

Abstract
Glutamate uptake by astrocytes controls the time course of glutamate in the extracellular space and affects neurotransmission, synaptogenesis, and circuit development. Astrocytic glutamate uptake has been shown to undergo post-natal maturation in the hippocampus, but has been largely unexplored in other brain regions. Notably, glutamate uptake has never been examined in the developing neocortex. In these studies, we investigated the development of astrocytic glutamate transport, intrinsic membrane properties, and control of neuronal NMDA receptor activation in the developing neocortex. Using astrocytic and neuronal electrophysiology, immunofluorescence, and Western blot analysis we show that: 1) glutamate uptake in the neonatal neocortex is slow relative to neonatal hippocampus; 2) astrocytes in the neonatal neocortex undergo a significant maturation of intrinsic membrane properties; 3) slow glutamate uptake is accompanied by lower expression of both GLT-1 and GLAST; 4) glutamate uptake is less dependent on GLT-1 in neonatal neocortex than in neonatal hippocampus, and 5) the slow glutamate uptake we report in the neonatal neocortex corresponds to minimal astrocytic control of neuronal NMDA receptor activation. Taken together, our results clearly show fundamental differences between astrocytic maturation in the developing neocortex and hippocampus, and corresponding changes in how astrocytes control glutamate signaling.
Keywords: GLT-1, GLAST, EAAT1, EAAT2, postnatal development, membrane resistance, developmental plasticity, glial development
INTRODUCTION
Astrocyte maturation is a complex process intimately associated with synapse and circuit development. Mature astrocytes are characterized by low membrane resistance (Steinhauser, Berger, Frotscher, and Kettenmann, 1992), hyperpolarized membrane potential (Kuffler, Nicholls, and Orkand, 1966; Kuffler and Nicholls, 1966), robust glutamate uptake (Bergles and Jahr, 1997; Danbolt, Storm-Mathisen, and Kanner, 1992; Schmidt and Wolf, 1988; Danbolt, 2001; Thomas, Tian, and Diamond, 2011), ramified morphology (Halassa, Fellin, Takano, Dong, and Haydon, 2007; Freeman, 2010; Bushong, Martone, and Ellisman, 2004), and gap-junction coupling (Theis, Sohl, Eiberger, and Willecke, 2005; Nagy, Li, Rempel, Stelmack, Patel, Staines, Yasumura, and Rash, 2001; Sohl, Odermatt, Maxeiner, Degen, and Willecke, 2004). Manifestation of these mature phenotypes allows astrocytes to spatially and temporally control neurotransmission (Piet, Vargova, Sykova, Poulain, and Oliet, 2004; Oliet, Piet, and Poulain, 2001), and maintain low extracellular potassium levels (Kofuji, Ceelen, Zahs, Surbeck, Lester, and Newman, 2000; Karus, Mondragão, Ziemens, and Rose, 2015).
A critical function of astrocytes is their ability to remove extracellular glutamate. There are 5 excitatory amino acid transporters (EAATs) which mediate glutamate uptake. Robust uptake ensures low extracellular glutamate levels, high temporal and spatial fidelity of synaptic transmission, and conservation of biochemical resources via glutamate/glutamine shuttling (Danbolt, 2001; Lieth, LaNoue, Berkich, Xu, Ratz, Taylor, and Hutson, 2001; Sibson, Mason, Shen, Cline, Herskovits, Wall, Behar, Rothman, and Shulman, 2001; Tani, Dulla, Huguenard, and Reimer, 2010; Tani, Dulla, Farzampour, Taylor-Weiner, Huguenard, and Reimer, 2014). In the neocortex and hippocampus, astrocytes express GLT-1 (EAAT2) and GLAST (EAAT1) (Lehre, Levy, Ottersen, Storm-Mathisen, and Danbolt, 1995) which, together, are responsible for almost all glutamate transport in these regions (Holmseth, Dehnes, Huang, Follin-Arbelet, Grutle, Mylonakou, Plachez, Zhou, Furness, and Bergles, 2012). Understanding how glutamate clearance develops across brain regions is a critical part of understanding synaptic and circuit level brain maturation.
Biochemical experiments and immunohistochemical/immunofluorescence analysis suggest that in the neonatal neocortex and hippocampus, GLT-1 protein and mRNA levels are very low or undetectable and that GLAST predominates (Benediktsson, Marrs, Tu, Worley, Rothstein, Bergles, and Dailey, 2012; Ullensvang, Lehre, Storm-Mathisen, and Danbolt, 1997). These approaches compare the expression levels of glutamate transporters, but provide limited information on transporter activity. To quantify transport activity, a number of studies have measured glutamate transporter currents (TCs) (e.g. (Bergles and Jahr, 1997; Diamond, 2005; Diamond, Bergles, and Jahr, 1998) and shown that glutamate uptake becomes more robust in the hippocampus as animals mature (Diamond, 2005) in agreement with the developmental increase in GLT-1 and GLAST expression (Voutsinos-Porche, Knott, Tanaka, Quairiaux, Welker, and Bonvento, 2003; Ullensvang, Lehre, Storm-Mathisen, and Danbolt, 1997; Furuta, Rothstein, and Martin, 1997). Similar approaches show that GLT-1 removes the bulk of released extracellular glutamate in the mature neocortex and hippocampus, and that GLAST plays a less significant role (Tanaka, Watase, Manabe, Yamada, Watanabe, Takahashi, Iwama, Nishikawa, Ichihara, Kikuchi, Okuyama, Kawashima, Hori, Takimoto, and Wada, 1997; Haugeto, Ullensvang, Levy, Chaudhry, Honore, Nielsen, Lehre, and Danbolt, 1996; Tanaka, Watase, Manabe, Yamada, Watanabe, Takahashi, Iwama, Nishikawa, Ichihara, Kikuchi, Okuyama, Kawashima, Hori, Takimoto, and Wada, 1997; Danbolt, 2001; Rothstein, Martin, Levey, Dykes-Hoberg, Jin, Wu, Nash, and Kuncl, 1994; Watanabe, Morimoto, Hirao, Suwaki, Watase, and Tanaka, 1999; Holmseth, Dehnes, Huang, Follin-Arbelet, Grutle, Mylonakou, Plachez, Zhou, Furness, and Bergles, 2012). Although the hippocampus has been studied in detail, little is known about the maturation of these processes in the neocortex.
Here we show using electrophysiological quantification of TCs that the rate at which glutamate is taken up by astrocytes increases massively during neocortical development. In comparison to the neonatal hippocampus, glutamate uptake in the neonatal cortex is substantially slower. We show that the developmental increase in the neocortex is accompanied by an increased reliance on GLT-1, as expected from immunochemical studies (Voutsinos-Porche, Knott, Tanaka, Quairiaux, Welker, and Bonvento, 2003; Ullensvang, Lehre, Storm-Mathisen, and Danbolt, 1997; Furuta, Rothstein, and Martin, 1997). We further show that GLT-1 and GLAST expression is significantly lower during development in the neocortex than in the hippocampus. We go on to show that intrinsic electrical properties of astrocytes in the neocortex undergo significant post-natal development not seen in hippocampal astrocytes. Finally, we demonstrate that differential transporter activity has functional consequences on the regulation of NMDA receptor activation. Taken together, the data reveal a previously unappreciated developmental and regional heterogeneity in astrocyte maturation. This has consequences for our understanding of the control of NMDA receptor activation during a critical developmental window.
MATERIALS AND METHODS
Preparation of Brain Slices
Cortical brain slices containing sensorimotor cortex (400 μm thick) were prepared from Sprague-Dawley rats (P2-P60). Briefly, rats were anesthetized (isofluorane), decapitated, and the brains were rapidly removed and placed in chilled (4°C) low-Ca, low-Na slicing solution consisting of (in mM): 234 sucrose, 11 glucose, 24 NaHC03, 2.5 KCl, 1.25 NaH2PO4, 10 MgSO4 and 0.5 CaCl2, equilibrated with a mixture of 95% O2:5% CO2. The brain was glued to the slicing stage of a Leica VT1 vibratome and slices were cut in a coronal orientation. The slices were then incubated in 32°C oxygenated aCSF (in mM: NaCl, 126, KCl, 2.5, NaH2PO4, 1.25, MgSO4, 1, CaCl2, 2, glucose, 10, NaHCO2, 26) for 1 hour, and then held at room temperature. Subsequent recordings were performed at 32°C. All guidelines of Tufts University’s Institutional Animal Care and Use Committee were followed.
Western Blot Analysis
Expression of GLT-1 and GLAST were analyzed via Western blot. Neocortex and hippocampus from P3, P7, and P14 rat brain slices was dissected and homogenized using RIPA buffer (150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) and protease inhibitor cocktail. Samples were then centrifuged at 13,200 rpm for 15 min at 4°C. 15 μg of protein was loaded onto a 10% SDS-PAGE gel and analyzed via electrophoresis. Proteins were detected using antibodies for GLT-1 (1:1000, Abcam) and GLAST (Batch Ab#314 (Holmseth, Scott, Real, Lehre, Leergaard, Bjaalie, and Danbolt, 2009). Anti-β-actin (1:2000, Sigma) was used to confirm equal loading. Protein bands were visualized using enhanced chemiluminescence and imaged with a LAS-3000 imaging system. Quantification of protein expression was conducted using ImageJ (NIH) and expressed in relation to β-actin expression.
Immunofluorescence
Rats at ages P6-P32 were anesthetized, perfused with saline followed by PFA, then decapitated, and brains were dissected and placed in 4% formaldehyde for 24 hours. Fixed brains were then cryoproteced in a high sucrose solution (in mM: 818 sucrose, 77 anhydrous sodium phosphate, 23 sodium phosphate), and then sectioned at 40 μm using a Thermo Fisher Microm HM 525 cryostat. Brain sections were placed in blocking buffer (5% normal goat serum, 1% bovine serum albumin, in PBS) for 1 hr at room temperature. Either GLT-1 (1:1000, Ab#360, concentration 200 μg/ml (Holmseth, Zhou, Follin-Arbelet, Lehre, Bergles, and Danbolt, 2012) or GLAST (1:500, Batch Ab#314, concentration 600 μg/ml (Holmseth, Scott, Real, Lehre, Leergaard, Bjaalie, and Danbolt, 2009)) antibodies were diluted in PBS with 2% Triton-X 100 and 5% blocking buffer. Brain sections were incubated with a diluted primary antibody for 12 hours at 4°C. Brain sections were then rinsed three times in PBS, and secondary antibodies (goat anti-rabbit Alexa 488, Jackson Labs; diluted 1:500 in PBS with 5% blocking buffer) were added to the sections for 2 hours at room temperature. Slices were mounted using Vectashield (Vector Labs) and imaged with a Nikon A1R upright confocal microscope with a 10x dry objective (Olympus).
For image analysis, regions of interest (ROIs) were extracted from cortex layer 5 and hippocampal CA1 stratum radiatum. Cell bodies that were unlabeled by GLT-1 or GLAST antibodies (i.e. neuronal cell bodies) were masked and excluded from the intensity analysis (Fig. 4C, F). Fluorescence density was calculated as the total fluorescence intensity over the ROI divided by the area of the ROI (excluding masked cell bodies). Excluding neuronal cell bodies accounted for differences in tissue architectures in neocortex and hippocampus. The fluorescence density of each cortical ROI was then normalized to the fluorescence density of the hippocampal ROI from the same image.
Figure 4. Immunofluorecence studies show lower GLT-1 and GLAST expression in neonatal cortex compared to hippocampus.

A. Confocal images of representative coronal sections stained for GLT-1 at 10x magnification. Scale bars are 500 μm. B. In GLT-1 stained sections, the fluorescence density from cortical ROIs normalized to the fluorescence density from CA1 stratum radiatum ROIs within each slice (P7, n = 3; P14, n = 4; P28, = 4). C. Examples from within ROIs in cortex and hippocampus at P28 showing the areas masked and excluded from the fluorescence density analysis of GLT-1 expression. Scale bars are 100 μm. D. Representative sections stained for GLAST. E. In GLAST stained sections, the fluorescence density from cortical ROIs normalized to the fluorescence density from CA1 stratum radiatum ROIs within each slice (P7, n = 4; P14, n = 4; P28, n = 3 cells). F. Examples from within regions of interest in cortex and hippocampus at P28 showing the areas masked and excluded from the fluorescence density analysis of GLAST expression. Scale bars are 100 μm. Error bars indicate SEM,*p<0.05, **p<0.01, ***p<0.001, one-sample t-test.
Glutamate Transporter Currents
In order to patch clamp astrocytes in cortical slices, acute brain slices were loaded with sulforhodamine 101 (SR-101), a dye which selectively loads astrocytes (Nimmerjahn, Kirchhoff, Kerr, and Helmchen, 2004). Immediately after preparation, brain slices were incubated with 0.5 μM SR-101 in aCSF for 5 minutes at 32 °C. Slices were then transferred into SR-101-free aCSF and allowed to recover for 1 hour as normal. Brain slices were then placed into a submersion recording chamber of an Olympus BX51 microscope. Brain slices were perfused with standard aCSF at 32°C containing CPP (10 μM) and DNQX (20 μM) to block NMDA and AMPA receptors respectively, GABAzine (10 μM) to block GABAA receptors, and 100 μM MNI-glutamate. Astrocytes in layer V of somatosensory cortex or in stratum radiatum of hippocampus area CA1 were identified by their small, round cell bodies and SR-101 loading (560 nm excitation, 630 nm emission filters set = 49008 ET, Chroma). Astrocytes were then whole-cell patch clamped using borosilicate glass electrodes (8–12 MΩ). Patch pipette internal solution contained (in mM): potassium gluconate, 120, HEPES, 20, EGTA 10, magnesium ATP, 2, sodium GTP, 0.2. Patched cells were voltage clamped and held at −80 mV. Astrocyte identity was confirmed electophysiologically by hyper-polarized membrane potential (−70 to −80 mV) and passive membrane properties. Membrane properties were assessed by injecting current into patched cells in 250 ms steps from −300 to 650 pA (Fig. 2A). A cell was considered to have only passive membrane properties if the change in membrane voltage was approximately linear across current steps and the cell did not fire action potentials when depolarized. Glutamate transporter currents were then evoked by focally flashing a 355 nm UV laser (spot size of 10 μm full width at half maximum) for 1 ms onto the astrocyte cell body while voltage clamping the astrocyte at −80mV. A 5 mV, 50 ms depolarizing pulse was applied 500 ms before uncaging in order to normalize evoked transporter currents for changes in access resistance commonly associated with recording from astrocytes (Diamond, Bergles, and Jahr, 1998). Additionally, astrocytic recordings with access resistances greater than 25 MΩ were excluded. The resulting glutamate transporter current’s decay kinetics were fit using a bi-exponential decay, and the time constant of the fast decay was used as a measure of glutamate transporter function (Bergles and Jahr, 1997). In order to isolate the glutamate transporter currents, in a subset of recordings we applied 100 μM DL-threo-β-Benzyloxyaspartic acid (TBOA) to inhibit glutamate transporters (Shimamoto, Lebrun, Yasuda-Kamatani, Sakaitani, Shigeri, Yumoto, and Nakajima, 1998) (Fig. 1C) and TBOA-subtracted currents were calculated. For analysis involving transporter current amplitude or charge transfer (Fig. 1F–G), membrane and input resistance were corrected for on a cell-by-cell basis (Armbruster, Hampton, Yang, and Dulla, 2014). Dihydrokainate (DHK; 300 μM), a GLT-1-specific antagonist (Arriza, Fairman, Wadiche, Murdoch, Kavanaugh, and Amara, 1994) was applied in a separate subset of cells to isolate the GLT-1-mediated current.
Figure 2. Electrophysiological properties of cortical and hippocampal astrocytes through development.

A. Representative whole cell current clamp recordings in hippocampal (upper panels) and cortical astrocytes (lower panels) at P3-P28. Injected current ranged from 300 pA to 650 pA in 50 pA steps. B. Current-voltage relationship for cortical astrocytes at ages P3 (black filled diamonds, n = 4 cells), P7 (grey filled diamonds, n = 8 cells), P14 (black open diamonds, n = 4 cells), P21 (grey open diamonds, n = 8 cells), and P28 (grey-filled black diamonds, n = 6 cells) calculated from the current steps shown in A. C. Hippocampal current-voltage relationship arranged as in B (P3: n = 9; P7 n = 12; P14 n = 8; P21 n = 15; P28 n = 17 cells). D. Quantification of I-V slopes shown in B. and C. for hippocampal (grey bars) and cortical astrocytes (black bars) at ages P3-P28. E. Membrane resistance (Rm) of hippocampal (grey bars) and cortical astrocytes (black bars) at ages P3-P28. Rm was obtained by subtracting access resistance from input resistance. Access and input resistances were calculated from voltage clamp recordings of 5mV steps. Error bars indicate SEM, *p<0.05, **p<0.01, ***p<0.001, two-sample t test.
Figure 1. Glutamate transporter currents are slower in neonatal cortex than in hippocampus.

A. Representative glutamate transporter current traces from hippocampal (grey) and cortical astrocytes (black) at ages P3-P28 normalized by amplitude. B. Similar to A. arranged by cortex (left panel) and hippocampus (right panel), showing ages P3 (black), P7 (grey), P14 (black dashed), P21 (light grey), and P28 (grey dashed). C. Representative example of a glutamate transporter current from a P7 cortical astrocyte before 100 μM TBOA wash on (black line), after TBOA wash on (light grey line), and showing the original trace with the TBOA trace subtracted (dark grey line). D. Quantification of fast decay constants (τFast) at P3 (nCor = 15, nHipp = 14 cells) P7 (nCor = 17, nHipp = 25 cells), P14 (nCor = 15, nHipp= 18 cells). P21 (nCor = 18, nHipp = 17 cells), and P28 (nCor = 9, nHipp = 19 cells). E. Quantification of TC centroids (<t>). F. Quantification of transporter current charge transfer from the onset of the laser pulse to the return of the current to baseline. G. Quantification of the maximum transporter current amplitude after normalization for membrane and input resistance. Charge transfer and amplitude are normalized to membrane and input resistance. D,G. Two sample t-test, *p<0.05, **p<0.01, ***p<0.001. F. p > 0.05, one way ANOVA. All error bars indicate SEM.
Neuronal recordings
Slices were prepared as described above. For NMDAR-mediated EPSC recordings DNQX (20 μM) and GABAzine (10 μM) were included in ACSF. For other recordings MNI-glutamate (100 μM) was included initially and DNQX, CPP, and GABAzine were washed in. For recording uncaging evoked changes in neuronal membrane potential, slices were visualized as described above and neurons in area CA1 of hippocampus were located with a 60x water immersion objective (Olympus). Neurons were patched with borosilicate glass electrodes containing (in mM): KGluconate 120, KCl 11, HEPES 10, EGTA 10, MgCl2 2, magnesium ATP, 2, sodium GTP, 0.3, pH = 7.3, mOsm = 270. Glutamate uncaging and current injections were carried out as described above for astrocyte recordings. For recording NMDAR-mediated EPSCs, slices were visualized with a 10x water immersion objective (Olympus) and a bipolar stimulating electrode (FHC, Cat. #CBARC73) was placed in the subcortical white matter or on the Schaeffer collateral pathway. Pyramidal neurons in layer V of cortex and CA1 of hippocampus were identified by sight using a 60x water immersion objective (Olympus) and DIC optics. Pyramidal neurons were patched with borosilicate glass electrodes (4–6 MΩ) containing (in mM): cesium methanesulfonate, 140, HEPES, 10, EGTA 0.2, NaCl, 5, magnesium ATP, 2, sodium GTP, 0.3, pH = 7.22, mOsm = 290. Once patched, their identity was verified by their resting membrane potential. Neurons were then voltage clamped at resting potential and slowly increased to +40mV and allowed to equilibrate over approximately 2 minutes. Single stimulations were delivered using a TTL-triggered stimulus isolator (A.M.P.I., ISO-Flex) with a 50 μs stimulus duration. The stimulation intensity necessary to evoke the maximum NMDA current was determined (increasing the stimulus amplitude until an evoked current of 20–50pA was achieved, failures were no longer observed, and the current remained constant as stimulation intensity was increased). The intensity of stimulation was then reduced until the current was approximately 50% of the maximum observed. Stimulation intensities were typically between 0.01 mA and 0.1 mA. NMDA currents were then evoked by trains of 5 stimulations (50 μs duration) separated by 10 ms (100 Hz) in aCSF, DNQX (20μM) and GABAzine (10 μM). Trains of stimuli were used in order to evoke a glutamate transient with synaptic and extrasynaptic components. TBOA was then washed on and NMDA currents were again evoked. To verify that the measured currents were mediated by NMDA receptors, CPP (10 μM) was then added and the 100 Hz stimulation was repeated. Access resistance was monitored throughout the experiment and cells with more than a 25% change were excluded from analysis.
Statistical Analysis
For comparison between two experimental groups, a student’s t-test was used. For three or more experimental groups, a one-way ANOVA with the Tukey post-hoc test was used. Values of p<0.05 were considered statistically significant.
RESULTS
Astrocyte glutamate transporter currents mature later in the neocortex compared to hippocampus
A number of groups have pioneered the use of glutamate transporter current (TC) decay kinetics as a relative measure of an individual astrocyte’s capacity to internalize extracellular glutamate (Bergles and Jahr, 1997; Bergles and Jahr, 1998; Bergles, Dzubay, and Jahr, 1997; Diamond, 2005; Diamond, Bergles, and Jahr, 1998). We carried out similar studies using whole-cell patch clamping of astrocytes in layer V of somatosensory cortex and in the stratum radiatum of area CA1 of the hippocampus. Glutamate uptake was evoked by spot UV-photolysis of 100 μM MNI-glutamate and TCs were recorded in voltage-clamped (−80 mV) astrocytes from P3 to P28 rats. AMPA and NMDA receptor antagonists (DNQX and CPP respectively) were included in the bath to prevent activation of neurons. Upon photolysis of MNI-glutamate, large inward currents were seen in patched astrocytes (Fig. 1A–C). To confirm that the inward currents were TCs, 100 μM TBOA (Anderson, Bridges, Chamberlin, Shimamoto, Yasuda–Kamatani, and Swanson, 2001), a wide-spectrum inhibitor of EAATs, was applied in a subset of TC recordings. TBOA eliminated the fast component of the TC and confirmed the specificity of the current (Fig. 1C). TBOA-insensitive currents primarily contributed to the slow phase of the uncaging-evoked current, and thus did not affect the calculation of TC decay time (see methods). To confirm that uncaging did not activate neuronal ionotropic glutamate receptors, whole-cell patch clamp recordings from neurons were also performed. In the presence of CPP, DNQX, and Gabazine, glutamate uncaging did not cause any change in neuronal membrane potential (n=5). When CPP, DNQX, and Gabazine were omitted from the perfusate, uncaging evoked large changes in membrane potential in all neurons (Fig. S1). Therefore, astrocytic recordings made in the presence of ionotropic receptor blockers are free from contamination due to action potential-dependent neuronal potassium efflux.
Measuring TCs directly allows comparison of functional glutamate uptake in a more intact preparation than biochemical approaches. Through recording TCs, we were able to precisely quantify temporal properties of glutamate uptake. TCs evoked in neonatal cortical astrocytes had extremely slow decay kinetics, suggesting very little functional glutamate uptake (Fig. 1A–C). At P3 and P7 cortical TC fast decay constants were 437.46 ± 75.74 ms and 278 ± 28.24 ms respectively. In hippocampal astrocytes at similar ages, TCs had significantly faster decay kinetics (Fig. 1D, P3: 115.13 ± 12.06 ms, p<0.001; P7: 69.36 ± 6.14 ms, p<0.001), consistent with published reports (Thomas, Tian, and Diamond, 2011). We continued these studies at P14, P21, and P28 and found that TC decay kinetics were slower in the cortex as compared to the hippocampus through P14 (P14: p<0.05; P21: p=0.44; P28: p=0.30).
In addition to decay kinetics, we compared TC centroids (<t>, Fig. 1E). Centroids are affected by changes in rise and decay kinetics and thus provide a more complete measure of the rate of glutamate uptake. The centroids of the TCs were significantly larger in cortical astrocytes compared to hippocampal astrocytes at P3 (<t>Cortex = 375.04 ± 54.84 ms, <t>Hipp = 116.79 ± 12.42 ms, p<0.001), P7 (<t>Cortex = 250.98 ± 21.24 ms, <t>Hipp = 80.71 ± 7.54 ms, p<0.001), and P14 (<t>Cortex = 59.80 ± 7.20 ms, <t>Hipp = 36.51 ± 3.91 ms, p<0.01). By P21, however, centroids were similar for cortical and hippocampal TCs (<t>Cortex = 25.47 ± 3.41 ms, <t>Hipp = 25.43 ± 1.20 ms, p=0.99). The centroids of TCs from cortical astrocytes were also significantly larger at P3 compared to P7 (p<0.05), at P7 compared to P14 (p<0.001) and at P14 compared to P21 (p<0.001). Developmental changes in hippocampal centroids were smaller than in cortex though still significant. Centroids in hippocampus were larger at P3 compared to P7 (p<0.05), at P7 compared to P14 (p<0.001), and at P14 compared to P21 (p<0.05). While the decay kinetics of glutamate clearance sped up with age, the total amount of glutamate uptake, quantified as the charge transfer of the TC, was similar across ages and brain regions (Fig. 1F, p=0.38, one way ANOVA). Furthermore, cortical astrocytes had smaller amplitude TCs than hippocampal astrocytes at P3 (Ctx: 46 ± 7 pA, Hipp: 204 ± 37 pA, p<0.001) and P7 (Ctx: 77 ± 14 pA, Hipp: 369 ± 55 pA, p<0.001), reflecting a lower density of glutamate transporters on neonatal cortical astrocytes (Fig. 1G).
TC kinetics have been reported to be unaffected by changes in glutamate concentrations within a sub-saturating range (Diamond and Jahr, 2000). However, it is possible that in neonatal neocortex, the glutamate released by uncaging saturates the especially low level of transport, forcing glutamate to diffuse further, and prolonging the TC decay kinetics. To examine this possibility we varied the power of the UV laser while uncaging glutamate in neonatal slices. In both neocortex and hippocampus at P7 and P14, reducing the laser power by 50% with a neutral density filter reliably decreased the TC amplitude by approximately 50% (Fig. S2A–B, P7hipp: p<0.001, P7cor: p<0.001, P14hipp: p<0.001, P14cor: p<0.001). However, reducing the laser power had no effect on TC decay kinetics (Fig. S2C–D, P7hipp: p=0.23, P7cor: p=0.55, P14hipp: p=0.13, P14cor: p=0.54). This indicates that even in young neocortex, when glutamate transport is very slow, the kinetics of uptake are uniform across a range of glutamate concentrations.
Electrophysiological characterization of astrocyte membrane properties in the developing neocortex and hippocampus
Mature astrocytes typically have low membrane resistances, passive membrane properties, and therefore linear and relatively flat current-voltage (I-V) slopes. However, reported morphological changes in astrocytes over development (Bushong, Martone, and Ellisman, 2004; Morel, Higashimori, Tolman, and Yang, 2014) and developmental differences in astrocyte ion channel expression (Seifert, Huttmann, Binder, Hartmann, Wyczynski, Neusch, and Steinhauser, 2009) led us to hypothesize that astrocytic membrane properties maybe significantly different during early postnatal development. To test this hypothesis, we made whole-cell current clamp recordings of astrocytes in layer V of cortex and stratum radiatum of hippocampal area CA1. Examination of the current voltage (I-V) relationship showed that both cortical and hippocampal astrocytes maintained passive membrane properties and linear I-V slopes throughout development (Fig. 2A–C), however, the I-V slopes of cortical astrocytes decreased significantly from P7 to P14 (p<0.001, Fig. 2B, D) while they remained constant for hippocampal astrocytes (Fig. 2C, D). The I-V slopes of cortical astrocytes were significantly steeper than those of hippocampal astrocytes at P3 (p<0.001) and P7 (p<0.001), but by P14 the I-V slopes of cortical and hippocampal astrocytes were comparable (p=0.67, Fig. 2D). Additionally, we used voltage clamp experiments to confirm that membrane resistance (Rm) in the neocortex decreased significantly from P7 to P14 (p<0.01, Fig. 2E). Hippocampal astrocytes, on the other hand, showed no developmental changes in Rm. Rm was significantly higher in the neocortex as compared to hippocampus at P3 (p<0.001) and P7 (p<0.001, Fig. 2E) corresponding to differences in the I-V slopes between cortical and hippocampal astrocytes at those time points.
No developmental differences in resting membrane potential were found in either brain region nor were there significant differences in resting membrane potential between neocortex and hippocampus (data not shown). To exclude the possibility that changes in access resistance contributed to the developmental changes in TC decay kinetics, we examined the relationship between access resistance and TC decay kinetics (data not shown). We found no correlation between TC decay kinetics and access resistance for hippocampal (RHipp = −0.0159) or cortical astrocytes (RCor = −0.0157) making it especially unlikely that differences in TC kinetics were artifacts of poor access resistance.
To confirm that differences in Rm were not responsible for differences in TC kinetics we performed centroid analysis at P7 as described in detail previously (Diamond, 2005). Briefly, since the measured TC is a convolution of the glutamate clearance waveform with the waveform of the filter imposed by the membrane properties of the astrocyte, electronic filtering, and other properties of the system, it is important to isolate the kinetics of the clearance waveform from the kinetics of the filter. To isolate <t>Clearance from <t>Filter we compared TC kinetics (<t>Cortex, <t>Hippocampus) to the inverse of the rising slope of the TC before and after partially blocking transporters with 10 μM TBOA (Fig. S3A). Inverse TC rising slopes in TBOA were normalized to the slopes before TBOA wash-on, yielding the inverse slope ratio (Slope Ratio−1) and the resulting relationship between <t>Cortex/Hippocampus and Slope Ratio−1 was linearly fit (R2Hippocampus = 0.615, R2Cortex = 0.695). The slope of the fit for cortical TCs was significantly steeper than for hippocampal TCs at P7 (SlopeCortex = 360.01± 87.42, SlopeHippocampus = 118.36 ± 33.91, p<0.05, nCells = 4–5, Fig. S3B). These data demonstrate that the differences we observe between <t>Cortex and <t>Hippocampus at young ages are due to differences in <t>Clearance and are not an artifact of the parallel developmental changes in Rm.
Expression of GLT-1 and GLAST is higher in the developing hippocampus as compared to neocortex
One likely cause of slow TC decay time in the young neocortex is a lower abundance of the glutamate transporter proteins in young neocortex (Voutsinos-Porche, Knott, Tanaka, Quairiaux, Welker, and Bonvento, 2003; Ullensvang, Lehre, Storm-Mathisen, and Danbolt, 1997; Furuta, Rothstein, and Martin, 1997). To explore this possibility, we directly compared GLT-1 and GLAST protein expression in whole neocortical and hippocampal homogenate using Western blot analysis (Fig. 3). Tissue samples were prepared from rat neocortex and hippocampus at P3, P7, and P14. At P3 and P7, GLT-1 (Fig. 3C) and GLAST (Fig. 3D) protein expression in the neocortex was significantly lower than in hippocampus (p<0.05 at both time points). By P14, however, the expression of GLT-1 and GLAST protein was similar in whole hippocampus and neocortex.
Figure 3. GLT-1 and GLAST protein expression are lower in neonatal rat cortex compared to hippocampus.

A. Representative Western blots from cortical and hippocampal homogenates showing protein expression of GLT-1 at P3 (nCortex = 6, nHippo = 6), P7 (nCortex = 7, nHippo = 7) and P14 (n = 6) B. Protein expression of GLAST at P3, P7 and P14 (n = 5) C–D. Protein expression was quantified by taking the ratio of protein to loading control (β-Actin), and compared between cortical and hippocampal samples for each time-point. Values were normalized to protein expression of P3 cortex to show fold change. Error bars indicate SEM, * p<0.05, two-sample t-test.
To compare developmental GLT-1 and GLAST expression between more specific regions of the neocortex and hippocampus, we used an immunofluorescence approach. Fixed sections were stained with antibodies raised against GLT-1 or GLAST and confocal images including both neocortex and hippocampus were collected (Fig. 4 A, D) at P7, 14, and 28. We verified that background fluorescence was negligible by imaging sections processed without a primary antibody (Fig. S4). All fluorescence values were evaluated in the deep layers of the neocortex (IV-VI) and in stratum radiatum of hippocampal area CA1. Fluorescence density values were normalized based on hippocampal immunostaining on an image by image basis after masking and excluding neuronal cell bodies (Fig. 4C, F, see methods). Though GLT-1 and GLAST expression are low in neonatal hippocampus and increase with age (Thomas, Tian, and Diamond, 2011) we found that GLT-1 was still substantially less abundant in neocortex than in hippocampus at P7 (52.5% ± 6.7%, p<0.05) and also at P14 (62.8% ± 2.5%, p<0.001). By P28 GLT-1 expression was similar between neocortex and hippocampus (p=0.77, Fig. 4A–B). GLAST was also less abundant in neocortex than in hippocampus at P7 (44.1% ± 5.6%, p<0.01) and P14 (55.6% ± 8.9%, p<0.05) and by P28 GLAST was similarly abundant in neocortex and hippocampus (p=0.39) (Fig. 4D, E).
Increased uptake kinetics is associated with more reliance on GLT-1-mediated transport
In order to examine the relative functional contribution of GLT-1 to TCs during development, we used the selective GLT-1 blocker, DHK (300 μM) (Bridges, Kavanaugh, and Chamberlin, 1999). Application of DHK during glutamate uncaging experiments caused TC kinetics to slow as transport by GLT-1 was blocked (Fig. 5A–B). The remaining current reflects the relative contribution of non-GLT-1 transporters, presumably GLAST, and the degree of slowing upon DHK wash in reflects the relative contribution of GLT-1 to the whole TC. This approach is especially powerful as it gives a functional characterization of the GLT-1 mediated uptake relative to other EAATs. We found that the relative contribution of GLT-1 TCs increased during postnatal development (Fig. 5C). At P3 and P7, DHK had a substantially greater effect in hippocampus than in neocortex, suggesting a larger functional contribution of GLT-1 in hippocampus compared to neocortex (P3: p<0.05; P7: p<0.01). By P14, however, the effect of DHK was similar between neocortex and hippocampus (p = 0.3) and comparable to the effect of DHK at older ages. Surprisingly, we found a significant effect of DHK in neocortex and hippocampus at all time points, implying that GLT-1 contributes appreciably to glutamate uptake even in the neonatal neocortex where GLT-1 protein expression is extremely low (Fig. 5B). When the kinetics of the DHK-insensitive current were compared we found that the decay was still significantly faster in hippocampus than in neocortex at P3-P14 (Fig. 5B, P3: p<0.001, P7: p<0.001, P14: p<0.01) showing that decreased DHK-insensitive transport also likely contributes to the slower TC decay kinetics in young neocortex.
Figure 5. GLT1-mediated glutamate uptake is present but extremely low in the neonatal cortex.

A. Representative transporter current traces from cortical and hippocampal astrocytes normalized by amplitude at ages P3-P14 in normal aCSF (black traces) and following the wash in of DHK (300 μM, gray traces). Scale bars = 100ms. B. Decay constants for transporter currents in astrocytes from cortex and hippocampus at P3 (nCor = 5, nHipp = 6 cells), P7 (nCor = 9, nHipp = 13 cells), and P14 (nCo r = 7, nHipp = 8 cells) before and after DHK wash in. C. Ratios of transporter current decay constants after the wash-in of DHK (post-DHK) to decay constants before the wash-in of DHK (pre-DHK) in hippocampal (black bars) and cortical astrocytes (grey bars) from P3-P14. Error bars indicate SEM, *p<0.05, **p<0.01, ***p<0.001, two-sample t test.
Differential maturation of astrocyte uptake correlates with functional effects on neuronal NMDA currents
Glutamate transporter currents evoked by glutamate uncaging may not accurately reflect how astrocytes modulate synaptically released glutamate. In order to determine whether the changes in glutamate uptake we report affect synaptically released glutamate transients, we recorded stimulus-evoked neuronal NMDA currents. NMDA receptor mediated currents provide a readout of extracellular glutamate dynamics from a neuronal perspective and are more useful than AMPA receptor-mediated currents in this context because NMDA receptors are less prone to desensitization than AMPA receptors (Trussell and Fischbach, 1989) and NDMA receptors are present in synaptic and extrasynaptic regions (Harris and Pettit, 2007). To perform these recordings, hippocampal CA1 and cortical layer V pyramidal neurons were patch clamped and held at +40 mV. A train of 5 electrical stimulations at 100 Hz was delivered to either the Schaeffer collateral (hippocampus) or white matter (cortex) axons. Trains of stimuli were used in order to evoke a glutamate transient with synaptic and extrasynaptic components, as it has been demonstrated that extrasynaptic NMDA receptor-mediated currents are affected by glutamate transport (Scimemi, Tian, and Diamond, 2009; Diamond, 2001). We then measured the charge transfer through NMDA receptors before and after blocking astrocytic glutamate uptake with 100 μM TBOA (Fig. 6A). The calibration of the stimulus intensity likely results in different numbers of activated synapses, reflecting changing synapse numbers through development. Differences in the number of activated synapses are accounted for by normalizing the traces after the wash on of TBOA to the traces before the wash on of TBOA on a cell-by-cell basis. The charge transfer ratio (CTR) was calculated by dividing the total charge transfer after TBOA by the charge transfer before TBOA. In hippocampus, the CTR was consistent from P7-P28 (Fig. 6B, P7: 2.77 ± 0.41; P14 2.63 ± 0.37; P28: 2.47 ± 0.23). In the neocortex, however, the CTR increased significantly between P7 (1.27 ± 0.03) and P28 (2.67 ± 0.47, p<0.05) and was significantly lower in neocortex than in hippocampus at P7 and P14 (P7: p<0.05, P14: p<0.05). The difference in the NMDA receptor-mediated charge transfer ratio between neocortex and hippocampus at P7 and P14 indicates that astrocytic glutamate uptake plays a larger role shaping endogenous glutamate signals in the adult neocortex and in the hippocampus than it does in the neonatal neocortex. While this developmental change is correlated with the changes we observe in TC kinetics, it also potentially reflects developmental differences in other synaptic properties such as glial ensheathment of synapses or probability of release.
Figure 6. Differential maturation of astrocyte uptake correlates with functional effects on neuronal NMDA currents.
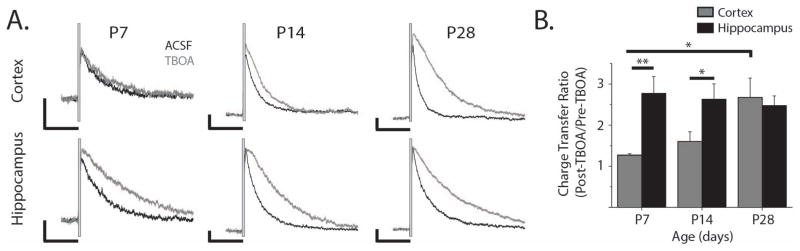
A. Representative NMDA receptor-mediated EPSCs evoked by 5 electrical stimulations 50 μs long at 100 Hz (stimulus artifacts blanked) in aCSF (black) and after wash-in of 100 μM TBOA (grey). Vertical scale bars are 50 pA, horizontal scale bars are 500 ms. B. Charge transfer ratio (calculated as the total integrated NMDAR-mediated EPSC after TBOA wash-in divided by the integrated EPSC before TBOA wash in) for cortical (grey bars) and hippocampal neurons (black bars) at P7 (nCor = 4, nHipp = 4 cells), P14 (nCor = 7, nHipp = 7 cells), and P28 (nCor = 7, nHipp= 3 cells). Error bars indicate SEM, *p<0.05, **p<0.01, two-sample t test.
DISCUSSION
These studies were undertaken to better understand the relationship between astrocytic glutamate uptake and the control of glutamatergic synaptic activity in the dynamic environment of the maturing neocortex. The importance of extracellular glutamate regulation is demonstrated by studies showing that focal glutamate induces the formation of new dendritic spines (Kwon and Sabatini, 2011), coordinated glutamatergic activity is necessary for the development of long-term network connectivity (Allene, Cattani, Ackman, Bonifazi, Aniksztejn, Ben-Ari, and Cossart, 2008; Allene and Cossart, 2010; Crepel, Aronov, Jorquera, Represa, Ben-Ari, and Cossart, 2007), and eliminating GLT-1-dependent astrocytic glutamate uptake causes seizures and death (Tanaka, Watase, Manabe, Yamada, Watanabe, Takahashi, Iwama, Nishikawa, Ichihara, Kikuchi, Okuyama, Kawashima, Hori, Takimoto, and Wada, 1997). Given the importance of astrocytic glutamate uptake in all brain regions, surprisingly little is known about the maturation of glutamate uptake in the neocortex. Although developmental up-regulation of EAATs has been well demonstrated (Furuta, Rothstein, and Martin, 1997; Ullensvang, Lehre, Storm-Mathisen, and Danbolt, 1997), our study is the first to directly compare the development of EAATs between neocortex and hippocampus. We were surprised to see slow TC kinetics in the neonatal neocortex. In fact, in the neonatal neocortex (at P3) TC decay times are an order of magnitude slower than in the juvenile neocortex (at P14) and 20-fold slower than in adult neocortex or hippocampus. This is in contrast to postnatal hippocampal development where we find that functional uptake and EAAT expression increase to a lesser degree than in neocortex, in agreement with Thomas et al. (Thomas, Tian, and Diamond, 2011). The finding that uptake of exogenously applied glutamate was unexpectedly slow in neonatal neocortex led us to question whether synaptically released glutamate would be similarly handled.
To directly address this question, we recorded NMDA receptor-mediated evoked EPSCs in neocortical and hippocampal neurons during development. We found that in the neonatal neocortex, glutamate uptake does little to limit the activation of NMDA receptors by synaptically-released glutamate. Over development, however, uptake begins to play a larger role in regulating glutamate signaling. In the hippocampus, unlike the neocortex, uptake is robust from a young age and provides consistent control of NMDA receptor activation throughout development. EAATs are known to control glutamate diffusion, NMDA receptor activation, and synaptic fidelity in adults. Our findings show that this control has not been established in the neonatal neocortex, implying a permissive environment for extrasynaptic receptor activation and synaptic crosstalk in the neonatal neocortex. It is important to note that first, train stimuli used in our experiments likely results in NMDA currents generated by a mix of synaptic, peri-synaptic, and extra-synaptic receptors, and second, developmental changes NMDA receptors subunit-specific expression and localization are known to occur. By calculating the charge transfer ratio on a cell-by-cell basis, however, we were able to account for any developmental changes in NMDA current properties and directly address NMDA current regulation by EAATs. Further studies are needed to address the spatial, and sub-unit specific heterogeneity of NMDA receptor control by EAAT-dependent glutamate uptake more directly.
Along with changes in glutamate uptake and neuronal modulation, we also report that the membrane properties of neocortical astrocytes undergo significant post-natal development. Neonatal cortical astrocytes have a higher membrane resistance than aged-matched hippocampal astrocytes, and over cortical development, astrocyte membrane resistance decreases to match hippocampal astrocytes. As astrocytes mature their processes ramify, increasing the total membrane surface area (Bushong, Martone, and Ellisman, 2004; Morel, Higashimori, Tolman, and Yang, 2014). It is likely that different morphological maturation of cortical versus hippocampal astrocytes contributes to the difference in astrocyte membrane resistance. Additionally, potassium channels Kir4.1, TWIK-1, and TREK-1 contribute to the passive membrane properties of astrocytes (Zhou, Xu, Xie, Zhang, Schools, Ma, Kimelberg, and Chen, 2009; Seifert, Huttmann, Binder, Hartmann, Wyczynski, Neusch, and Steinhauser, 2009) and may be involved in the developmental changes we observe (Bordey and Sontheimer, 2000; Higashimori and Sontheimer, 2007). Kir4.1 expression has been shown to increase in the hippocampus and in the neocortex during development (Gupta and Kanungo, 2013; Seifert, Huttmann, Binder, Hartmann, Wyczynski, Neusch, and Steinhauser, 2009). However, developmental regulation of TWIK-1 and TREK in the hippocampus and neocortex has, to our knowledge, not been determined. It is unlikely that developmental differences in gap junction coupling are responsible for the observed changes in membrane resistance, as astrocyte membrane resistance has been shown to be an intrinsic cellular property (Schools, Zhou, and Kimelberg, 2006). Interestingly, we found that the membrane properties of cortical astrocytes appear to reach their mature phenotype by approximately two weeks of age. This occurs simultaneously with the largest developmental change in glutamate uptake (P7-14). Whether similar or different cues drive the maturation of glutamate uptake and membrane properties is unknown.
Our findings highlight both the complexity of the neuron-astrocyte relationship and our limitations in understanding of the spatiotemporal properties of glutamate transients in the developing brain. For example, glutamate dynamics in the synaptic cleft are likely different than we report here, and are known to be driven by diffusion (Thomas, Tian, and Diamond, 2011; Diamond, 2005). Furthermore, glial ensheathment of synapses is not uniform (Lehre and Danbolt, 1998; Ventura and Harris, 1999) and areas that lack astrocytic coverage and the presence of EAATs may have significantly less control of extracellular glutamate (Oliet, Piet, and Poulain, 2001). In light of this, it is likely that rates of glutamate uptake during development may not be entirely dependent on EAAT expression. Factors like the extracellular space fraction, astrocyte surface area and morphology, astrocyte density, glial ensheathment of synapses, and transporter distribution are also likely playing important roles in the developmental differences that we observe. These factors may also account for the differences seen in our studies between developmental transporter expression (Fig. 3 and 4) and functional glutamate uptake (Fig. 1). Changes in trafficking, surface motility, and peri-synaptic localization of EAATs combined with the complexity of brain tissue topology may lead to non-linear relationships between expression and function. This could be especially relevant during pathological states such as stroke, seizure, and brain injury when glutamate levels are extremely high, astrocytic morphology is changed, and neurotransmission loses spatial fidelity (During and Spencer, 1993; Oberheim, Tian, Han, Peng, Takano, Ransom, and Nedergaard, 2008; Hinzman, Thomas, Quintero, Gerhardt, and Lifshitz, 2012).
In summary, our studies reveal significant regional heterogeneity in astrocyte development and provide a first glimpse into the functional maturation of cortical astrocytes. Less astrocytic uptake in the neocortex may prolong extracellular glutamate transients and promote glutamate-driven spontaneous early network oscillations, activation of extrasynaptic and metabotropic receptors, and glutamate-driven synaptogenesis. Thus, later maturation of astrocytes in the neocortex could allow for an early period of developmental plasticity. This is highly likely as our studies directly show that immature astrocytes less tightly regulate NMDA receptors, which are known to guide synaptic maturation (Paoletti, Bellone, and Zhou, 2013). This highlights an important question regarding the relationship between astrocyte development and synaptic maturation (Corty and Freeman, 2013). Do astrocytes play an instructive or permissive role in synaptic maturation? Astrocyte secreted factors are known to drive synaptogenesis in the neonatal neocortex (Clarke and Barres, 2013), consistent with an instructive role for astrocytes in synaptic maturation. Given that glutamate directly induces synapse formation at a similar time point (Kwon and Sabatini, 2011), our findings suggest that the relatively low amount of glutamate uptake in the neonatal neocortex may create a permissive environment for glutamate-driven synaptic maturation.
Supplementary Material
Figure S1: Neurons are not activated by photolysis of MNI-glutamate in the presence of CPP, DNQX, and Gabazine. Current-clamp recordings from a representative pyramidal neuron in area CA1 of P29 rat hippocampus before and after wash-on of the glutamate blockers CPP and DNQX and the GABA blocker Gabazine. A. Left panel shows 5 overlaid traces before during and after a 1 ms laser pulse on the soma. Bath included only aCSF and MNI-glutamate. Current injection in right panel shows the ability of the neuron to fire action potentials when injected with positive current in the absence of glutamate and GABA receptor blockers. (Timing of uncaging indicated by tick mark in left panel, vertical scale bars = 20 mV, horizontal scale bars = 50 ms). B. Glutamate receptor blockers DNQX and CPP and the GABA receptor blocker Gabazine were washed into the bath and uncaging was repeated (as in A). Membrane potential did not shift in response to laser uncaging on the soma. Current injection in right panel shows that action potential generation is unaffected by glutamate and GABA receptor blockers. (Timing of uncaging indicated by tick mark in left panel, vertical scale bars = 20 mV, horizontal scale bars = 50 ms).
Figure S2: Uptake kinetics are unaffected by the amount of glutamate uncaged. A. Representative glutamate transporter current traces from hippocampal and cortical astrocytes normalized to input resistance at P7 and P14. Currents were evoked by a 1 ms laser pulse with 100% laser power (black) or with 50% laser power (grey), modulated by a neutral density filter. Scale bars = 100 ms. B. Peak TC amplitudes after uncaging at 100% and 50% laser power normalized to 100% laser power at P7 (nCor = 15, nHipp = 13 cells) and P14 (nCor = 18, nHipp = 20 cells). C. Peak-normalized traces from A. D. Quantification of TC decay kinetics after uncaging at 100% or 50% laser power in cortex and hippocampus at P7 and P14. Error bars indicate SEM, *p<0.05, **p<0.01, ***p<0.001, two-sample t test.
Figure S3: Centroid analysis confirms larger <t>Clearance in cortical astrocytes compared to hippocampal astrocytes. A. Inverse slope ratios (calculated as the inverse of the rising TC slope in low dose TBOA (10 μM) normalized to the rising slope before TBOA wash on) plotted against <t> on a cell-by-cell basis. Solid circles = pre-TBOA individual cells, open circles = same cells in 10 μM TBOA, solid squares = mean pre-TBOA values with Y error bars showing SEM, open squares = mean post-TBOA values with X and Y error bars showing SEM. (nCor = 5, nHipp = 4 cells) B. Slopes for linear fits shown in A. Error bars indicate SEM, *p<0.05, t test.
Figure S4: Background fluorescence is negligible in sections stained with only Alexa 488-conjugated secondary. Coronal sections from a P29 rat were prepared for immunofluorescence as described in Methods. Primary antibodies were excluded from the primary incubation step. The sections were then incubated with goat-anti-rabbit Alexa 488 secondary, mounted with DAPI-containing mounting media, and imaged with confocal microscopy as described in Methods. Laser and device settings for the confocal imaging of Alexa 488 fluorescence were identical to those used for imaging GLT-1 expression at P28 (Fig. 4A) A. DAPI fluorescence in a no-primary section showing cortex and hippocampus. B. Alexa 488 fluorescence in the same no-primary section showing low background labeling.
Main Points.
Astrocytes take up glutamate more slowly in the developing neocortex than in hippocampus.
NMDA receptor activation is less constrained by astrocytic glutamate uptake in the developing neocortex.
Membrane properties mature later in cortical astrocytes.
Acknowledgments
This work was supported by the Epilepsy Foundation (CD), National Institute of Neurological Disease and Stroke R01-NS076885 (CD), Synapse Neurobiology Training Grant (EH, NINDS T32-NS061764, M. Jacob, P.I.), the National Institute of General Medical Sciences (K12GM074869, DC), Tufts Center for Neuroscience Research (P30 NS047243), and The Research Council of Norway (240844, NCD). The authors would like to thank David Hampton for his assistance and Dr. Yongjie Yang and Dr. Haruki Higashimori for their helpful discussions.
Reference List
- Allene C, Cattani A, Ackman JB, Bonifazi P, Aniksztejn L, Ben-Ari Y, Cossart R. Sequential generation of two distinct synapse-driven network patterns in developing neocortex. J Neurosci. 2008;28:12851–12863. doi: 10.1523/JNEUROSCI.3733-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allene C, Cossart R. Early NMDA receptor-driven waves of activity in the developing neocortex: physiological or pathological network oscillations? J Physiol. 2010;588:83–91. doi: 10.1113/jphysiol.2009.178798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Bridges RJ, Chamberlin AR, Shimamoto K, Yasuda–Kamatani Y, Swanson RA. Differing effects of substrate and non–substrate transport inhibitors on glutamate uptake reversal. J Neurochem. 2001;79:1207–1216. doi: 10.1046/j.1471-4159.2001.00668.x. [DOI] [PubMed] [Google Scholar]
- Armbruster M, Hampton D, Yang Y, Dulla CG. Laser-scanning astrocyte mapping reveals increased glutamate-responsive domain size and disrupted maturation of glutamate uptake following neonatal cortical freeze-lesion. Frontiers in cellular neuroscience. 2014:8. doi: 10.3389/fncel.2014.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benediktsson AM, Marrs GS, Tu JC, Worley PF, Rothstein JD, Bergles DE, Dailey ME. Neuronal activity regulates glutamate transporter dynamics in developing astrocytes. Glia. 2012;60:175–188. doi: 10.1002/glia.21249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Dzubay JA, Jahr CE. Glutamate transporter currents in bergmann glial cells follow the time course of extrasynaptic glutamate. Proc Natl Acad Sci U S A. 1997;94:14821–14825. doi: 10.1073/pnas.94.26.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in the hippocampus. J Neurosci. 1998;18:7709–7716. doi: 10.1523/JNEUROSCI.18-19-07709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997;19:1297–1308. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H. Ion channel expression by astrocytes in situ: comparison of different CNS regions. Glia. 2000;30:27–38. doi: 10.1002/(sici)1098-1136(200003)30:1<27::aid-glia4>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Bridges RJ, Kavanaugh MP, Chamberlin AR. A pharmacological review of competitive inhibitors and substrates of high-affinity, sodium-dependent glutamate transport in the central nervous system. Current pharmaceutical design. 1999;5:363–379. [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Ellisman MH. Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. International Journal of Developmental Neuroscience. 2004;22:73–86. doi: 10.1016/j.ijdevneu.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nature Reviews Neuroscience. 2013;14:311–321. doi: 10.1038/nrn3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corty MM, Freeman MR. Architects in neural circuit design: Glia control neuron numbers and connectivity. The Journal of cell biology. 2013;203:395–405. doi: 10.1083/jcb.201306099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepel V, Aronov D, Jorquera I, Represa A, Ben-Ari Y, Cossart R. A parturition-associated nonsynaptic coherent activity pattern in the developing hippocampus. Neuron. 2007;54:105–120. doi: 10.1016/j.neuron.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Storm-Mathisen J, Kanner BI. An [Na+ + K+]coupled L-glutamate transporter purified from rat brain is located in glial cell processes. Neurosci. 1992;51:295–310. doi: 10.1016/0306-4522(92)90316-t. [DOI] [PubMed] [Google Scholar]
- Diamond JS. Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. 2001;21:8328–8338. doi: 10.1523/JNEUROSCI.21-21-08328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS. Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J Neurosci. 2005;25:2906–2916. doi: 10.1523/JNEUROSCI.5125-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Bergles DE, Jahr CE. Glutamate release monitored with astrocyte transporter currents during LTP. Neuron. 1998;21:425–433. doi: 10.1016/s0896-6273(00)80551-6. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol. 2000;83:2835–2843. doi: 10.1152/jn.2000.83.5.2835. [DOI] [PubMed] [Google Scholar]
- During MJ, Spencer DD. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. The lancet. 1993;341:1607–1610. doi: 10.1016/0140-6736(93)90754-5. [DOI] [PubMed] [Google Scholar]
- Freeman MR. Specification and morphogenesis of astrocytes. Science. 2010;330:774–778. doi: 10.1126/science.1190928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta A, Rothstein JD, Martin LJ. Glutamate transporter protein subtypes are expressed differentially during rat CNS development. The Journal of Neuroscience. 1997;17:8363–8375. doi: 10.1523/JNEUROSCI.17-21-08363.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Kanungo M. Glial molecular alterations with mouse brain development and aging: up-regulation of the Kir4. 1 and aquaporin-4. Age. 2013;35:59–67. doi: 10.1007/s11357-011-9330-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG. Synaptic islands defined by the territory of a single astrocyte. J Neurosci. 2007;27:6473–6477. doi: 10.1523/JNEUROSCI.1419-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AZ, Pettit DL. Extrasynaptic and synaptic NMDA receptors form stable and uniform pools in rat hippocampal slices. The Journal of physiology. 2007;584:509–519. doi: 10.1113/jphysiol.2007.137679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honore T, Nielsen M, Lehre KP, Danbolt NC. Brain glutamate transporter proteins form homomultimers. J Biol Chem. 1996;271:27715–27722. doi: 10.1074/jbc.271.44.27715. [DOI] [PubMed] [Google Scholar]
- Higashimori H, Sontheimer H. Role of Kir4. 1 channels in growth control of glia. Glia. 2007;55:1668–1679. doi: 10.1002/glia.20574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinzman JM, Thomas TC, Quintero JE, Gerhardt GA, Lifshitz J. Disruptions in the regulation of extracellular glutamate by neurons and glia in the rat striatum two days after diffuse brain injury. Journal of neurotrauma. 2012;29:1197–1208. doi: 10.1089/neu.2011.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmseth S, Scott HA, Real K, Lehre KP, Leergaard TB, Bjaalie JG, Danbolt NC. The concentrations and distributions of three C-terminal variants of the GLT1 (EAAT2; slc1a2) glutamate transporter protein in rat brain tissue suggest differential regulation. Neurosci. 2009;162:1055–1071. doi: 10.1016/j.neuroscience.2009.03.048. [DOI] [PubMed] [Google Scholar]
- Holmseth S, Dehnes Y, Huang YH, Follin-Arbelet VV, Grutle NJ, Mylonakou MN, Plachez C, Zhou Y, Furness DN, Bergles DE. The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. The Journal of Neuroscience. 2012;32:6000–6013. doi: 10.1523/JNEUROSCI.5347-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmseth S, Zhou Y, Follin-Arbelet VV, Lehre KP, Bergles DE, Danbolt NC. Specificity Controls for Immunocytochemistry The Antigen Preadsorption Test Can Lead to Inaccurate Assessment of Antibody Specificity. Journal of Histochemistry & Cytochemistry. 2012;60:174–187. doi: 10.1369/0022155411434828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karus C, Mondragão MA, Ziemens D, Rose CR. Astrocytes restrict discharge duration and neuronal sodium loads during recurrent network activity. Glia. 2015 doi: 10.1002/glia.22793. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000;20:5733–5740. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuffler SW, Nicholls JG. The physiology of neuroglial cells. Ergeb Physiol. 1966;57:1–90. [PubMed] [Google Scholar]
- Kuffler SW, Nicholls JG, Orkand RK. Physiological properties of glial cells in the central nervous system of amphibia. J Neurophys. 1966;29:768–787. doi: 10.1152/jn.1966.29.4.768. [DOI] [PubMed] [Google Scholar]
- Kwon HB, Sabatini BL. Glutamate induces de novo growth of functional spines in developing cortex. Nature. 2011;474:100–104. doi: 10.1038/nature09986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15:1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC. The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. The Journal of Neuroscience. 1998;18:8751–8757. doi: 10.1523/JNEUROSCI.18-21-08751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieth E, LaNoue KF, Berkich DA, Xu B, Ratz M, Taylor C, Hutson SM. Nitrogen shuttling between neurons and glial cells during glutamate synthesis. J Neurochem. 2001;76:1712–1723. doi: 10.1046/j.1471-4159.2001.00156.x. [DOI] [PubMed] [Google Scholar]
- Morel L, Higashimori H, Tolman M, Yang Y. VGluT1+ Neuronal Glutamatergic Signaling Regulates Postnatal Developmental Maturation of Cortical Protoplasmic Astroglia. J Neurosci. 2014 Aug 13;34(33):10950–10962. doi: 10.1523/JNEUROSCI.1167-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Li X, Rempel J, Stelmack G, Patel D, Staines WA, Yasumura T, Rash JE. Connexin26 in adult rodent central nervous system: demonstration at astrocytic gap junctions and colocalization with connexin30 and connexin43. J Comp Neurol. 2001;441:302–323. doi: 10.1002/cne.1414. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nature Methods. 2004;1:31–37. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Tian GF, Han X, Peng W, Takano T, Ransom B, Nedergaard M. Loss of astrocytic domain organization in the epileptic brain. The Journal of Neuroscience. 2008;28:3264–3276. doi: 10.1523/JNEUROSCI.4980-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Piet R, Poulain DA. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science. 2001;292:923–926. doi: 10.1126/science.1059162. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nature Reviews Neuroscience. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]
- Piet R, Vargova L, Sykova E, Poulain DA, Oliet SH. Physiological contribution of the astrocytic environment of neurons to intersynaptic crosstalk. Proc Natl Acad Sci U S A. 2004;101:2151–2155. doi: 10.1073/pnas.0308408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Schmidt W, Wolf G. High-affinity uptake of L-[3H]glutamate and D-[3H]aspartate during postnatal development of the hippocampal formation: a quantitative autoradiographic study. Exp Brain Res. 1988;70:50–54. doi: 10.1007/BF00271846. [DOI] [PubMed] [Google Scholar]
- Schools GP, Zhou M, Kimelberg HK. Development of gap junctions in hippocampal astrocytes: evidence that whole cell electrophysiological phenotype is an intrinsic property of the individual cell. J Neurophys. 2006;96:1383–1392. doi: 10.1152/jn.00449.2006. [DOI] [PubMed] [Google Scholar]
- Scimemi A, Tian H, Diamond JS. Neuronal transporters regulate glutamate clearance, NMDA receptor activation, and synaptic plasticity in the hippocampus. 2009;29:14581–14595. doi: 10.1523/JNEUROSCI.4845-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert G, Huttmann K, Binder DK, Hartmann C, Wyczynski A, Neusch C, Steinhauser C. Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4. 1 subunit. The Journal of Neuroscience. 2009;29:7474–7488. doi: 10.1523/JNEUROSCI.3790-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T. DL-threo-beta-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol. 1998;53:195–201. doi: 10.1124/mol.53.2.195. [DOI] [PubMed] [Google Scholar]
- Sibson NR, Mason GF, Shen J, Cline GW, Herskovits AZ, Wall JE, Behar KL, Rothman DL, Shulman RG. In vivo (13)C NMR measurement of neurotransmitter glutamate cycling, anaplerosis and TCA cycle flux in rat brain during. J Neurochem. 2001;76:975–989. doi: 10.1046/j.1471-4159.2001.00074.x. [DOI] [PubMed] [Google Scholar]
- Sohl G, Odermatt B, Maxeiner S, Degen J, Willecke K. New insights into the expression and function of neural connexins with transgenic mouse mutants. Brain Res Brain Res Rev. 2004;47:245–259. doi: 10.1016/j.brainresrev.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Steinhauser C, Berger T, Frotscher M, Kettenmann H. Heterogeneity in the Membrane Current Pattern of Identified Glial Cells in the Hippocampal Slice. Eur J Neurosci. 1992;4:472–484. doi: 10.1111/j.1460-9568.1992.tb00897.x. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tani H, Dulla CG, Huguenard JR, Reimer RJ. Glutamine is required for persistent epileptiform activity in the disinhibited neocortical brain slice. J Neurosci. 2010;30:1288–1300. doi: 10.1523/JNEUROSCI.0106-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani H, Dulla CG, Farzampour Z, Taylor-Weiner A, Huguenard JR, Reimer RJ. A Local Glutamate-Glutamine Cycle Sustains Synaptic Excitatory Transmitter Release. Neuron. 2014;81:888–900. doi: 10.1016/j.neuron.2013.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M, Sohl G, Eiberger J, Willecke K. Emerging complexities in identity and function of glial connexins. Trends Neurosci. 2005;28:188–195. doi: 10.1016/j.tins.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Thomas CG, Tian H, Diamond JS. The relative roles of diffusion and uptake in clearing synaptically released glutamate change during early postnatal development. The Journal of Neuroscience. 2011;31:4743–4754. doi: 10.1523/JNEUROSCI.5953-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trussell LO, Fischbach GD. Glutamate receptor desensitization and its role in synaptic transmission. Neuron. 1989;3:209–218. doi: 10.1016/0896-6273(89)90034-2. [DOI] [PubMed] [Google Scholar]
- Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC. Differential developmental expression of the two rat brain glutamate transporter proteins GLAST and GLT. Eur J Neurosci. 1997;9:1646–1655. doi: 10.1111/j.1460-9568.1997.tb01522.x. [DOI] [PubMed] [Google Scholar]
- Ventura R, Harris KM. Three-dimensional relationships between hippocampal synapses and astrocytes. The Journal of Neuroscience. 1999;19:6897–6906. doi: 10.1523/JNEUROSCI.19-16-06897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voutsinos-Porche B, Knott G, Tanaka K, Quairiaux C, Welker E, Bonvento G. Glial glutamate transporters and maturation of the mouse somatosensory cortex. Cerebral Cortex. 2003;13:1110–1121. doi: 10.1093/cercor/13.10.1110. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Morimoto K, Hirao T, Suwaki H, Watase K, Tanaka K. Amygdala-kindled and pentylenetetrazole-induced seizures in glutamate transporter GLAST-deficient mice. Brain Res. 1999;845:92–96. doi: 10.1016/s0006-8993(99)01945-9. [DOI] [PubMed] [Google Scholar]
- Zhou M, Xu G, Xie M, Zhang X, Schools GP, Ma L, Kimelberg HK, Chen H. TWIK-1 and TREK-1 are potassium channels contributing significantly to astrocyte passive conductance in rat hippocampal slices. J Neurosci. 2009;29:8551–8564. doi: 10.1523/JNEUROSCI.5784-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Neurons are not activated by photolysis of MNI-glutamate in the presence of CPP, DNQX, and Gabazine. Current-clamp recordings from a representative pyramidal neuron in area CA1 of P29 rat hippocampus before and after wash-on of the glutamate blockers CPP and DNQX and the GABA blocker Gabazine. A. Left panel shows 5 overlaid traces before during and after a 1 ms laser pulse on the soma. Bath included only aCSF and MNI-glutamate. Current injection in right panel shows the ability of the neuron to fire action potentials when injected with positive current in the absence of glutamate and GABA receptor blockers. (Timing of uncaging indicated by tick mark in left panel, vertical scale bars = 20 mV, horizontal scale bars = 50 ms). B. Glutamate receptor blockers DNQX and CPP and the GABA receptor blocker Gabazine were washed into the bath and uncaging was repeated (as in A). Membrane potential did not shift in response to laser uncaging on the soma. Current injection in right panel shows that action potential generation is unaffected by glutamate and GABA receptor blockers. (Timing of uncaging indicated by tick mark in left panel, vertical scale bars = 20 mV, horizontal scale bars = 50 ms).
Figure S2: Uptake kinetics are unaffected by the amount of glutamate uncaged. A. Representative glutamate transporter current traces from hippocampal and cortical astrocytes normalized to input resistance at P7 and P14. Currents were evoked by a 1 ms laser pulse with 100% laser power (black) or with 50% laser power (grey), modulated by a neutral density filter. Scale bars = 100 ms. B. Peak TC amplitudes after uncaging at 100% and 50% laser power normalized to 100% laser power at P7 (nCor = 15, nHipp = 13 cells) and P14 (nCor = 18, nHipp = 20 cells). C. Peak-normalized traces from A. D. Quantification of TC decay kinetics after uncaging at 100% or 50% laser power in cortex and hippocampus at P7 and P14. Error bars indicate SEM, *p<0.05, **p<0.01, ***p<0.001, two-sample t test.
Figure S3: Centroid analysis confirms larger <t>Clearance in cortical astrocytes compared to hippocampal astrocytes. A. Inverse slope ratios (calculated as the inverse of the rising TC slope in low dose TBOA (10 μM) normalized to the rising slope before TBOA wash on) plotted against <t> on a cell-by-cell basis. Solid circles = pre-TBOA individual cells, open circles = same cells in 10 μM TBOA, solid squares = mean pre-TBOA values with Y error bars showing SEM, open squares = mean post-TBOA values with X and Y error bars showing SEM. (nCor = 5, nHipp = 4 cells) B. Slopes for linear fits shown in A. Error bars indicate SEM, *p<0.05, t test.
Figure S4: Background fluorescence is negligible in sections stained with only Alexa 488-conjugated secondary. Coronal sections from a P29 rat were prepared for immunofluorescence as described in Methods. Primary antibodies were excluded from the primary incubation step. The sections were then incubated with goat-anti-rabbit Alexa 488 secondary, mounted with DAPI-containing mounting media, and imaged with confocal microscopy as described in Methods. Laser and device settings for the confocal imaging of Alexa 488 fluorescence were identical to those used for imaging GLT-1 expression at P28 (Fig. 4A) A. DAPI fluorescence in a no-primary section showing cortex and hippocampus. B. Alexa 488 fluorescence in the same no-primary section showing low background labeling.