

Abstract
Wnt signaling plays an essential role in developmental and regenerative myelination of the CNS, therefore it is critical to understand how the factors associated with the various regulatory layers of this complex pathway contribute to these processes. Recently, Apcdd1 was identified as a negative regulator of proximal Wnt signaling, however its role in oligodendrodcyte (OL) differentiation and reymelination in the CNS remain undefined. Analysis of Apcdd1 expression revealed dynamic expression during OL development, where its expression is upregulated during differentiation. Functional studies using ex vivo and in vitro OL systems, revealed that Apcdd1 promotes OL differentiation, suppresses Wnt signaling, and associates with β-catenin. Application of these findings to white matter injury (WMI) models revealed that Apcdd1 similarly promotes OL differentiation after gliotoxic injury in vivo and acute hypoxia ex vivo. Examination of Apcdd1 expression in white matter lesions from neonatal WMI and adult Multiple Sclerosis revealed its expression in subsets of oligodendrocyte precursors. These studies describe, for the first time, the role of Apcdd1 in OLs after WMI and reveal that negative regulators of the proximal Wnt pathway can influence regenerative myelination, suggesting a new therapeutic strategy for modulating Wnt signaling and stimulating repair after WMI.
Keywords: Apcdd1, Oligodendrocyte, White matter injury, Wnt signaling
Introduction
Oligodendrocytes (OLs) produce and assemble myelin sheaths that insulate axons in the CNS and are required for saltatory conduction. Loss or dysfunction of OLs, coupled with a failure to regenerate myelin, results in white matter injury (WMI) and subsequent neuronal impairment and loss (Chang et al., 2000; Compston and Coles, 2008; Kuhlmann et al., 2008). The neurologic consequences of WMI in humans are demyelinating disorders such as Multiple Sclerosis (MS) in adults and Cerebral Palsy (CP) in infants (Back and Rosenberg, 2014; Lassmann, 2014). In these conditions, the ability of oligodendrocyte precursors (OLPs) to remyelinate white matter lesions is often impaired, as “stalled” OLP populations are found within sites of WMI in humans (Chang et al., 2000; Emery, 2010; Franklin RJ, 2002; Kuhlmann et al., 2008). Therefore a comprehensive understanding of the regulatory factors that contribute to the inhibition of OLP differentiation and remyelination after WMI may provide new insight into the pathogenesis of these devastating neurological disorders.
Recent studies revealed that chronic activation of canonical Wnt signaling in OLPs results in delayed remyelination due to inhibition of OLP differentiation (Fancy et al., 2009; Fancy et al., 2011). For example, activated Tcf4/β-catenin signaling acts as a negative regulator of OL differentiation during developmental and regenerative myelination (Fancy et al., 2009). In addition Axin2, a transcriptional target of canonical Wnt singnaling negatively feeds back on the pathway, resulting in β-catenin degradation, thus loss of Axin2 results in delayed OL differentiation during development and after WMI (Fancy et al., 2011). These observations implicate Wnt signaling in the “failure to remyelinate” manifest in chronic MS and newborn brain injury (Guo, et al 2015). Given these links between the activation of Wnt signaling and OLP differentiation in human WMI, a deeper understanding of how Wnt signaling contributes to these disorders is likely to reveal critical insight into the pathology and treatment of WMI.
While these prior studies were the first to implicate the Wnt pathway in human WMI, they focused solely on how the distal, transcriptional components of the Wnt pathway influenced OLP differentiation. Given the complexity of Wnt signaling, which is comprised of numerous components and several regulatory layers, it is likely that other components of this pathway also contribute to the regulation of OLP differentiation (Bilić et al., 2007; Pan et al., 2008; Fancy et al., 2009; Ye, et al. 2010; Azim and Butt, 2011). Binding of Wnt ligands to its cell surface receptors consisting of Frizzled (Fz) and low-density lipoprotein receptor-related protein 6 (LRP6) results in stabilization of β-catenin and subsequent distal transcriptional events. While proximal ligand/receptor interactions are crucial for Wnt signaling, how these relationships influence OLP differentiation remainpoorly defined. Indeed, several studies have demonstrated that disruption of Wnt ligand-receptor interaction by secreted protein DKK1 disrupts LRP6/Fz interactions, resulting in decreased Wnt/β-catenin signaling and inhibition of Wnt receptor clustering mechanisms can promote OLP differentiation and remyelination (Bafico et al., 2001; Mao et al., 2001; Semënov et al., 2001; Lee at al. 2015). These observations suggest that manipulating the inhibitory components of Wnt signaling could influence OLP differentiation. Importantly, these proximal regulators of the Wnt pathway represent a potentially more tractable therapeutic target than distal transcriptional components because they function at the cell membrane and are more accessible for pharmacological manipulation.
Previously we demonstrated that Apcdd1 is expressed in glial precursors during embryonic spinal cord development, however its role in OLP differentiation remains undefined (Kang et al., 2012). Interestingly, other groups have shown that Apcdd1 binds LRP6 receptors and antagonizes canonical Wnt signaling in hereditary hypotrichosis simplex (Shimomura et al., 2010). This observation, put together with the expression of Apcdd1 in glial lineages, raises the possibility that it may function to antagonize Wnt signaling and promote OLP differentiation. Using this premise as our starting point, we found that Apcdd1 is expressed in the OL lineage in the developing and adult spinal cord, and in human WMI lesions. Because Apcdd1 is a Wnt antagonist, we focused on the more clinically relevant overexpression studies, finding that Apcdd1 is sufficient to promote OLP differentiation in vitro and after WMI. Mechanistically, we found that Apcdd1 associates with key regulators of Wnt signaling and antagonizes Wnt-activation. These functional studies, taken together with our characterization in human WMI, are the first to identify Apcdd1 as a critical regulator of canonical Wnt signaling during OL differentiation and regenerative myelination in the CNS.
Material and Methods
Constructs
Apcdd1 constructs were sub-cloned into the GFP tagged FUIGW virus backbone following as previously described (Kang et al., 2012). Tagged cDNA for Apcdd1 was cloned into mammalian expression vectors (3xFlag or Myc-pcDNA). All of the plasmid constructs were confirmed by DNA sequencing.
In Situ Hybridization and Immunohistochemistry
Spinal cords were fixed in 4% paraformaldehyde, and then cryoprotected in 20% sucrose. Mouse spinal cord was analyzed using colorimetric in situ hybridization as previously described (Kang et al., 2012). Immunohistochemistry: mouse anti-MBP (Covance 1:500), mouse anti-PLP (1:500), rabbit anti-Olig2 (Abcam 1:1000), rabbit anti-GFAP (DAKO 1:1000), rabbit anti-Iba1 (Wako 1:600).
To perform the two-color fluorescent in situ hybridization using the TSA-FITC/TSA-Cy5 system, we generated FITC labeled Apcdd1 probes, and combined with the probes of DIG labeled oligodendrocyte lineage markers (PDGFRα, PLP). Day 1 of the two-color fluorescent in situ hybridization was performed the same as for the colorimetric in situ. For day 2 of in situ, the slides were washed with 0.1X SSC and endogenous peroxidase activity was quenched by incubation in 3% H2O2 for 20 minutes. After quenching step, the slides were blocked with 0.5% of a blocking reagent containing TN (100 mM Tris-HCl, pH 7.5, 150 mM NaCl). The following antibodies were used to detect either FITC or DIG labeled probes; anti-FITC-POD, along with FITC-Tyramide; anti-DIG-POD with Cy5-Tyramide in Amplification Reagent (TSA™ kit).
Human WMI staining
All human tissue was collected in accordance with guidelines established by Baylor College of Medicine (H-35483) and the University of California San Francisco Committee on Human Research (H11170-19113-07). A retrospective review of neonatal autopsy cases for a pathologic diagnosis of periventricular leukomalacia, telencephalic leukoencephalopathy, or hypoxic-ischemic encephalopathy (HIE) was performed. Hematoxylin and eosin-stained slides were reviewed to confirm the diagnosis. The brain was previously fixed in 20% formalin for two weeks, cut in the coronal plane and representative sections were fixed in 20% formalin for an additional 2 days. Brain tissue was then paraffin-embedded and cut into 5-um-thick sections. The procurement of tissue and its pathological diagnosis has been described in detail previously (Fancy et al., 2011). The neonatal sample included in figure 6A–B is from a 12 week old, former 27 week gestational age (GA) with multiple complications of prematurity. Pathology in this case shows telencephalic leukoencephalopathy with diffuse white matter gliosis. We have assessed Apcdd1 expression from three similar cases, all between 27–29 week GA, as well as 38 week HIE case, and all demonstrate similar patterns of expression as shown in fig. 6A–B. The human MS tissue was provided by the Rocky Mountain Multiple Sclerosis Center Tissue Bank. The sample included in figure 6C–D is from a 63 year-old male, with clinical MS and lesions within the periventricular white matter. We assessed Apcdd1 expression in one additional periventricular white matter lesion from a 71 year-old female, and found similar staining as shown in fig. 6C–D.
Figure 6. Expression of Apcdd1 in human white matter injury.

(A–B) In situ hybridization of Apcdd1 and immunostaining for Olig2 in lesions from human neonatal white matter injury (WMI). (C–D) Anlysis of Apcdd1 and Olig2 co-labeling in adult Multiple Sclerosis lesions. In both cases, Apcdd1 demonstrates heterogeneous expression in oligodendrocyte precursors in human WMI tissue; it demonstrates very high- (black, filled arrows) or very low- (unfilled arrows) levels of expression in subpopulations of Olig2-expressing cells. Moreover, there are several Olig2-expressing cells that do not exhibit any Apcdd1 expression (red arrows).
Cell Culture, Immunoprecipitation, Western Blotting and Reporter Assay
Human HEK293 was maintained in DMEM in the presence of 10% FBS. For the IP transfections, 5 ug of Myc-Apcdd1 and Flag-β-catenin were transfected in HEK293 cells using calcium phosphate transfection methods. We performed the immuno-precipitation using antibodies for the protein tags, then analyzed by SDS PAGE/immunoblotting.
For the luciferase reporter assay, Oli-Neu cells were maintained in DMEM in the presence of 5% horse serum and 10% FBS with N2 supplement, and transfected with Top-flash reporter construct (30 ng) and Apcdd1 (50 ng, 150 ng) and CMV-β-galactosidase vector (50 ng) using lipofecatamine 2000 transfection reagent (Invitrogen). Cells were harvested 48hrs later and lysates analyzed for luciferase activity. β-galactosidase was used to normalize for transfection efficiency.
Cerebellar ex vivo Slice culture
Slice culture was performed as described previously (Birgbauer et al., 2004; Fancy et al., 2011). Briefly, the sagittal slices of postnatal day 0–1 mouse cerebellum were cut at 350 uM using a McIlwain tissue chopper, the slices were plated on Millicell-CMTM culture inserts (Millipore, 0.4μm) in slice culture medium (50% MEM with Earle’s salts, 35% Earle’s balanced salt solution, 25% heat-inactivated horse serum, glutamax, fungizone, penicillin-streptomycin, and glucose) for up to 14–28 days at 37 °C.
The hypoxic slice cultures were exposed to 1% O2 conditions for 48hrs after 2 days in culture and then returned to normal culture conditions. Either Apcdd1-lentivirus or control virus was added following hypoxia exposure. The hypoxic slices were fixed at 18 days in vitro.
For immunostaining of slice cultures, slices were fixed in 4% PFA for 1h while attached to membranes. Slices were then rinsed in PBS, blocked with 3% heat-inactivated horse serum, 2% bovine serum albumin, and 0.3% Triton X-100 in PBS, then incubated overnight at 4°C in primary antibody diluted in block solution. Primary antibodies used were mouse monoclonal anti-MBP (Covance 1:500).
Oligodendrocyte precursor culture
Oligosphere cultures were performed as previously described (Fancy et al., 2012; Harrington et al., 2010). For viral infection of OLPs, cells were dissociated and plated on Poly-D-Lysine-coated coverslips at a density of 1.5 × 104cells/cm2 in Oligodendrocyte Precursor Media (OPM) media subsequently infected with Apcdd1-lenti-GFP or control GFP fluorescent protein virus for 14 hours.
Lysolecithin-induced white matter demyelination model
Lysolecithin induced demyelinated lesions were generated in the white matter of the ventrolateral spinal cord of 8~10 week old mice. For viral misexpression studies, the animal was injected with lysolecithin in white matter lesion of spinal cord, followed by secondary focal injection of the lesioned region with either a Apcdd1-containing lentivirus or control lentivirus at 3 day post lesion (dpl). For these viral experiments, mice were perfused at 10 dpl.
Results
Apcdd1 is expressed in oligodendrocytes during CNS development
Previously we found that Apcdd1 expression is induced in ventricular zone (VZ) populations during the onset of gliogenesis in the developing spinal cord (Kang et al., 2012). To further examine its expression characteristics during developmental gliogenesis, we performed in situ hybridization during gliogenic stages and found that Apcdd1 is expressed in migrating cell populations in the mantle regions that likely represent presumptive migrating glial populations (Fig. 1A–B). Moreover, we found that Apcdd1 is expressed in endothelial cells that occupy the spinal cord.
Figure 1. Apcdd1 is dynamically expressed in the oligodendrocyte lineage.

(A–B) In situ hybridization of Apcdd1 expression in the embryonic spinal cord at E12.5 and E14.5; note that Apcdd1 is expressed throughout the dorsal-ventral extent of the ventricular zone at E12.5, filled arrows. Unfilled arrows denote endothelial cell expression. (C) Quantification of the number of PDGFRα (black bars), RIP-1 (blue bar), and PLP (red bars) cells that co-express Apcdd1 at P7 and in the adult. (D–I) Double in situ hybridization with Apcdd1 and PDGFRα in the spinal cord at P7 and in the adult (~3 months); unfilled arrows denote non-overlapping expression of Apcdd1 and PDGFRα. (J–O) Double in situ hybridization with Apcdd1 and PLP in the spinal cord at P7 and in the adult; white arrows denote overlapping expression of Apcdd1 and PLP. (P–R) (J–O) Double labeling with Apcdd1 and RIP-1 in the adult; white arrows denote overlapping expression of Apcdd1 and PLP. Each double staining experiment was executed at least three times and images are representative. Quantification in C is derived from at least three sections from each of the three independent labeling experiments. Error bars are SD.
The gross patterns of Apcdd1 expression suggest that it is expressed in migrating glial populations, and is consistent its role in astrocyte precursor migration (Kang, et al. 2012). Interestingly, Apcdd1 is also expressed throughout the dorsal ventral axis of the spinal cord during the formative stages of gliogenesis, including the pMN domain, which contains oligodendrocyte precursors (OLPs). Given its expression in this key population of cells during early glial development, we sought to define the expression dynamics of Apcdd1 during oligodendrocyte (OL) lineage development by assessing its co-expression with OLP markers using double in situ hybridization (ISH). Double labeling with the OLP marker, PDGFRα revealed nominal co-localization with Apcdd1, indicating that it is very lowly expressed in OLP populations in the early post-natal and adult spinal cord (Fig. 1D–I; C). Next, we assessed Apcdd1 expression in intermediate and mature OL populations, finding that it is co-localized with PLP and expressed in mature OL populations in both the post-natal and adult spinal cord and strongly co-localizes with the intermediate lineage marker RIP-1 (Fig. 1J–R; C). Put together our double ISH staining experiments indicate that Apcdd1 demonstrates a dynamic pattern of expression in the OL lineage, where it is very lowly expressed in OLP populations and is upregulated as these cells differentiate into mature OLs.
Apcdd1 promotes OLP differentiation
The forgoing observation that Apcdd1 is lowly expressed in OLPs and upregulated in mature OL populations is consistent with its putative role as a negative regulator of Wnt signaling. These expression dynamics, coupled with the link between inhibition of Wnt signaling and OLP differentiation, raises the question of whether overexpression of Apcdd1 suppresses Wnt signaling and promotes OLP differentiation. To address this we investigated the effects of Apcdd1 overexpression on OLPs differentiation using mouse cerebellar slice cultures. Slices of P0 neonatal cerebellum were cultured in the presence of either GFP tagged Apcdd1 virus and the effects on OLP differentiation in the cerebellar white matter were assess two weeks later. Our analysis revealed that overexpression of Apcdd1 led to enhanced OLP differentiation, as indicated by an increase in number of MBP positive OLs and presence of elaborated processes, compared to the GFP control (Fig. 2A–E).
Figure 2. Overexpression of Apcdd1 suppresses oligodendrocyte differentiation.
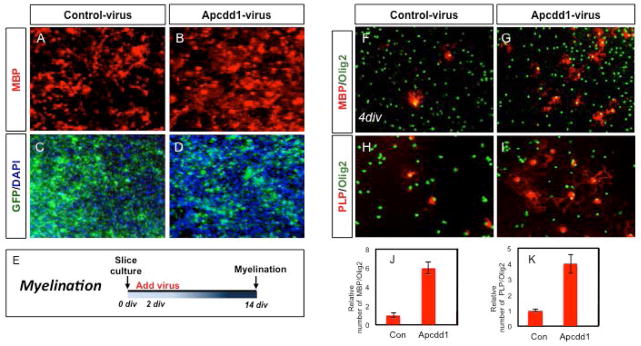
(A–E) Overexpression of Apcdd1 in ex vivo cerebellar slice culture. Cultures were infected with either GFP (A,C) or Apcdd1 (B,D) viruses and oligodendrocyte differentiation was analyzed through staining for MBP. (E) Schematic of experimental paradigm, virus was added to culture after 2 days in vitro (DIV) and analyzed 12 days later (i.e. at 14 DIV). (F–K) Oligodendrocyte progenitor (OLP) culture experiments. OLPs were generated and infected with GFP (F,H) or Apcdd1 (G,I) viruses and oligdodendrocyte differentiation was analyzed through staining for MBP and PLP. OLPs were infected with virus and allowed to differentiate two days later; differentiation was analyed four days later. Graphs in J–K are derived from three independent experiments, performed in triplicate (t-test, p=0.007).
Next, we complemented these ex vivo, slice culture experiments with in vitro OLP culture studies. Here we generated OLP cultures and after mitogen withdrawal provoked differentiation, infected with Apcdd1 or GFP viruses, assessing the extent of OL differentiation four days later. Our analysis revealed that overexpression of Apcdd1 resulted in a significant increase in the number of MBP, PLP, expressing cells without any effect on the expression of OLP marker Olig2 (Fig. 2F–K). Collectively, these ex vivo and in vitro studies indicate that Apcdd1 is sufficient to promote OLP differentiation.
Apcdd1 suppresses Wnt signaling via association with β-catenin
Previous studies demonstrated that Apcdd1 overexpression antagonizes Wnt3A induction of a Wnt-reporter (TOP-FLASH) (Shimomura et al., 2010). To extend these studies to the OL lineage, we performed analogous reporter assays in an oligodendrocyte cell line, Oli-Neu cells, and found that Apcdd1 similarly antagonizes transcriptional activity induced by Wnt3a (Fig. 3A). To better understand how Apcdd1 suppresses Wnt activation, we examined its ability antagonize LRP6 and β-catenin dependent Wnt induction, finding that it similarly inhibits Wnt induction in the presence of LRP6 and β-catenin (Fig. 3A). That Apcdd1 interferes with LRP6 and Wnt3a induction of Wnt activity is not surprising, given that it associates with LRP6 at the Wnt receptor complex, however its ability to antagonize β-catenin induction was unexpected. To further investigate the relationship between Apcdd1 and β-catenin, we determined whether they associate by performing co-immunoprecipitation (co-IP) experiments in 293 cells. Overexpression of tagged versions of Apcdd1 and β-catenin resulted in co-IP of the tagged proteins, indicating that they can associate under these conditions (Fig. 3B). This biochemical phenomenon, coupled with our TOP-FLASH reporter data, suggest that Apcdd1 antagonizes Wnt signaling through its association with β-catenin and functions as a bridge between the Wnt receptor complex and the transcriptional effectors that mediate signal activation.
Figure 3. Apcdd1 antagonizes Wnt activity and associates with β -catenin.

(A) Top-FLASH reporter assay in Oli-Neu cells. Apcdd1 antagonizes Wnt3a, LRP6, and β-catenin induction of Wnt activity. Error bars represent standard deviation, of at least three independent experiments, performed in triplicate (t-test, p=0.05). (B) Apcdd1 co-immunoprecipitates with β-catenin when overexpressed in 293 cells. Gel images are representative of three independent experiments.
Apcdd1 promotes OLP differentiation after gliotoxic white matter injury
The foregoing data indicate that Apcdd1 is expressed in oligodendrocytes and promotes OLP differentiation, therefore we next investigated whether it similarly promotes OLP differentiation after white matter injury (WMI). To test this possibility, we first examined its expression in the adult spinal cord after lysolecithin lesioning, a well-established gliotoxic injury model that allows for the precise evaluation of remyelination repair kinetics (Fancy et al., 2009; Fancy et al., 2011). As indicated Fig. 4A–C, Apcdd1 is co-expressed with the pan-glial marker, NFIA at 7 days post lesion (7dpl), indicating that it is present in both glial populations within these mouse lesions.
Figure 4. Overexpression of Apcdd1 accelerates oligodendrocyte differentiation after lysolecithin lesioning of the adult spinal cord.

(A–C) Co-expression of Apcdd1 and NFIA at 7dpl in a mouse lysolecithin lesion. (D) Schematic of the viral manipulation paradigms used in the lesioning experiments. (E–P) Viral overexpression of Apcdd1 (K–M) promotes the expression of MBP and PLP markers at 9dpl compared to infection with a GFP-control virus (E–G). Overexpression of Apcdd1 in lesions did not affect the number of oligodendrocyte precursors (H v. N; see S) or the presence of reactive astrocytes or immune cells (I–J, O–P). Dashed lines in A–P indicate lesion borders, identified, in part, through hypercellularity indicated by DAPI staining. (Q–S) Quantification of marker expression; experiments were performed on at least 4 animals per condition, and quantification involved at least 8 sections from three animals demonstrating the highest expression of the respective viral transgene (GFP or Apcdd1). Error bars in all graphs are SD. (*p<0.005; **p<0.01)
To determine whether Apcdd1 promotes OLP differentiation during remyelination, we focused on gain of function paradigms, reasoning that elevated levels of Apcdd1 in white matter lesions are likely to have higher clinical and therapeutic impact. Towards this, we injected adult spinal cord white matter with lysolecithin, followed by secondary focal injection of the lesioned region with either a GFP tagged Apcdd1-containing lentivirus or GFP-control virus at 3 dpl to catch the OLPs as they are recruited into the lesion. We then assessed OL differentiation at 10 dpl (7 days after viral infection, see Fig. 4D) when OLP populations are poised to differentiate. Analysis of OLP differentiation in Apcdd1 lentivirus injected mice revealed an 8-fold increase in the number of MBP and PLP expressing cells compared to control virus injection (Fig. 4Q–R; F,G v. L,M). Importantly, there was no effect on the number of Olig2 expressing cells, indicating that the increase in number of differentiating OLPs is not the result of increased OLP production (Fig. 4S). Additionally, we assessed the reactive astrocyte and macrophage responses by staining for GFAP and Iba-1 and found that Apcdd1 overexpression did not influence the constituency of these cell populations within the lesion (Fig. 4I–J v. O–P). Together, these data indicate that overexpression of Apcdd1 promotes OLP differentiation but does not affect OLP recruitment during remyelination in a lysolecithin model of WMI in the adult spinal cord.
Apcdd1 promotes OLP differentiation following acute hypoxia
In addition to lysolecithin induced remyelination, the ex vivo cerebellar system can be used to model WMI following hypoxia (Fancy et al., 2011). To test the role of Apcdd1 in OL differentiation after hypoxia, cerebellar slices were established and maintained at 1% oxygen for two days, after which there was a complete loss of mature OL populations (Fig. 5A). After this acute hypoxia treatment, the cultures were returned to normoxia and infected with either the Apcdd1-GFP or GFP-control virus (Fig. 5D–M). Subsequent analysis of OL regeneration revealed that Apcdd1-overexpression demonstrated increased expression of MBP and PLP compared to the GFP control (Fig. 5D–H v. I–M), indicating that OL differentiation is enhanced in the presence of Apcdd1. These findings provide evidence that Apcdd1 promotes OL differentiation after hypoxia and further reinforces the contributions of this gene to repair after WMI.
Figure 5. Apcdd1 stimulates oligodendrocyte differentiation after acute hypoxia.

(A) Schematic of ex vivo, acute hypoxia experimental paradigm. (D–M) Overexpression of Apcdd1 in ex vivo cerebellar slice culture after acute hypoxia. Slice cultures were infected with either GFP (D–H) or Apcdd1 (I–M) viruses and oligodendrocyte differentiation was analyzed through staining for MBP at 10- or 18- DIV. Overexpression of Apcdd1 resulted in increased MBP expression at both 10- and 18- DIV compared to the GFP control (D,E v. I,J) and increased PLP expression at 18-DIV (G,H v. L,M). Images are representative of least five independent experiments, per timepoint, thus the general phenomenon was observed at least ten times. Cerebellar slices that were maintained at normoxia (i.e. not exposed to hypoxia) are shown for the sake of comparison in B–C.
Collectively, our findings indicate that Apcdd1 promotes OLP differentiation during regenerative myelination after gliotoxic injury and acute hypoxia. To determine whether Apcdd1 is present in human WMI we investigated its expression in OLP populations in white matter lesions in the subcortical white matter of the human neonatal brain and in adult MS lesions. Neonatal WMI and human MS lesions were diagnosed based on both clinical and neuropathological correlations (see methods). Using in situ hybridization for human Apcdd1 (Fig. 6A), in combination with immunostaining for Olig2 (Fig. 6B), we found that Apcdd1 is expressed in OLP populations in areas of gliotic subcortical white matter. Interestingly, it demonstrates heterogeneous expression within OLPs in these lesions, with some Olig2-expressing OLPs demonstrating high Apcdd1 expression (Fig. 6B-filled arrow), others demonstrating no expression (red arrows) of Apcdd1. Moreover, we observed several Apcdd1-expressing cells that did not express Olig2 (unfilled arrows). Next we performed a similar analysis in human MS lesions from the periventricular white matter and found a very similar pattern of expression, where Apcdd1 is highly expressed in subsets of OLP populations, and in non-OLP populations within the lesion (Fig. 6C–D). These findings, together with our functional studies after WMI, implicate Apcdd1 in the OLP regenerative response to WMI.
Discussion
Here we describe the expression and role of Apcdd1 during developmental and regenerative myelination. Apcdd1 demonstrates dynamic expression over the course of oligodendrocyte development, where it is lowly expressed in OLPs and upregulated as these cells differentiate into mature OLs. Consistent with its expression dynamics, functional studies revealed that it is sufficient to promote OL differentiation and suppresses β-catenin dependent Wnt activation, which is mediated by an association between Apcdd1 and β-catenin. Examination of the role of Apcdd1 after white matter injury (WMI) revealed that it similarly promotes OL differentiation after gliotoxic injury in the adult spinal cord and hypoxia in ex vivo slices. Finally, we apply these findings to human WMI by demonstrating that Apcdd1 is expressed in OLPs in neonatal HIE and MS tissue.
Our previous studies on Apcdd1 implicated it in astrocyte precursor migration during embryonic development, thus these studies represent the first characterization of Apcdd1 in the OL lineage (Kang et al., 2012). That Apcdd1 is expressed in mature OL populations and promotes their differentiation, while also playing a role in astrocyte precursor migration, suggests that it is playing diverse roles across these lineages. These distinct functions can likely be attributed to the complexities of Wnt signaling, where canonical and non-canonical pathways regulate diverse biological processes (Clevers and Nusse, 2012; De Ferrari et al., 2006; Inestrosa et al., 2012; Logan et al., 2004; Moon et al., 2004). For example, our previous studies found that Apcdd1 influences membrane localization of Rho-GTPase, which plays a key role in non-canonical Wnt signaling and cell migration (Kang et al., 2012). Here we show that Apcdd1 antagonizes canonical Wnt signaling via β-catenin, and these functions are consistent with our observations that it functions to promote OL differentiation. Together, these data suggest that in astrocyte precursors, Apcdd1 functions through the planar cell polarity (PCP), non-canonical Wnt pathway to promote cell migration, while in the OL lineage, it functions to suppress the canonical Wnt pathway to promote OL differentiation. It will be important to further discern these distinct functions of Apcdd1 in these glial sub-lineages, and how its role toggles between these distinct Wnt pathways. Future studies will include astrocyte- and oligodendrocyte-specific deletion of Apcdd1, to determine whether it functions through distinct- or unified-Wnt mechanisms in glial sub-lineages. Another interesting feature of Apcdd1 in the CNS is its expression in endothelial cells (Lippmann et al., 2012). Recently, the interplay between oligodendrocytes and endothelial cells during development and remyelination has been linked to canonical Wnt signaling (Pham, et al. 2012; Yuen et al., 2013). Given that Apcdd1 is expressed in both populations and is associated with the regulatory pathway mediating this relationship, it is possible that Apcdd1 also contributes to this facet of CNS development, which is also an important area for future conditional mouse deletion studies.
Canonical Wnt signaling is an extremely complex pathway, with multiple regulatory layers ranging from extracellular ligands and inhibitors to intracellular transcriptional effectors. Here we found a negative regulator of Wnt signaling, that functions through the receptor complex to promote OL differentiation during developmental and regenerative myelination. Importantly, that Apcdd1 suppresses β-catenin dependent Wnt activation and associates with both β-catenin and LRP6, suggests that it functions as a bridge between the proximal Wnt receptor complex and the distal nuclear effectors (Shimomura et al., 2010). Given that sequestration of β-catenin in the cytoplasm is a critical regulatory event in the canonical Wnt pathway, modulating Apcdd1 levels or activity may be a way to shuttle β-catenin and regulate the transcriptional outputs of the Wnt pathway. While our studies point to Apcdd1 as playing a key role in this process, it will be important to further delineate its relationship with other components of the Wnt pathway that regulate β-catenin localization. Understanding how Apcdd1 fits into the broader regulatory biology surrounding β-catenin has very important therapeutic implications for human WMI as inhibition of Wnt signaling stimulates remyelination across a variety of animal models, and β-catenin is a key effector of this transcriptional program that inhibits this process (Fancy et al., 2009; Guo, et al. 2015). Because pharmacologically inhibiting transcriptional components of a pathway is challenging and given that Apcdd1 bridges the nuclear and receptor components, it could serve as an alternative therapeutic target in this context. Indeed, our studies after gliotoxic WMI in vivo and acute hypoxia ex vivo support the notion that increasing Apcdd1 levels stimulate remyelination. Future studies will be geared towards delineating the biochemical basis for Apcdd1 inhibition of β-catenin function and Wnt signaling in the OL lineage during development and after WMI.
Because Acpdd1 inhibits Wnt signaling, we reasoned that its overexpression would stimulate remyelination after injury, therefore we focused our studies on gain of function paradigms after WMI. Towards this we used a viral approach and overexpressed Apcdd1 in lysolecithin lesions, finding that it promotes OL differentiation after injury. While we cannot rule out the possibility that Apcdd1 is acting in a cell non-autonomous manner to promote OL differentiation after injury, viral over expression in vitro and in slice culture also resulted in enhanced OL differentiation. Together these results suggest that the effects on differentiation we witness after lysolecithin injury are specific to the OL lineage, though additional conditional mouse deletion studies will be required to definitively prove this. The effects of Apcdd1 on OL differentiation after injury and acute hypoxia led us to examine its expression in neonatal WMI that resulted from HIE, revealing that Apcdd1 is expressed in OLP populations within these lesions. This is different from what we observed during mouse development and in the adult CNS, where Apcdd1 was very lowly expressed in these OLP populations. The elevated expression of Apcdd1 in OLPs in human WMI is likely a reflection of the disease state, where internal OLP programs are inducing Apcdd1 expression in an attempt to dampen Wnt signaling and provoke OL differentiation. Nevertheless, in spite of these attempts, it is likely that the physiological elevation of Apcdd1 expression in these cells is short-circuited by chronic transcriptional activation of Wnt effectors manifest in these OLP populations in WMI (Fancy et al., 2014). The net result being stalled OLP populations with elevated levels of Apcdd1 that are incapable to titrating out the transcriptional Wnt effectors. This highlights an interesting “tug-of-war” between the distal transcriptional effectors- and proximal receptor complex components of the Wnt pathway, where chronic activation of transcription can override the transduction mechanisms. However, as suggested by our reporter assays, increasing levels of Apcdd1 can override transcription activation by β-catenin activation, suggesting that intervention via these proximal pathways may serve as a therapeutic intervention. Importantly, further dissecting how Apcdd1 fits into the existing framework overseeing β-catenin activation will be important for developing these therapeutic strategies.
Currently there are no available therapies that promote repair of human WMI, thus a comprehensive understanding of the regulatory factors associated with this process and how they become dysregulated is critical for developing these much needed therapies (Franklin and Gallo, 2014). The Wnt pathway plays a central role in the suppression of OLP differentiation in human WMI, MS in adults and CP infants. Thus understanding the contributions of the various components of this pathway towards white matter repair is a critical step in the development of therapeutics for these devastating neurological disorders. We have demonstrated a critical role for Apcdd1 in OL differentiation after WMI that points to a potential therapeutic approach for inhibiting Wnt signaling in these disorders.
Main Points.
We found that Apcdd1 is a negative regulator of Wnt signaling that stimulates myelin repair and is expressed in human white matter injury. These studies suggest a new therapeutic strategy for modulating Wnt signaling and stimulating myelin repair.
Acknowledgments
We thank Mauro Costa-Mattioli for use of his McIlwain tissue chopper, and Jason Yustein for the use of his hypoxic chamber. This work was supported by grants from the National Multiple Sclerosis Society (RG 4623A1/2 to BD, TA 3054-A-1 to HL), the Sontag Foundation (BD), Cancer Prevention Research Institute of Texas (RP140102 to WZ), and the National Institutes of Health (R01 NS071153 to BD, R01NS078294 to BA, 5-T32HL092332-08 to DL). This work was also supported in part by the Baylor College of Medicine IDDRC (HD024064). Human MS tissue was provided by the Rocky Mountain Multiple Sclerosis Center Tissue Bank.
References
- Azim K, Butt AM. GSK3β negatively regulates oligodendrocyte differentiation and myelination in vivo. Glia. 2011 Apr;59(4):540–5. doi: 10.1002/glia.21122. [DOI] [PubMed] [Google Scholar]
- Back SA, Rosenberg PA. Pathophysiology of glia in perinatal white matter injury. Glia. 2014 Nov;62(11):1790–815. doi: 10.1002/glia.22658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bafico A, Guizhong L, Yaniv A, Gazit A, Aaronson SA. Novel mechanism of Wnt signaling inhibition mediated by Dickkopf-1 interaction with LRP6/Arrow. Nat Cell Biol. 2001;3:683–686. doi: 10.1038/35083081. [DOI] [PubMed] [Google Scholar]
- Bilić J, Huang Y, Davidson G, Zimmermann T, Cruciat C, Bienz M, Niehrs C. Wnt induces LRP6 signalosomes and promotes Dishevelled-dependent LRP6 phosphorylation. Science. 2007;316:1619–1622. doi: 10.1126/science.1137065. [DOI] [PubMed] [Google Scholar]
- Birgbauer E, Rao TS, Webb M. Lysolecithin induces demyelination in vitro in a cerebellar slice culture system. J Neurosci. 2004;78:157–166. doi: 10.1002/jnr.20248. [DOI] [PubMed] [Google Scholar]
- Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med. 2000;346:165–173. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- De Ferrari GV, Moon RT. The ups and downs of Wnt signaling in prevalent neurological disorders. Oncogene. 2006;25:7545–7553. doi: 10.1038/sj.onc.1210064. [DOI] [PubMed] [Google Scholar]
- Emery B. Regulation of oligodendrocyte differentiation and myelination. Science. 2010;330:779–782. doi: 10.1126/science.1190927. [DOI] [PubMed] [Google Scholar]
- Fancy SP, Baranzini SE, Zhao C, Yuk DI, Irvine KA, Kaing S, Sanai N, Franklin RJ, Rowitch DH. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009;23:1571–1585. doi: 10.1101/gad.1806309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Harrington EP, Baranzini SE, Silbereis JC, Shiow LR, Yuen TJ, Huang EJ, Lomvardas S, Rowitch DH. Parallel states of pathological Wnt signaling in neonatal brain injury and colon cancer. Nat Neurosci. 2014;17:506–512. doi: 10.1038/nn.3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, Franklin RJ, Rowitch DH. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat Neurosci. 2011;14:1009–1016. doi: 10.1038/nn.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Glasgow SM, Finley M, Rowitch DH, Deneen B. Evidence that nuclear factor IA inhibits repair after white matter injury. Ann Neur. 2012;72:224–233. doi: 10.1002/ana.23590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJ. Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci. 2002;3:705–714. doi: 10.1038/nrn917. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Gallo V. The translational biology of remyelination: past, present, and future. Glia. 2014 Nov;62(11):1905–15. doi: 10.1002/glia.22622. [DOI] [PubMed] [Google Scholar]
- Guo F, Lang J, Sohn J, Hammond E, Chang M, Pleasure D. Canonical Wnt signaling in the oligodendroglial lineage-puzzles remain. Glia. 2015 Mar 18; doi: 10.1002/glia.22813. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Harrington EP, Zhao C, Fancy SP, Kaing S, Franklin RJ, Rowitch DH. Oligodendrocyte PTEN is required for myelin and axonal integrity, not remyelination. Ann Neurol. 2010;68:703–716. doi: 10.1002/ana.22090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inestrosa NC, Montecinos-Oliva C, Fuenzalida M. Wnt signaling: role in Alzheimer disease and schizophrenia. J Neuroimmune Pharmacol. 2012;7:788–807. doi: 10.1007/s11481-012-9417-5. [DOI] [PubMed] [Google Scholar]
- Kang P, Lee HK, Glasgow SM, Finley M, Donti T, Gaber ZB, Graham BH, Foster AE, Novitch BG, Gronostajski RM, Deneen B. Sox9 and NFIA coordinate a transcriptional regulatory cascade during the initiation of gliogenesis. Neuron. 2012;74:79–94. doi: 10.1016/j.neuron.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann T, Miron V, Cui Q, Wegner C, Antel J, Brück W. Differentiation block of oligo-dendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 2008;131:1749–1758. doi: 10.1093/brain/awn096. [DOI] [PubMed] [Google Scholar]
- Lassmann H. Mechanisms of white matter damage in multiple sclerosis. Glia. 2014 Nov;62(11):1816–30. doi: 10.1002/glia.22597. [DOI] [PubMed] [Google Scholar]
- Lee HK, Chaboub LS, Zhu W, Zollinger D, Rasband MN, Fancy SF, Deneen B. Daam2-PIP5K is a novel regulatory pathway for Wnt signaling and therapeutic target for remyelination in the CNS. Neuron. 2015;85(6):1227–1243. doi: 10.1016/j.neuron.2015.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, Palecek SP, Shusta EV. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol. 2012;30:783–791. doi: 10.1038/nbt.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, Niehrs C. LDL receptor-related protein 6 is a receptor for Dickkopf proteins. Nature. 2001;411:321–325. doi: 10.1038/35077108. [DOI] [PubMed] [Google Scholar]
- Moon RT, Kohn AD, De Ferrari GV, Kaykas A. Wnt and beta-catenin signaling: diseases and therapies. Nat Rev Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- Pan W, Choi SC, Wang H, Qin Y, Volpicelli-Daley L, Swan L, Lucast L, Khoo C, Zhang X, Li L, Abrams CS, Sokol SY, Wu D. Wnt3a-mediated formation of phosphatidylinositol 4,5-bisphosphate regulates LRP6 phosphorylation. Science. 2008;321:1350–1353. doi: 10.1126/science.1160741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham LD, Hayakawa K, Seo JH, Nguyen MN, Som AT, Lee BJ, Guo S, Kim KW, Lo EH, Arai K. Crosstalk between oligodendrocytes and cerebral endothelium contributes to vascular remodeling after white matter injury. Glia. 2012 May;60(6):875–81. doi: 10.1002/glia.22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semënov MV, Tamai K, Brott BK, Kühl M, Sokol S, He X. Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr Biol. 2001;11:951–961. doi: 10.1016/s0960-9822(01)00290-1. [DOI] [PubMed] [Google Scholar]
- Shimomura Y, Agalliu D, Vonica A, Luria V, Wajid M, Baumer A, Belli S, Petukhova L, Schinzel A, Brivanlou AH, Barres BA, Christiano AM. APCDD1 is a novel Wnt inhibitor mutated in hereditary hypotrichosis simplex. Nature. 2010;464:1043–1047. doi: 10.1038/nature08875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Hu Q, Liu H, Yan Y, D’ercole AJ. beta-catenin mediates insulin-like growth factor-I actions to promote cyclin D1 mRNA expression, cell proliferation and survival in oligodendroglial cultures. Glia. 2010 Jul;58(9):1031–41. doi: 10.1002/glia.20984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, Fancy SP, Zahed H, Maltepe E, Rowitch DH. Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell. 2013;158:383–396. doi: 10.1016/j.cell.2014.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]