
Table 2. Pathways enriched for metabolic changes in MQ-treated Jurkat cells compared to chemotherapeutic-treated Jurkat cells.
Metabolites contributing to the significance of each pathway, along with their fold change across all time points, are included in Table S5.
| Pathway | FDR-corrected p-value |
|---|---|
| Propanoate metabolism | 0.000257 |
| Pyrimidine metabolism | 0.000257 |
| beta-Alanine metabolism | 0.000257 |
| Pantothenate and CoA biosynthesis | 0.000257 |
| Glycerophospholipid metabolism | 0.000649 |
| Sphingolipid metabolism | 0.002883 |
| Pentose phosphate pathway | 0.00919 |
| Alanine, aspartate and glutamate metabolism | 0.029828 |
| Glyoxylate and dicarboxylate metabolism | 0.029828 |
| Butanoate metabolism | 0.030357 |
| Arginine and proline metabolism | 0.030357 |
| Pentose and glucuronate interconversions | 0.030357 |
Phosphoethanolamine is significantly accumulated in MQ-treated Jurkat cells
The analyte with the most significant ANOVA p-value was phosphoethanolamine (PE). A detailed analysis of PE levels reveals that it is accumulated specifically in MQ-treated Jurkat cells as early as 24 h (approximately two-fold increase compared to vehicle-treated controls) and with quite high induction at 48 h and 72 h (approximately five-fold increase compared to controls), but this is not observed in chemotherapeutic-treated Jurkat cells or MQ-treated and control lymphoblast cells (Fig. 4). This suggests a mechanism specific to MQ-mediated cell death since extensive accumulation of PE does not happen until antiproliferative activity is observed while the same patterns are not observed in identically treated lymphoblasts. PE was the only analyte measured to display such a unique phenotype. To confirm the identity of PE, we obtained a pure standard and verified its retention time and mass spectrum in our samples.
Fig. 4. PE levels in MQ-treated (A) Jurkat and (B) lymphoblast cells.
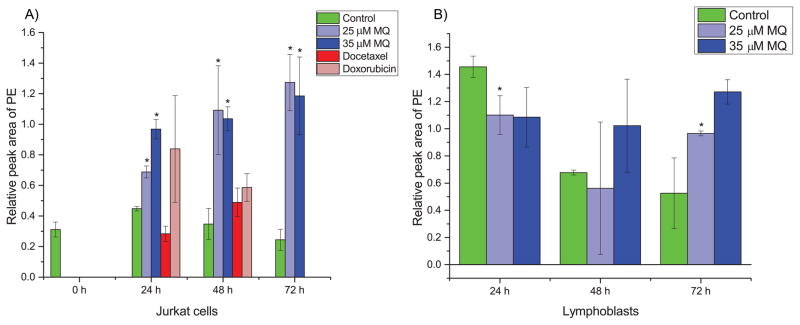
(A) PE is statistically significantly accumulated in MQ-treated Jurkat cells as early as 24 hours, with substantial accumulation by later times. Chemotherapeutic-treated cells show insignificant changes with slightly increasing trends much smaller than the magnitude of accumulation due to MQ treatment. (B) In lymphoblast cells, there is a suppression of PE levels at early time points due to MQ treatment, and at later time points usually insignificant changes with slightly increasing trends much less than the magnitude of accumulation in Jurkat cells. Error bars represent one standard deviation. Green bars represent control samples, light blue represents 25 μM MQ treatment, dark blue represents 35 μM MQ treatment, dark red represents docetaxel treatment, and light red represents doxorubicin treatment. * indicates statistical significance (p < 0.05, two tailed t-tests).
PE stimulates apoptosis in Jurkat cells but not in lymphoblasts
Based on this unique profile across all samples, we then hypothesized that PE itself may have similar activity to MQ. We identified from previous literature a concentration range over which PE was expected to show antiproliferative activity29. We treated both Jurkat cells and lymphoblasts with two different doses of PE from this range, 8 mM and 12 mM. The results, shown in Fig. 5, support that PE has apoptosis-inducing activity on Jurkat cells but not lymphoblasts, as indicated by both annexin and caspase 3/7 assays.
Fig. 5. PE stimulates apoptosis in Jurkat cells.

Cells were assayed for apoptotic markers after 72 hours of treatment. (A) In Jurkat cells, PE induces apoptosis based on both annexin and caspase 3/7 assays. (B) In lymphoblasts, PE does not induce apoptosis and may even slightly prevent apoptosis in cells present at 72 hours. Error bars represent one standard deviation. Light blue bars are treatment with 8 mM PE, dark blue are treatment with 12 mM PE, and green bars are control. * indicates significance at p < 0.05 and ** indicates significance at p < 0.005 (two-tailed t-tests).
MQ treatment also increases PE levels during antiproliferative effects on another cancer cell line
To test for the generality of the link between MQ and PE, we treated the ovarian cancer cell line OVCAR-3 with 100 μM MQ (based on a literature report30) and measured the resulting PE levels (Fig. 6). We found that the levels of PE went up after treatment with MQ, and corresponded with substantial antiproliferative activity of MQ (see Fig. S4). This suggests that the link between MQ and PE is not unique just to Jurkat cells, but may generalize to other types of cancer; if true, this would in turn suggest that therapeutics that leverage this connection could potentially be useful for a range of cancer types.
Fig. 6. PE also accumulates in MQ-treated ovarian cancer cells.

The accumulation of PE due to MQ treatment was also present in a totally different cell type, the OVCAR-3 ovarian cancer cell line. Levels of PE increased approximately nine-fold due to MQ treatment compared to control. * indicates statistical significance (p < 0.05, one-tailed t-test).
Discussion
We have used GCxGC-MS metabolomics to characterize the anticancer activity of menaquinone (MQ) in a leukemia model cell line (Jurkat). Our previous work used analysis of transcriptional data from Jurkat cells and noncancerous lymphoblasts to predict that MQ levels would be lower in Jurkat cells8. It was further hypothesized that if MQ was in fact depleted in Jurkat cells, then supplementing with MQ could in fact have an antiproliferative effect on the cells by pushing them back towards a more normal metabolic state, and thus potentially allowing for the induction of apoptosis or preventing proliferation. Initial experiments suggested this was true, and so we sought to confirm those findings and characterize the phenomenon in more detail. We found that MQ does in fact have an antiproliferative effect, at lower concentrations than previously found, and with some selectivity for Jurkat cells over lymphoblasts (particularly at those lower concentrations). In fact, at low concentrations MQ actually induces a modest but statistically insignificant increase in proliferation of lymphoblasts. We also demonstrated via two different well-known markers, annexin and caspase 3/7, that at anti-proliferative dosages, MQ induces apoptosis in Jurkat cells.
In further characterizing the metabolic impacts of MQ supplementation using GCxGC-MS metabolomics, we found that it causes significant, system-wide changes in the metabolism of a Jurkat model of leukemia, but causes fairly few changes in the metabolism of noncancerous lymphoblasts. Moreover, the metabolic changes identified in MQ-treated Jurkat cells are unique to MQ relative to two other known chemotherapeutic agents. These results are supportive of the hypothesis that MQ-induced apoptosis is somehow metabolically mediated, which is a logical extension of the prediction by our original computational model.
Among the annotated metabolites measured via GCxGC-MS, there were numerous noteworthy differences between the MQ-treated Jurkat cells, MQ-treated lymphoblasts, and all of the control cells. In terms of individually significant analytes between MQ-treated and chemotherapeutic-treated Jurkat cells, a number of metabolites have previously been implicated in apoptosis. For example, hydroxyproline (higher in MQ treatment) and one of the enzymes acting on it, hydroxyproline oxidase, have been implicated in apoptosis through the generation of reactive oxygen species (ROS)31. Similarly, hypotaurine has been shown to serve an antioxidant role32. In addition, asparagine has been shown to play a role in preventing apoptosis in glutamine-starved conditions in cancer cells33. Taken together, these unique aspects of MQ treatment suggest the possibility of substantial differences in terms of oxidative stress response and overall mechanisms of apoptosis, particularly as related to metabolic causes and protection against apoptosis.
There were also a number of pathways with significant enrichment for changes in MQ-treated Jurkat cells relative to chemotherapeutic-treated cells. Pathway-level analysis allows for the identification of where changes in metabolite levels may be clustered within metabolism, which can suggest potential relationships or functional links to previously identified metabolic changes in cancer. Pathway enrichment analysis also allows for the identification of metabolic changes via the combination of smaller alterations, rather than by focusing only on individually significant alterations. For example, the pathway analysis presented in Table 2 compared the MQ- and chemotherapeutic-treated Jurkat cells without considering that the samples within each group were at different time points, which inherently introduces significant within-class variability. While only seven metabolites were individually significant based on t-tests for this analysis (FDR < 0.05), enrichment of pathway-level changes could still be identified. Among the pathways identified as being enriched for changes, proline metabolism is widely known to be implicated in cancerous proliferation and is considered a potential target for treatment34–36. Similarly, metabolites in alanine, aspartate, and glutamine metabolism have been implicated as potential early markers for induction of apoptosis in certain cell lines37. Pyrimidine metabolism, and in general nucleotide metabolism, is widely known to play an important role in cancerous proliferation, as a sufficient pool of nucleotides is necessary for the requisite DNA replication of mitosis during proliferation38. Changes were also identified in lipid metabolism pathways; lipid metabolism is known to be implicated in cancer, and lipid signaling mediates many cellular mechanisms, including apoptosis39. Changes in lipid pathways could be indicative of some direct effect of menaquinone, an indirect effect mediated by phosphoethanolamine, or an even more indirect effect as the result of induced apoptosis. Taken together, though, these pathway-level metabolic differences suggest the basis for a more detailed exploration of the origin and function of the changes induced by MQ treatment.
One of the most interesting insights that directly emerged from the metabolomics analysis of MQ treatment was the metabolic profile of phosphoethanolamine (PE) across the different treatments and cell types. Phosphoethanolamine was significantly accumulated (approximately two-fold increase compared to vehicle-treated controls) starting at 24 h in the MQ-treated Jurkat cells, but not in the noncancerous lymphoblasts. By 72 h of MQ treatment, PE levels in the Jurkat cells were five-fold higher compared to vehicle-treated controls, while the lymphoblasts still showed no MQ-induced accumulation of PE. Similarly, treatment of Jurkat cells with two broad spectrum chemotherapeutics did not cause statistically significant increases in PE compared to vehicle-treated control; this indicates that PE accumulation is not just a common result of cell death, but instead is largely specific to MQ treatment. The specificity of PE accumulation, and the fact that this accumulation occurred earlier than the induction of apoptosis was observed, suggested that PE may be playing a causal role.
Based on this, we further hypothesized that PE may itself have an antiproliferative effect. Subsequent characterization identified that both PE and MQ treatment induce caspase 3/7-mediated apoptosis, indicating that at least part of the mechanistic pathway for PE and MQ antiproliferative activity is shared in common. Recently published work supports our predictions and experimental evidence of the pro-apoptotic potential of exogenous phosphoethanolamine for leukemic cell models29 and other types of cancer40, 41 (though the previous study did not characterize the effect of PE on noncancerous lymphoblasts). It is worth noting that the treatment levels of PE in our experiments were rather high (8–12 mM), at what may not be physiologically relevant serum concentration levels. However, these levels are consistent with previous literature observations and may be necessary to offset the fact that phosphoethanolamine (without a strong nonpolar moiety but containing a phosphate group) is likely inefficiently taken up by the cells. Also, the induction of apoptosis by PE was moderate (in less than 20% of intact cells, a roughly two-fold increase) relative to the results of MQ treatment (in approximately 30% of intact cells, a roughly three-fold increase), but it is still statistically significant. The difference in efficiency of apoptosis induction could indicate that the metabolites are exerting their effects via different pathways, or it could be due to differences in metabolite uptake as previously discussed.
We also showed that the intersection of MQ and PE may be a more general phenomenon beyond just the Jurkat model of leukemia. Using a cell line from a different type of cancer (OVCAR-3, an ovarian epithelial cancer cell line), we found that MQ had antiproliferative activity and induced increased levels of PE after treatment. In this model, the increase of PE levels caused by MQ treatment was even more substantial, yielding approximately nine-fold higher levels than in the vehicle-treated controls. This suggests that the relationship we discovered between MQ and PE is not specific to one cell-line, or even to one cell type.
Looking towards the possible mechanistic basis for this connection, MQ’s known functional roles do not provide immediate insight. MQ’s role in blood coagulation and bone mineralization is well characterized9–11. All forms of vitamin K (including MQ) function as cofactors for γ-glutamyl carboxylase (GGCX), catalyzing the γ-carboxylation (a post-translational modification) of substrate proteins, which is an essential step for the function of substrates such as blood coagulation factors42 and osteocalcin43. Vitamin K is reduced to Vitamin K hydroquinone and, upon carboxylation, is oxidized to Vitamin K epoxide (VKO). VKO is reduced back to Vitamin K for reuse42. MQ’s potential role in cancer is comparatively poorly understood. Previous studies have shown that MQ can have antiproliferative effects: it can trigger both apoptotic and autophagic programmed cell death, as well as both intrinsic and extrinsic pathways of apoptosis, in a variety of cell types including colon cancer12, acute myeloid leukemia13, hepatocellular carcinoma14, 15, prostate cancer16, and cervical carcinoma17. MQ is also known to bind directly to BAK (Bcl2 antagonist or killer, a pro-apoptotic protein) and is associated with apoptosis17. It induces posttranslational modifications and forms a BAK isoform by covalent binding of its intracellular metabolite VKO to a cysteine residue of BAK17. Adding the free radical scavenger α-tocopherol prevents BAK isoform formation and apoptosis upon treatment with MQ17, 44. However, when the essential function of MQ, γ-carboxylation, was blocked by γ-glutamyl carboxylase (GGCX) knockdown in the LNCaP prostate cancer cell line, this also abrogated the pro-apoptotic activity of MQ treatment45. This shows that the mechanism of MQ-triggered apoptosis is complex and interacts with cell death pathways at many levels. To our knowledge our study is the first to use a metabolomics approach to characterize MQ-triggered apoptosis and to identify the intersection of PE elevation and cancer cell death in response to MQ treatment.
PE also has some well-characterized functions and roles in cells, though none are directly related to MQ. PE is a central precursor for biosynthesis of phosphatidylethanolamine, a membrane phospholipid which is an essential, major membrane constituent and plays vital roles in cell signaling, membrane fusion, cell division and apoptosis46. Products of phosphatidylethanolamine metabolism (e.g. fatty acids, diacyl glycerol and phosphatidic acids) also act as second messengers in many other signaling pathways47, 48, suggesting a possible function of modulation of PE levels in initiating apoptosis.
However, none of this prior knowledge about PE’s and MQ’s cellular functions, nor knowledge of the metabolic pathways of PE and MQ, suggests how or why these two would be linked. The fact that the relationship between MQ and PE is evident in multiple cancer types, and that it was not associated with cancer cell death caused by chemotherapeutics, suggests that there may in fact be a functional link between the two. However, intracellular modulation of PE levels during MQ treatment is an extremely difficult task, and thus we cannot conclude here, based only on the presented data, that PE accumulation mediates the effects of MQ treatment. Identification of a functional link between MQ and PE is thus a particularly interesting direction for future exploration. In that regard, a state of the art virtual ligand screening approach49, FINDSITEcomb, suggests that both MQ and PE putatively bind stAR-related lipid transfer protein 13 isoform beta (DLC2), which is implicated in tumor suppression50, providing one candidate mechanistic link for future investigation.
We note that metabolites measured by GCxGC-MS may be identified based on their mass spectra and retention time, but due to the incompleteness of mass spectral databases and limited availability of analytical standards, numerous analytes may not be assigned a metabolite identity even though they may be tracked from sample to sample. Some of the conservative choices in our data processing pipeline, meant to minimize the number of false metabolite identity annotations, likely contribute to the number of unknown analytes tracked in our studies. As such, while we only consider the biological implications of annotated metabolites, we still include the unannotated analytes in our considerations of clustering and multivariate analysis. We also note that even despite our conservative choices in the data processing pipeline, some annotated metabolites may still be misannotated.
Also, while the idea of metabolite-based therapeutics is promising in the long term, this current model of MQ in ALL does not have as large a therapeutic window as would be desirable, which could make complete elimination of treatment side effects difficult. However, given the known significant side effects of most chemotherapeutics and the fact that MQ is essentially an unoptimized lead, it is at least a promising start. If nothing else, it helps us to identify unique aspects of metabolism that could lead to the discovery of even more selective molecules or targets.
In conclusion, our work is the first-reported identification of the link between MQ and PE in antileukemic activity and the first to quantify the semi-selectivity of each analyte in its respective pro-apoptotic activity. It suggests the potential importance of decoupling MQ treatment from PE production to identify whether PE plays a functional role in the MQ-mediated induction of apoptosis. Further work in this area may help to clarify whether either molecule could be useful as a therapeutic lead, or may help identify relevant targets for future therapeutic development.
Supplementary Material
Acknowledgments
The authors acknowledge the NIH for funding via R21CA167500, thank Robert Dromms for performing LOESS correction, and thank Megan Cole for a critical review of the manuscript.
References
- 1.Hanahan D, Weinberg RA. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Chen Z, Lu W, Garcia-Prieto C, Huang P. J Bioenerg Biomembr. 2007;39:267–274. doi: 10.1007/s10863-007-9086-x. [DOI] [PubMed] [Google Scholar]
- 3.Siegel R, Ma J, Zou Z, Jemal A. CA: a cancer journal for clinicians. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 4.Tan SY, Poh BK, Nadrah MH, Jannah NA, Rahman J, Ismail MN. Journal of human nutrition and dietetics : the official journal of the British Dietetic Association. 2013;26(Suppl 1):23–33. doi: 10.1111/jhn.12074. [DOI] [PubMed] [Google Scholar]
- 5.Bobrovnikova-Marjon E, Hurov JB. Annual review of medicine. 2014;65:157–170. doi: 10.1146/annurev-med-092012-112344. [DOI] [PubMed] [Google Scholar]
- 6.Locasale JW. Nature reviews Cancer. 2013;13:572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reynolds CP, Maurer BJ, Kolesnick RN. Cancer letters. 2004;206:169–180. doi: 10.1016/j.canlet.2003.08.034. [DOI] [PubMed] [Google Scholar]
- 8.Arakaki AK, Mezencev R, Bowen NJ, Huang Y, McDonald JF, Skolnick J. Mol Cancer. 2008;7:57. doi: 10.1186/1476-4598-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cranenburg EC, Schurgers LJ, Vermeer C. Thrombosis and haemostasis. 2007;98:120–125. [PubMed] [Google Scholar]
- 10.Groenen-van Dooren MM, Soute BA, Jie KS, Thijssen HH, Vermeer C. Biochemical pharmacology. 1993;46:433–437. doi: 10.1016/0006-2952(93)90519-3. [DOI] [PubMed] [Google Scholar]
- 11.Mochizuki M, Shimizu S, Kitazawa T, Umeshita K, Goto K, Kamata T, Aoki A, Hatayama K. The Journal of toxicological sciences. 2008;33:307–314. doi: 10.2131/jts.33.307. [DOI] [PubMed] [Google Scholar]
- 12.Kawakita H, Tsuchida A, Miyazawa K, Naito M, Shigoka M, Kyo B, Enomoto M, Wada T, Katsumata K, Ohyashiki K, Itoh M, Tomoda A, Aoki T. International journal of molecular medicine. 2009;23:709–716. doi: 10.3892/ijmm_00000184. [DOI] [PubMed] [Google Scholar]
- 13.Yokoyama T, Miyazawa K, Naito M, Toyotake J, Tauchi T, Itoh M, Yuo A, Hayashi Y, Georgescu MM, Kondo Y, Kondo S, Ohyashiki K. Autophagy. 2008;4:629–640. doi: 10.4161/auto.5941. [DOI] [PubMed] [Google Scholar]
- 14.Yao Y, Li L, Zhang H, Jia R, Liu B, Zhao X, Zhang L, Qian G, Fan X, Ge S. Oncology letters. 2012;4:163–167. doi: 10.3892/ol.2012.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li L, Qi Z, Qian J, Bi F, Lv J, Xu L, Zhang L, Chen H, Jia R. Molecular and cellular biochemistry. 2010;342:125–131. doi: 10.1007/s11010-010-0476-8. [DOI] [PubMed] [Google Scholar]
- 16.Samykutty A, Shetty AV, Dakshinamoorthy G, Kalyanasundaram R, Zheng G, Chen A, Bosland MC, Kajdacsy-Balla A, Gnanasekar M. Evidence-based complementary and alternative medicine : eCAM. 2013;2013:287358. doi: 10.1155/2013/287358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karasawa S, Azuma M, Kasama T, Sakamoto S, Kabe Y, Imai T, Yamaguchi Y, Miyazawa K, Handa H. Molecular pharmacology. 2013;83:613–620. doi: 10.1124/mol.112.082602. [DOI] [PubMed] [Google Scholar]
- 18.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Gao D, Chen Z, Li S, Gao C, Cao D, Liu F, Liu H, Jiang Y. PLoS One. 2013;8:e63572. doi: 10.1371/journal.pone.0063572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sellick CA, Hansen R, Maqsood AR, Dunn WB, Stephens GM, Goodacre R, Dickson AJ. Anal Chem. 2009;81:174–183. doi: 10.1021/ac8016899. [DOI] [PubMed] [Google Scholar]
- 21.Sellick CA, Hansen R, Stephens GM, Goodacre R, Dickson AJ. Nat Protoc. 2011;6:1241–1249. doi: 10.1038/nprot.2011.366. [DOI] [PubMed] [Google Scholar]
- 22.Kind T, Wohlgemuth G, Lee do Y, Lu Y, Palazoglu M, Shahbaz S, Fiehn O. Anal Chem. 2009;81:10038–10048. doi: 10.1021/ac9019522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vermeersch KA, Wang L, McDonald JF, Styczynski MP. BMC Syst Biol. 2014;8:134. doi: 10.1186/s12918-014-0134-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vermeersch KA, Wang L, Mezencev R, McDonald JF, Styczynski MP. PLoS One. 2015;10:e0118262. doi: 10.1371/journal.pone.0118262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei XL, Shi X, Koo I, Kim S, Schmidt RH, Arteel GE, Watson WH, McClain C, Zhang X. Bioinformatics. 2013;29:1786–1792. doi: 10.1093/bioinformatics/btt275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunn WB, Broadhurst D, Begley P, Zelena E, Francis-McIntyre S, Anderson N, Brown M, Knowles JD, Halsall A, Haselden JN, Nicholls AW, Wilson ID, Kell DB, Goodacre R, HSMHC Nature Protocols. 2011;6:1060–1083. doi: 10.1038/nprot.2011.335. [DOI] [PubMed] [Google Scholar]
- 27.Xia J, Mandal R, Sinelnikov IV, Broadhurst D, Wishart DS. Nucleic acids research. 2012;40:W127–133. doi: 10.1093/nar/gks374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menendez JA, Oliveras-Ferraros C, Cufi S, Corominas-Faja B, Joven J, Martin-Castillo B, Vazquez-Martin A. Cell cycle. 2012;11:2782–2792. doi: 10.4161/cc.20948. [DOI] [PubMed] [Google Scholar]
- 29.Ferreira AK, Santana-Lemos BA, Rego EM, Filho OM, Chierice GO, Maria DA. British journal of cancer. 2013;109:2819–2828. doi: 10.1038/bjc.2013.510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sibayama-Imazu T, Fujisawa Y, Masuda Y, Aiuchi T, Nakajo S, Itabe H, Nakaya K. J Cancer Res Clin Oncol. 2008;134:803–812. doi: 10.1007/s00432-007-0349-z. [DOI] [PubMed] [Google Scholar]
- 31.Cooper SK, Pandhare J, Donald SP, Phang JM. The Journal of biological chemistry. 2008;283:10485–10492. doi: 10.1074/jbc.M702181200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aruoma OI, Halliwell B, Hoey BM, Butler J. The Biochemical journal. 1988;256:251–255. doi: 10.1042/bj2560251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Fan J, Venneti S, Cross JR, Takagi T, Bhinder B, Djaballah H, Kanai M, Cheng EH, Judkins AR, Pawel B, Baggs J, Cherry S, Rabinowitz JD, Thompson CB. Molecular cell. 2014;56:205–218. doi: 10.1016/j.molcel.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW, Phang JM. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:8983–8988. doi: 10.1073/pnas.1203244109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phang JM, Donald SP, Pandhare J, Liu Y. Amino acids. 2008;35:681–690. doi: 10.1007/s00726-008-0063-4. [DOI] [PubMed] [Google Scholar]
- 36.Phang JM, Liu W, Hancock C. Epigenetics : official journal of the DNA Methylation Society. 2013;8:231–236. [Google Scholar]
- 37.Halama A, Riesen N, Moller G, Hrabe de Angelis M, Adamski J. Journal of internal medicine. 2013;274:425–439. doi: 10.1111/joim.12117. [DOI] [PubMed] [Google Scholar]
- 38.Aird KM, Zhang R. Cancer letters. 2015;356:204–210. doi: 10.1016/j.canlet.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang C, Freter C. Int J Mol Sci. 2015;16:924–949. doi: 10.3390/ijms16010924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferreira AK, Freitas VM, Levy D, Ruiz JL, Bydlowski SP, Rici RE, Filho OM, Chierice GO, Maria DA. PLoS One. 2013;8:e57937. doi: 10.1371/journal.pone.0057937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ferreira AK, Meneguelo R, Pereira A, Filho OM, Chierice GO, Maria DA. Biomed Pharmacother. 2013;67:481–487. doi: 10.1016/j.biopha.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 42.Stafford DW. J Thromb Haemost. 2005;3:1873–1878. doi: 10.1111/j.1538-7836.2005.01419.x. [DOI] [PubMed] [Google Scholar]
- 43.Hauschka PV, Lian JB, Gallop PM. Proceedings of the National Academy of Sciences of the United States of America. 1975;72:3925–3929. doi: 10.1073/pnas.72.10.3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shibayama-Imazu T, Sonoda I, Sakairi S, Aiuchi T, Ann WW, Nakajo S, Itabe H, Nakaya K. Apoptosis. 2006;11:1535–1543. doi: 10.1007/s10495-006-7979-5. [DOI] [PubMed] [Google Scholar]
- 45.Akamatsu S, Takata R, Haiman CA, Takahashi A, Inoue T, Kubo M, Furihata M, Kamatani N, Inazawa J, Chen GK, Le Marchand L, Kolonel LN, Katoh T, Yamano Y, Yamakado M, Takahashi H, Yamada H, Egawa S, Fujioka T, Henderson BE, Habuchi T, Ogawa O, Nakamura Y, Nakagawa H. Nat Genet. 2012;44:426–429. S421. doi: 10.1038/ng.1104. [DOI] [PubMed] [Google Scholar]
- 46.Zhu L, Johnson C, Bakovic M. J Lipid Res. 2008;49:2197–2211. doi: 10.1194/jlr.M800259-JLR200. [DOI] [PubMed] [Google Scholar]
- 47.Bakovic M, Fullerton MD, Michel V. Biochem Cell Biol. 2007;85:283–300. doi: 10.1139/o07-006. [DOI] [PubMed] [Google Scholar]
- 48.Eyster KM. Adv Physiol Educ. 2007;31:5–16. doi: 10.1152/advan.00088.2006. [DOI] [PubMed] [Google Scholar]
- 49.Zhou H, Skolnick J. J Chem Inf Model. 2013;53:230–240. doi: 10.1021/ci300510n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ching YP, Wong CM, Chan SF, Leung TH, Ng DC, Jin DY, Ng IO. The Journal of biological chemistry. 2003;278:10824–10830. doi: 10.1074/jbc.M208310200. [DOI] [PubMed] [Google Scholar]
- 51.Storey JD, Tibshirani R. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.