

Abstract
Interleukin-37 (IL-37) is a recently identified cytokine with potent antiinflammatory and immunosuppressive functions. The objective of this study was to compare levels of IL-37 mRNA in immunological subgroups of chronic human immunodeficiency virus-1 (HIV-1)-infected individuals and noninfected controls, to determine IL-37’s association with biomarkers of inflammation and reservoir size. This was a cross-sectional study. The HIV-1-infected patients were categorized in three subgroups depending on their combination antiretroviral therapy (cART) treatment status and CD4+ T-cell count. Quantitative RT-PCR was used for the detection of IL-37 mRNA and HIV-1 DNA in peripheral blood mononuclear cells (PBMCs). Biomarkers in plasma were quantified by enzyme-linked immunosorbent assay (ELISA), whereas T-cell activation was determined by flow cytometry. Lastly, lipopolysaccharide (LPS) stimulations of patients PBMCs were carried out to determine differences in IL-37 mRNA response between the subgroups. Sixty HIV-1-infected patients and 20 noninfected controls were included in the study. Steady-state IL-37 mRNA levels in PBMCs were significantly higher in HIV-1-infected individuals compared with noninfected controls: 2.4-fold (p ≤ 0.01) cART-naïve subjects; 3.9-fold (p ≤ 0.0001) inadequate immunological responders; and 4.0-fold (p ≤ 0.0001) in immunological responders compared with non-infected controls. Additionally, levels of the monocyte inflammatory marker sCD14 correlated with IL-37 mRNA (p = 0.03), whereas there was no association with T-cell activation. Finally, we observed a significant correlation between total viral HIV-1 DNA and IL-37 mRNA in PBMCs (p < 0.0001). Collectively, our data shows that the level of IL-37 mRNA is affected by chronic HIV-1-infection. A relationship with the activation of the monocyte compartment is suggested by the correlation with sCD14 and, interestingly, IL-37 could be related to the size of the total viral HIV-1 reservoir.
INTRODUCTION
Lately, the newly described interleukin-37 has emerged as an important part of the antiinflammatory system affecting both innate and adaptive immunity. We know that within the interleukin-1 (IL-1) family, the proinflammatory members are tightly regulated to avoid excessive inflammation. Examples of this include the potent blocking of IL-1α and IL1-β by IL-1 receptor antagonist (1), IL-18 binding protein’s capacity to neutralize IL-18 (2) and IL-36 receptor antagonist’s ability to reduce IL-36 activity (3). However, IL-37, a new member of the IL-1 family, has unique properties because this cytokine broadly suppresses the function of other members of the IL-1 family as well as proinflammatory signals delivered by toll-like receptor (TLR) agonists (reviewed in [4]). IL-37 itself appears to be under tight regulatory control, as steady-state levels of mRNA are limited by an inherent instability sequence (5). Five transcript variants (IL-37a–e) have been described with the largest, IL-37b containing five of the six exons (6–10). In PBMCs, IL-37 protein is primarily expressed in monocytic cells (11).
Similar to IL-1α and IL-33, IL-37 has been shown to be active both extracellularly and intracellularly, where it is translocated to the nucleus and suppresses TLR-agonist-induced cytokines by a caspase-1-dependent mechanism (12,13). In addition, neutralizing antibodies to IL-37 have demonstrated an effect of extracellular IL-37, resulting in a higher proinflammatory load (12). Therefore, due to these antiinflammatory and immunosuppressive functions of IL-37 and to the potential for a future therapeutic use of this cytokine, diseases dominated by excess inflammation are becoming a subject of great interest. For instance, studies have reported higher levels of IL-37 expression in melanoma patients, systemic lupus erythematosus, colitis, rheumatoid arthritis, atopic dermatitis and morbid obesity (14–19). In addition, several studies are showing correlation between both increased and decreased amounts of IL-37 to the severity of the disease (reviewed in [20]). In a mouse model, a marked protection against LPS-induced shock was observed with transgenic expression of human IL-37 (IL-37tg) (17). In addition, IL-37tg mice subjected to chemically- induced colitis exhibited a lower level of colonic inflammation, reduced IL-1β and tumor necrosis factor alpha (TNFα) production, but increased levels of IL-10; effects of which proved to be myeloid-derived rather than epithelial cell-derived (16). IL-37 presumably mediates suppression of acquired immunity by the inhibition of dendritic cell maturation, thereby affecting the functional link between innate and adaptive immunity (17,21).
HIV-1 infection is characterized by excess immune activation (22,23). Although cART effectively suppresses viral replication, persistent HIV-1-associated immune activation is believed to lead to impaired immune function and premature immunologic aging, as well as an increased risk of cardiovascular disease and cancer (23–29). The underlying mechanisms are still poorly understood, but the persistence of low-level viremia despite effective cART and translocation of microbial products, such as LPS, across a compromised gastrointestinal mucosal surface has been suggested to play important roles (30–32).
Notably, HIV-1-infected patients with inadequate reconstitution of the CD4+ T-cell compartment upon initiation of cART, termed inadequate immunological responders (IIRs), exhibit higher levels of inflammatory biomarkers, greater T-cell activation and increased mortality compared with immunological responders (33–40). Yet, how innate immune responses influence these pathological processes remains unclear (41).
Thus, as HIV-1-associated inflammation negatively influences the health of infected patients despite suppressive cART and as IL-37 has emerged as a cytokine with unique immune regulatory effects, assessing IL-37 expression in the context of chronic HIV-1 infection is an important and unexplored area of research. Therefore, we determined IL-37 mRNA expression, inflammation and persistent HIV-1 DNA in an immunological mixed cohort of chronically HIV-1-infected patients.
MATERIALS AND METHODS
Study Design and Inclusion
This was a cross-sectional study in which three groups of HIV-1-infected patients were examined: (1) cART-naïve with no history of antiretroviral treatment; (2) cART-treated IIRs defined as having CD4+ T-cell counts continuously below 500 mm3 despite viral suppression with plasma HIV RNA <50 copies/mL for >3 years; (3) cART-treated immunological responders defined as having reached a CD4+ T-cell count of >500 mm3 within 3 years of cART-induced viral suppression with plasma HIV-1 RNA <50 copies/mL. Healthy non–HIV-1- infected persons were included as control population. Data on CD4 T-cell count, plasma HIV-1 RNA and use of cART were obtained from hospital databases and medical records. Subjects known to be pregnant, coinfected with Hepatitis B or C or diagnosed with any autoimmune or autoinflammatory disease were excluded. The study was conducted at the Department of Infectious Diseases, Aarhus University Hospital, Denmark. Written informed consent was obtained from each participant. The study was approved by The Regional Research Ethics Committee for the Region of Midtjylland and The Danish Data Protection Agency for the Region of Midtjylland.
Serum and plasma samples were obtained from each participant. Samples were stored at −80°C until analyses. PBMCs were isolated using Ficoll-Paque PLUS (GE Healthcare, Pittsburgh, PA USA) and stored at −180°C.
IL-37 mRNA in PBMCs
For each participant, total RNA was extracted from 1 million PBMCs to standardize input, using the High Pure Isolation kit (Roche, Basel, Switzerland) as described by the manufacturer. Synthesis of cDNA was performed using an Omniscript Reverse Transcription kit (Qiagen, Venlo, the Netherlands) with 5 μL RNA as template and a final volume of 20 μL. A mixture of 1 μL oligo dT primer (0.4 μg/L) and 1 μL random hexamers (0.4 μg/L) were used in addition to the standard protocol. The gene U6 snRNA (Table 1) was measured by real-time polymerase chain reaction (RT-PCR) as a quality control of the cDNA and for further use as an endogenous normalization control.
Table 1.
Primer sequences.
| Cytokine | Forward (5′→3′) | Reverse (5′→3′) | Length |
|---|---|---|---|
| IL-37 (1 round) | CTCCTGGGGGTCTCTAAAGG | GATGAACCATCCGGGGTGAG | 204 bp |
| U6 snRNA | GCTTCGGCAGCACATATACTAAAA | CGCTTCACGAATTTGCGTGTCAT | 84 bp |
A nested-PCR containing two amplification rounds was used to measure IL-37 levels in the cDNA samples. We used a full-length cloned IL-37 cDNA transcript in serial dilutions as positive control producing a final standard curve. Nontemplate controls and RNA samples not reverse transcribed were run alongside patient samples on every plate to ensure optimal control for possible contamination. Patient samples were mixed on all plates to ensure samples from each of the four groups were represented in every run.
The first amplification round was limited to 10 cycles with the following conditions (GeneAmp PCR System 9700, Applied Biosystems [Thermo Fisher Scientific Inc., Waltham, MA, USA]): 95°C for 10 min, 10 cycles of 94°C for 20 s, 55°C for 40 s and 72°C for 40 s and maintained at 4°C. We used 5 μL of cDNA from each participant completed with a PCR mixture consisting of 25 μL AmpliTaq Gold PCR Master Mix (Applied Biosystems [Thermo Fisher Scientific]), 2 μL of each 20 μmol/L IL-37 primer (see Table 1) and 16 μL RNase free water to complete a final reaction volume of 50 μL. All samples were run in duplicates. The IL-37 primer pair was designed to cover each of the five isoforms of IL-37(a–e). Once amplified, the product was 204 bp, crossing the exon(5)–exon(6) junction.
In the second round of nested-PCR, we used an IL-37 FAM-labeled Taqman Gene Expression Assay (Applied Biosystems [Thermo Fisher Scientific]) covering all five isoforms. All samples from the first round were assayed in duplicates, making a total of quadruplicate determinations per patient. The PCR-mix consisted of 10 μL 2× TaqMan Gene Expression MasterMix, 1 μL 20× TaqMan Gene Expression Assay (Hs00367201m1, Applied Biosystems [Thermo Fisher Scientific]) and 5 μL RNase free water. 4 μL template was added from the previous amplification, making a final 20 μL reaction volume. The RT-qPCR was performed using the following conditions: 95°C for 10 min, 45 cycles of 95°C for 15 s and 60°C for 1 min on a Bio-Rad CFX96 Real-Time system (Bio-Rad, Hercules, CA, USA). The relative ratio of IL-37 mRNA in the PBMCs was determined by the Pfaffl method using a standard curve. All patient values were related to the U6 snRNA as the reference control. Seventy-three patient samples were analyzed. The missing data on the last seven patients were due to a limited amount of cells.
Stimulation of PBMCs
We investigated the induction of IL-37 mRNA in PBMCs in response to 24 h of stimulation with LPS (100 ng/mL). Cryopreserved PBMCs were rapidly thawed and rested 3 h at 37°C, 5% CO2 in complete medium (RPMI 1640 supplemented with 10% Hi-FBS, 1% l-glutamine and 1% Penicillin-Streptomycin [Biowest, Nuaille, France]) in 96-well plates (250,000 cells/100 μL/well). After 3 h, quadruplicate wells were stimulated with 100 μL of LPS (final concentration 100 ng/mL) or control medium, resulting in a final volume of 200 μL per well. After 24 h at 37°C, 5% CO2, supernatants were collected, and the PBMCs washed and lysed directly in lysis buffer in the wells. The samples were frozen at −80°C. The RNA was purified using the High Pure Isolation kit (Roche). cDNA and subsequent nested PCR were carried out as above. Data are missing on eight patients in the RPMI and six patients in the LPS stimulation due to a limited amount of cells.
Soluble Markers
We measured the levels of soluble CD14, IL-6 and soluble CD163 in plasma. sCD14 and IL-6 were quantified using Quantikine High Sensitivity ELISA, both from R&D Systems (Minneapolis, MN, USA). Plasma levels of sCD163 were determined using an in-house assay. This sandwich ELISA assay is using an polyclonal rabbit anti-CD163 IgG as capture antibody and a monoclonal anti-CD163 as detection, described in more details previously (42). Data from one patient in the sCD163 analysis were excluded as an outlier.
T-Cell Markers
Cryopreserved PBMCs were rapidly thawed and transferred to 13 mL cold phosphate buffered saline solution (PBS). After washing, 106 cells were resuspended in FACS tubes with a staining buffer containing bovine serum albumin and incubated in the dark for 20 min with the following antibodies: α-CD3 conjugated PE-Cy7 (BD Biosciences, San Jose, CA, USA), α-CD4 conjugated APC-H7 (BD Biosciences) and α-CD38 conjugated PerCP-Cy5.5 (BioLegend, San Diego, CA, USA). Samples were washed with 2 mL FACS flow buffer (BD Biosciences) and analyzed on a FACSCanto 2 (BD Biosciences). The CD3+CD4− population was used as an approximation for CD8+ cells. Monocyte percentages were estimated with forward and side scatter. Data were analyzed using FlowJo v.7.6.5 (Tree Star, Ashland, OR).
Total Viral Reservoir
The total viral reservoir was determined in IIRs and Responders as copies of integrated HIV-1 DNA per 106 PBMCs using quantitative PCR. The frequency of cells carrying HIV-DNA was calculated from a standard curve of serially diluted DNA from 8E5 cells (43). Lower limit of detection in the assay was defined as 25 copies/106 PBMCs, below which the quantification was not considered reliable. Patients with lower HIV-DNA levels were assigned a random value around 25 ± 1.
Statistical Analysis
Characteristics for the groups were compared using ordinary one-way analysis of variance (ANOVA) or Kruskall-Wallis test. When only data from two groups were available, Rank sum test (Mann-Whitney) or Student t test were used as appropriate. Differences in laboratory results for the four groups were compared using Holm-Sidak multiple comparisons test or Dunn multiple comparisons test in relation to distribution of data. Paired data were analyzed using Wilcoxon signed rank test. Data distribution was assessed by inspection of histograms and normal quantile–quantile (q–q) plots. Pearson correlation and linear regression were used to explore correlations between parameters. Two-tailed P values below 0.05 were considered significant. Statistical analyses and graphical presentations were performed using GraphPad Prism, version 6.0 (GraphPad Software, Inc., La Jolla, CA, USA) and Stata 12 (StataCorp, College Station, TX, USA).
All supplementary materials are available online at www.molmed.org.
RESULTS
Study Population
We included a total of 60 HIV-1-infected patients (20 cART-naïve, 20 IIRs and 20 responders) as well as 20 noninfected healthy subjects. Their characteristics are shown in Table 2.
Table 2.
Characteristics for all participants.
| Characteristic | cART-naïve (n = 20) | cART-treated inadequate responders (n = 20) | cART-treated responders (n = 20) | Noninfected controls (n = 20) | P valuea |
|---|---|---|---|---|---|
| Age, years (IQRb) | 40.5 (34.3–52.8) | 52.0 (43.0–64.5) | 46.5 (43.0–51.0) | 47.5 (35.0–55.0) | 0.08 |
| Sex | |||||
| Male, no. | 17 | 14 | 12 | 16 | — |
| Female, no. | 3 | 6 | 8 | 4 | — |
| CD4+ cell count inclusion date, median cells/μL (IQR) | 555 (385–743) | 320 (280–378) | 790 (640–1000) | — | <0.0001 |
| Nadir CD4+ cell count, median cells/μL (IQR) | 480 (385–613) | 63 (20–139) | 197 (119–340) | — | <0.0001 |
| HIV RNA level, median log(copies/mL) (IQR) | 4.2 (3.6–5.0) | — | — | — | — |
| Duration of cART, median years (IQR) | — | 5.6 (4.4–7.2) | 10.1 (7.7–14.5) | — | 0.0005 |
| Previremic index, median log(copies/mL) (IQR) | — | 4.9 (4.3–6.1) | 5.0 (4.7–5.8) | — | 0.96 |
| Monocyte percentage, % (IQR) | 18.9 (12.2–25.6) | 22.4 (16.2–29.0) | 19.4 (15.4–22.9) | 20.2 (15.1–23.4) | 0.48 |
P values determined by ordinary ANOVA.
IQR, interquartile range.
Levels of IL-37 mRNA in PBMCs from HIV-1-Infected Patients
The steady state, constitutive levels of IL-37 mRNA expression per U6 copy in PBMCs were significantly higher among HIV-1-infected patients compared with noninfected controls. The difference was 2.4-fold (p ≤ 0.01) in cART-naïve subjects, 3.9-fold (p ≤ 0.0001) in IIRs and 4.0-fold (p ≤ 0.0001) in responders when compared with the levels in noninfected controls. However, there were no statistically significant differences in IL-37 mRNA levels between the HIV-1-infected groups (Figure 1). There were no significant differences in the proportions of monocytes between the subgroups (see Table 2). Further, when the levels of IL-37 mRNA were normalized to the monocyte proportion, we observed the same pattern of IL-37 mRNA distribution as in Figure 1, indicating that the increase in PBMC IL-37 mRNA reflected an increase in the amount of IL-37 mRNA per monocyte rather than increased numbers of monocytes (Figure 2).
Figure 1.
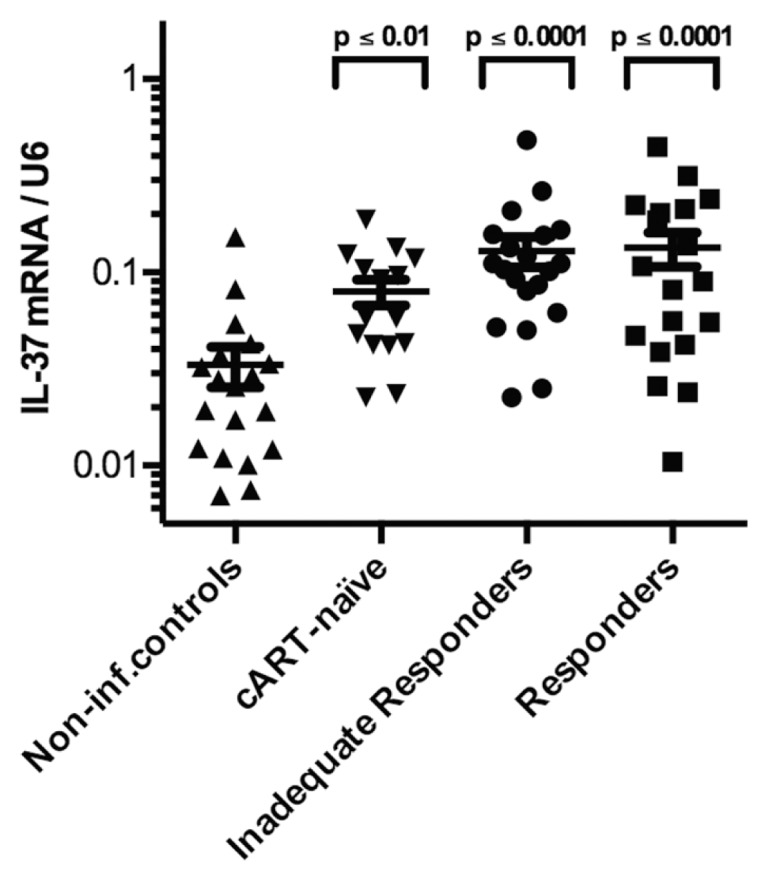
IL-37 mRNA levels in PBMCs of HIV-1-infected patients. Levels of mRNA per copies of U6 for each of the four patient groups (noninfected controls, cART-naïve, IIRs and responders). The P value above each group corresponds to the comparison made to noninfected controls using Holm-Sidak multiple comparisons test. Depicted are group mean with standard error of the mean (SEM).
Figure 2.
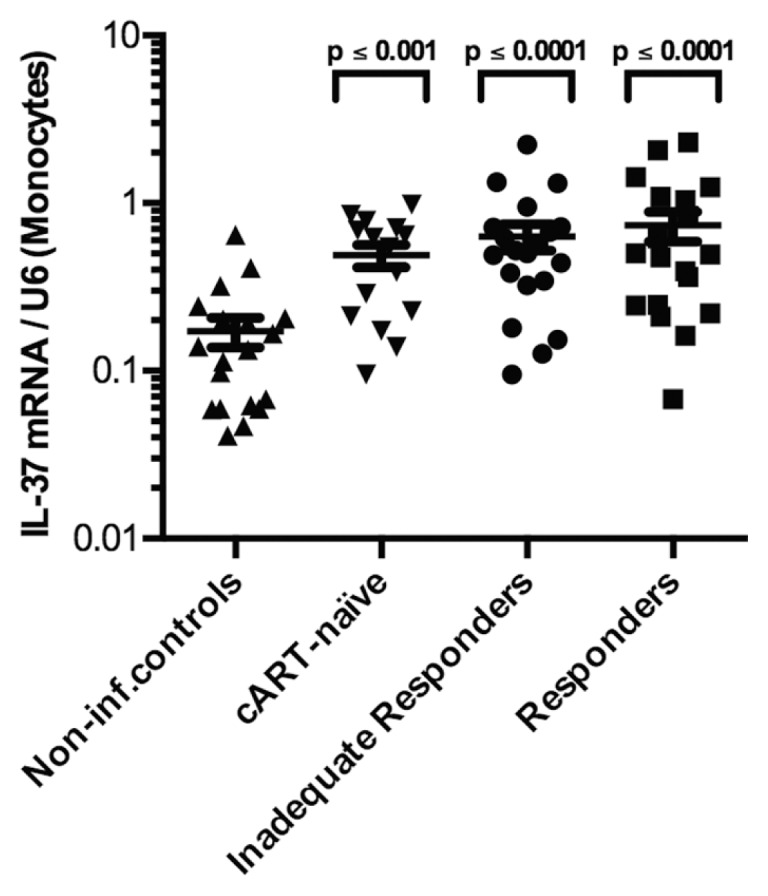
IL-37 mRNA levels in PBMCs of HIV-1-infected patients normalized to monocyte proportions. Levels of mRNA per copies of U6 for each of the four patient groups (noninfected controls, cART-naïve, IIRs and responders), normalized to monocyte percentages in the PBMCs. The P value above each group corresponds to the comparison made to noninfected controls using Holm-Sidak multiple comparisons test. Depicted are group mean with standard error of the mean (SEM).
IL-37 mRNA Response to LPS Stimulation
Next, we examined the induction of IL-37 mRNA in response to LPS stimulation in PBMCs. After stimulation, all HIV-1-infected groups showed significantly higher levels of IL-37 mRNA as compared with noninfected controls, both with and without LPS. Though the relative induction of IL-37 mRNA did not differ between groups, the absolute increase in IL-37 mRNA was higher in the HIV-1-infected groups (Figure 3). Also, the IIRs showed significantly higher levels of IL-37 than the cART-naïve group, when stimulated with LPS (p ≤ 0.01). The IL-37 mRNA induction in response to LPS stimulation compared with media alone was statistically significant in all groups, except for responders (p = 0.07), using Wilcoxon signed rank test (see Figure 3).
Figure 3.

IL-37 mRNA levels with- and without stimulation. Levels of mRNA per copies of U6 both with and without simulation for each of the four patient groups: noninfected (n = 19/20/20 [PBMC/RPMI/LPS]); cART-naïve (n = 15/19/18); IIRs (n = 20/14/14); and responders (n = 19/19/19). The P value corresponds to the comparison between the LPS-stimulated cART-naïve and IIRs. The significance levels indicated by asterisks (**p < 0.01; ***p < 0.001; ****p < 0.0001) correspond to the comparison made to non-infected controls all using Holm-Sidak multiple comparisons test. Depicted are group mean with SEM.
IL-37 mRNA in Relation to sCD14, IL-6 and sCD163
We observed significantly higher levels of circulating sCD14 in IIRs compared with noninfected controls. The other HIV-1-infected groups showed higher levels as well, though not significant (Table 3 and Figure 4A). Moreover, we identified a statistically significant positive correlation between IL-37 mRNA and sCD14 (p = 0.03; Figure 4B). Levels of sCD163 were significantly higher in cART-naïve and IIRs compared with noninfected controls (p ≤ 0.001; p ≤ 0.05), with the highest levels in the cART-naïve group. The cART-naïve group exhibited significantly higher levels than the responders (p ≤ 0.01) (see Table 3 and Figure 5A). As expected, sCD163 correlated with sCD14 (p = 0.03), but there was no correlation between sCD163 and IL-37 mRNA in PBMCs (Figure 5B). We found a 1.4-fold higher level of IL-6 in the plasma of HIV-1-infected individuals compared with noninfected controls, however, using multiple comparisons, no individual significant differences were observed between the groups (see Table 3). In contrast to sCD14, there was no correlation between circulating IL-6 and mRNA levels of IL-37 in PBMCs (p = 0.10).
Table 3.
Laboratory results for all participants.
| Characteristic | cART-naïve (n = 20)a | cART-treated inadequate responders (n = 20)a | cART-treated responders (n = 20)a | Noninfected controls (n = 20)a | Overall P value | |||
|---|---|---|---|---|---|---|---|---|
| IL-37 mRNA:U6 ratio per 106 PBMCs unstimulated (SD) | 0.080 (0.05) | ** | 0.129 (0.10) | **** | 0.134 (0.12) | **** | 0.033 (0.03) | <0.0001 |
| IL-37 mRNA:U6 ratio per 106 PBMCs stimulated with RPMI for 24 h (SD) | 0.559 (0.45) | **** | 0.815 (0.38) | **** | 0.658 (0.34) | **** | 0.140 (0.11) | <0.0001 |
| IL-37 mRNA:U6 ratio per 106 PBMCs stimulated with LPS 24h (SD) | 0.759 (0.62) | ** | 1.372 (0.76) | **** | 1.198 (1.40) | **** | 0.210 (0.13) | <0.0001 |
| sCD14 plasma, 106 pg/mL (SD) | 4.27 (1.05) | ns | 4.91 (1.07) | * | 3.65 (0.80) | ns | 2.62 (0.35) | 0.03 |
| sCD163 plasma μg/mL (SD) | 2.78 (1.30) | *** | 2.25 (0.91) | * | 1.66 (0.55) | ns | 1.52 (0.44) | <0.0001 |
| hsIL-6 plasma, pg/mL (SD) | 3.65 (2.94) | ns | 3.13 (2.79) | ns | 3.43 (2.90) | ns | 2.35 (2.49) | 0.17 |
| CD38 positive CD4+ T cells (SD) | 16.9 (6.5) | ns | 15.3 (9.1) | ns | 10.9 (4.3) | ns | 11.6 (4.9) | 0.01 |
| CD38 positive CD8+ T cells (SD) | 21.3 (10.6) | **** | 7.0 (6.0) | ns | 3.1 (1.9) | ns | 3.7 (2.0) | <0.0001 |
| Total viral reservoir, HIV-DNA per 106 PBMCs (SD) | — | — | 424.9 (623) | — | 582.6 (976) | — | — | 0.95 |
SD, standard deviation.
ANOVA with multiple comparisons analysis. Asterisks (*) correspond to the level of significance in relation to noninfected controls: *: p ≤ 0.05; **: p ≤ 0.01; ***: p ≤ 0.001; ****: p ≤ 0.0001; ns: p > 0.05. Right column corresponds to the overall comparison.
Figure 4.

Levels of sCD14 in plasma and correlation with IL-37 mRNA. (A) Levels of sCD14 in plasma for each of the four patient groups (noninfected controls, cART-naïve, IIRs and responders). The P value above each group corresponds to the comparison made to non-infected controls using Holm-Sidak multiple comparisons test (ns; nonsignificant). Depicted are group mean with SEM. (B) Levels of sCD14 in plasma compared with the levels of IL-37 mRNA in PBMCs using Spearman correlation (r = 0.25).
Figure 5.

Levels of sCD163 in plasma and correlation with IL-37 mRNA. (A) Levels of sCD163 in plasma for each of the four patient groups (noninfected controls, cART-naïve, IIRs and responders). The P value above each group corresponds to the comparison made to noninfected controls using Dunn’s multiple comparisons test (ns, nonsignificant). Depicted are group mean with SEM. (B) Levels of sCD163 in plasma compared with the levels of IL-37 mRNA in PBMCs using Pearson correlation (r = −0.06)
IL-37 and T-Cell Activation
To investigate T-cell activation, we analyzed the expression of CD38 on CD4+ and CD8+ T cells. As shown in Table 3, CD38 expression (both in CD4+ and CD8+ T cells) followed the pattern: cART-naïve > IIRs > noninfected controls > responders. We observed no correlation between IL-37 mRNA levels and CD38 expression when assessed in the total T-cell pool or in individual T-cell subtype (Supplementary Figures S1, S2).
IL-37 mRNA and the Size of the Total Viral Reservoir
We identified a significant positive correlation between the size of the total viral reservoir as measured by HIV-1 DNA and IL-37 mRNA levels in the PBMCs. This association was present for both LPS-induced IL-37 mRNA expression and unstimulated IL-37 mRNA expression (p < 0.0001 and p = 0.04; Figures 6A, B). There was no difference in HIV-1 DNA per 106 PBMCs between IIRs and responders (436.1, [95% CI: 148–724] for IIRs and 591.1, [95% CI: 137–1046] for responders; p = 0.95).
Figure 6.

Correlation between the total viral reservoir and IL-37 mRNA in PBMCs. Levels of IL-37 mRNA compared with the size of the total viral reservoir (HIV-1 DNA copies/106 PBMCs) both with (B) and without (A) LPS stimulation. P values corresponds to the correlation between IL-37 mRNA and HIV-1 DNA using Spearman correlation (r = 0.33 [PBMC]; r = 0.67 [LPS]).
DISCUSSION
To our knowledge, this study is the first to investigate the expression of IL-37 in HIV-1 infection. That the observed increased expression of IL-37 could play a regulatory role in HIV-1 by limiting inflammation is consistent with the role of other antiinflammatory members of the IL-1 family. For instance, elevated levels of the IL-1 receptor antagonist, soluble IL-1 receptor type 2 and IL-18 binding protein are often present in autoimmune and autoinflammatory diseases, and serve as indicators of disease severity.
We know that IL-37 is immunosuppressive and that it imposes a tolerogenic state on dendritic cells (21). However, trying to link the increased expression of IL-37 in HIV-1 as a causal factor for the immunosuppression of the disease does require consideration for the antiinflammatory effect of the cytokine. In a recent study on obesity-related inflammation, a higher expression of IL-37 mRNA in adipose tissue provided better insulin sensitivity, meaning a beneficial role for greater expression of IL-37 (44). Both concepts must be kept in mind, therefore, in the present study in HIV-1-infected patients.
The excessive inflammation in HIV-1 infection has been proposed to be associated with persistent but low level viremia as well as with translocation of microbial products, such as LPS, from a compromised gastrointestinal mucosal barrier (30,45–47). Since LPS stimulation of TLR4 on monocytes is an important signal for IL-37 production, acting by stabilizing IL-37 mRNA (5), we set out to study this. Clearly, LPS stimulation, as well as RPMI alone, upregulated IL-37 mRNA production to higher levels among HIV-1-infected patients compared with noninfected controls. Interestingly the relative inductions in all four groups were comparable, which could indicate that the capability of inducing IL-37 mRNA in HIV-1-infected patients is still intact. Thus the higher absolute levels could be a sign of a decreased capability of producing functional protein and/or a higher proinflammatory response to LPS in the HIV-1-infected patients. In this context, the significant higher level of IL-37 mRNA in the IIRs, but not the responders, compared with cART-naïve could be related to both outlined concepts.
Looking at the effect of plasma viral load on IL-37 mRNA, we did not observe any changes in the levels between the cART-naïve and cART-treated patients in unstimulated PBMCs, indicating that increased viral replication does not affect IL-37 transcription. Instead we propose that increased IL-37 expression may be due to ongoing low-level inflammation affecting the monocytes, and that despite effective cART, this inflammation is still not fully suppressed in terms of upregulated IL-37 transcription. The relevance for HIV-1-infected patients is that increased IL-37 expression may contribute to the immunosuppression of the disease, for example, response to neoantigens, to recall antigens or to the maintenance of cytotoxic T cells targeting HIV-1. Though whether IL-37 is a disease-specific biomarker in HIV-1 that predicts clinical outcomes remains an open question.
Another property of IL-37 that may affect HIV-1-infected subjects is the increase in IL-10 and TGFβ. In mice transgenic for human IL-37, increased production of IL-10 has been reported in models of colitis (16) and endotoxemia (48). In addition, TGFβ induces IL-37 production and is upregulated in HIV (49) and, more specifically, a high production of TGFβ in response to HIV has been observed to inhibit an appropriate IFNγ response (50). The antiinflammatory and immunosuppressive properties of these cytokines therefore seem closely related, although the precise interactions and their possible relations to HIV-1 pathogenesis require further experiments.
In this study, we observed a positive correlation between sCD14 and IL-37 mRNA. As sCD14 has formerly been associated with increased morbidity and mortality in HIV-1 infection (51–53), the correlation between IL-37 mRNA production and sCD14 levels might also indicate an important role of IL-37 in response to the ongoing low-level inflammation on cART, although this correlation was weak and obviously limiting the conclusions to be made here. Signs of a prognostic relevance for this cytokine does require further attention in future studies as a faulty inflammatory regulation has been observed to have prognostic implications with other members of the IL-1 family in different conditions (54–56).
That sCD14 burden is not lower in cART-naïve and cART-treated subjects mimics the pattern of IL-37 mRNA, that is, monocyte activation is not directly affected by plasma viral load. Instead, other mechanisms such as microbial translocation from the intestines contribute to the chronic inflammatory state as observed previously (57).
In line with previous reports, we observed higher levels of sCD163 among HIV-1-infected patients compared with noninfected controls, though not significant for the responders (58). However, sCD163 did not correlate with IL-37 mRNA in this cross-sectional cohort. This finding could reflect sCD163’s linkage as a more macrophage-specific activation marker (59).
Interestingly, we observed a positive correlation between IL-37 mRNA expression levels and the size of the total viral reservoir as measured by HIV-1 DNA in PBMCs. It is still unclear whether the size of the total viral reservoir is a predictor of, or a causal factor in, chronic inflammation. Whereas a recent study reported a consistent association between cell-based measures of viral persistence and immune activation (60), a similar investigation did not find any significant correlation between the levels of HIV-1 DNA in PBMCs and measures of immune activation (61). Whether there is a direct relationship between reservoir size and IL-37 remains elusive because the correlation is based on a limited amount of patients, but we speculate that IL-37 mRNA production and total viral load in this context both might be indicators of immune dysfunction.
This study is limited in the nature of a cross-sectional study preventing any conclusions on causality. Secondly, as we measured all isoforms of IL-37 mRNA collectively in this study, an isoform-specific analysis could have provided further insight into the internal regulation of IL-37. Thirdly, although these initial findings are based primarily on the amount of mRNA, and therefore not the functional cytokine, other studies in human PBMCs and in mice transgenic for human IL-37, have shown that the IL-37 protein is synthesized under inflammatory conditions such as TLR stimulation (12,17). We, and others, have observed considerable variation using the Adipogen IL-37 ELISA for (EDTA) plasma and serum levels in a cohort of healthy human subjects (ages 20 to 72). In addition, we observed an inability of the ELISA to detect IL-37 in LPS-stimulated monocyte supernatants and lysates, with the same samples being positive on a validated Western blotting with the same antibody used in the ELISA. Therefore, we must conclude that levels of mRNA are the most reliable data at present to evaluate the cytokine.
Lastly, we defined IIRs and responders based on the available literature (33,34). However, other studies have used lower cutoffs of CD4+ T-cell counts to define those who have been termed immunological nonresponders (40,62) as a subgroup of the IIRs. This reduces the immunological proximity between the groups and is likely to enhance differences between these subgroups of HIV-1 patients.
In conclusion, steady-state levels of IL-37 mRNA in PBMCs of HIV-1-infected individuals, both with and without LPS stimulation, were significantly higher than in noninfected controls. IL-37 mRNA correlated positively with circulating levels of the monocyte inflammatory marker sCD14, which could suggest a functional link between monocyte activation and IL-37. Moreover, a significant positive correlation between the size of the HIV-1 reservoir and mRNA levels of IL-37 in PBMCs was observed. Collectively, these data add to the understanding of the inflammatory processes observed with chronic HIV-1 infection and its possible relation to IL-37.
Supplemental Data
ACKNOWLEDGMENTS
We would like to acknowledge all the participating patients who made this study possible. Special gratitude is also extended for Lene Svinth Jøhnke and Erik Hagen for their help and guidance in the lab, and to the medical laboratory assistants in Q-lab. The work was supported by The Danish Research Council (FSS) by grant 0602-02300B to M Tolstrup and by the Augustinusfonden, Familien Hede Nielsens Fund and Ulla og Mogens Folmer Andersens Fund to JF Højen.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Cite this article as: Højen JF, et al. (2015) Interleukin-37 expression is increased in chronic HIV-1-infected individuals and is associated with inflammation and the size of the total viral reservoir. Mol. Med. 21:337–45.
REFERENCES
- 1.Arend WP. The balance between IL-1 and IL-1Ra in disease. Cytokine Growth Factor Rev. 2002;13:323–40. doi: 10.1016/s1359-6101(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 2.Novick D, et al. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity. 1999;10:127–36. doi: 10.1016/s1074-7613(00)80013-8. [DOI] [PubMed] [Google Scholar]
- 3.Towne JE, et al. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J Biol Chem. 2011;286:42594–602. doi: 10.1074/jbc.M111.267922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dinarello CA, Bufler P. Interleukin-37. Semin Immunol. 2013;25:466–8. doi: 10.1016/j.smim.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Bufler P, Gamboni-Robertson F, Azam T, Kim SH, Dinarello CA. Interleukin-1 homologues IL-1F7b and IL-18 contain functional mRNA instability elements within the coding region responsive to lipopolysaccharide. Biochem J. 2004;381:503–10. doi: 10.1042/BJ20040217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Busfield SJ, et al. Identification and gene organization of three novel members of the IL-1 family on human chromosome 2. Genomics. 2000;66:213–6. doi: 10.1006/geno.2000.6184. [DOI] [PubMed] [Google Scholar]
- 7.Kumar S, et al. Interleukin-1F7B (IL-1H4/IL-1F7) is processed by caspase-1 and mature IL-1F7B binds to the IL-18 receptor but does not induce IFN-gamma production. Cytokine. 2002;18:61–71. doi: 10.1006/cyto.2002.0873. [DOI] [PubMed] [Google Scholar]
- 8.Pan G, et al. IL-1H, an interleukin 1-related protein that binds IL-18 receptor/IL-1Rrp. Cytokine. 2001;13:1–7. doi: 10.1006/cyto.2000.0799. [DOI] [PubMed] [Google Scholar]
- 9.Smith DE, et al. Four new members expand the interleukin-1 superfamily. J Biol Chem. 2000;275:1169–75. doi: 10.1074/jbc.275.2.1169. [DOI] [PubMed] [Google Scholar]
- 10.Taylor SL, Renshaw BR, Garka KE, Smith DE, Sims JE. Genomic organization of the interleukin-1 locus. Genomics. 2002;79:726–33. doi: 10.1006/geno.2002.6752. [DOI] [PubMed] [Google Scholar]
- 11.Bufler P, et al. A complex of the IL-1 homologue IL-1F7b and IL-18-binding protein reduces IL-18 activity. Proc Natl Acad Sci U S A. 2002;99:13723–8. doi: 10.1073/pnas.212519099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bulau AM, et al. Role of caspase-1 in nuclear translocation of IL-37, release of the cytokine, and IL-37 inhibition of innate immune responses. Proc Natl Acad Sci U S A. 2014;111:2650–5. doi: 10.1073/pnas.1324140111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma S, et al. The IL-1 family member 7b translocates to the nucleus and down-regulates proinflammatory cytokines. J Immunol. 2008;180:5477–82. doi: 10.4049/jimmunol.180.8.5477. [DOI] [PubMed] [Google Scholar]
- 14.Luo Y, et al. IL-37, an inhibitor of innate and adaptive immunity, is elevated in the blood of melanoma patients [abstract] Pigment Cell Melanoma Res. 2013;26:951. [Google Scholar]
- 15.Song L, et al. Glucocorticoid regulates interleukin-37 in systemic lupus erythematosus. J Clin Immunol. 2012;33:111–7. doi: 10.1007/s10875-012-9791-z. [DOI] [PubMed] [Google Scholar]
- 16.McNamee EN, et al. Interleukin 37 expression protects mice from colitis. Proc Natl Acad Sci U S A. 2011;108:16711–6. doi: 10.1073/pnas.1111982108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nold MF, et al. IL-37 is a fundamental inhibitor of innate immunity. Nat Immunol. 2010;11:1014–22. doi: 10.1038/ni.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujita H, Inoue Y, Seto K, Komitsu N, Aihara M. Interleukin-37 is elevated in subjects with atopic dermatitis. J Dermatol Sci. 2012;69:173–5. doi: 10.1016/j.jdermsci.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 19.Moschen AR, et al. Adipose and liver expression of interleukin (IL)-1 family members in morbid obesity and effects of weight loss. Mol Med. 2011;17:840–5. doi: 10.2119/molmed.2010.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dinarello CA, Bufler P. Interleukin-37. Semin Immunol. 2013;25:466–8. doi: 10.1016/j.smim.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 21.Luo Y, et al. IL-37 suppresses contact hypersensitivity by inducing tolerogenic dendritic cells. Cytokine. 2013;63:283. [Google Scholar]
- 22.Hunt PW. HIV and inflammation: mechanisms and consequences. Curr HIV/AIDS Rep. 2012;9:139–47. doi: 10.1007/s11904-012-0118-8. [DOI] [PubMed] [Google Scholar]
- 23.Neuhaus J, et al. Markers of inflammation, coagulation, and renal function are elevated in adults with HIV infection. J Infect Dis. 2010;201:1788–95. doi: 10.1086/652749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuller LH, et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5:e203. doi: 10.1371/journal.pmed.0050203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Currier JS, et al. Epidemiological evidence for cardiovascular disease in HIV-infected patients and relationship to highly active antiretroviral therapy. Circulation. 2008;118:e29–35. doi: 10.1161/CIRCULATIONAHA.107.189624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet. 2007;370:59–67. doi: 10.1016/S0140-6736(07)61050-2. [DOI] [PubMed] [Google Scholar]
- 27.Marin B, et al. Non-AIDS-defining deaths and immunodeficiency in the era of combination antiretroviral therapy. AIDS. 2009;23:1743–53. doi: 10.1097/QAD.0b013e32832e9b78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deeks SG, Phillips AN. HIV infection, antiretroviral treatment, ageing, and non-AIDS related morbidity. BMJ. 2009;338:a3172. doi: 10.1136/bmj.a3172. [DOI] [PubMed] [Google Scholar]
- 29.Sogaard OS, et al. Hospitalization for pneumonia among individuals with and without HIV infection, 1995–2007: a Danish population-based, nationwide cohort study. Clin Infect Dis. 2008;47:1345–53. doi: 10.1086/592692. [DOI] [PubMed] [Google Scholar]
- 30.Brenchley JM, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–71. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 31.Mavigner M, et al. HIV-1 residual viremia correlates with persistent T-cell activation in poor immunological responders to combination anti-retroviral therapy. PloS One. 2009;4:e7658. doi: 10.1371/journal.pone.0007658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostrowski SR, Katzenstein TL, Pedersen BK, Gerstoft J, Ullum H. Residual viraemia in HIV-1-infected patients with plasma viral load ≤ 20 copies/ml is associated with increased blood levels of soluble immune activation markers. Scand J Immunol. 2008;68:652–60. doi: 10.1111/j.1365-3083.2008.02184.x. [DOI] [PubMed] [Google Scholar]
- 33.Gaardbo JC, Hartling HJ, Gerstoft J, Nielsen SD. Incomplete immune recovery in HIV infection: mechanisms, relevance for clinical care, and possible solutions. Clin Dev Immunol. 2012;2012:670957. doi: 10.1155/2012/670957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaufmann GR, et al. Characteristics, determinants, and clinical relevance of CD4 T cell recovery to < 500 cells/microL in HIV type 1-infected individuals receiving potent antiretroviral therapy. Clin Infect Dis. 2005;41:361–72. doi: 10.1086/431484. [DOI] [PubMed] [Google Scholar]
- 35.Piketty C, et al. Discrepant responses to triple combination antiretroviral therapy in advanced HIV disease. AIDS. 1998;12:745–50. doi: 10.1097/00002030-199807000-00011. [DOI] [PubMed] [Google Scholar]
- 36.Piconi S, et al. Immune activation, apoptosis, and Treg activity are associated with persistently reduced CD4+ T-cell counts during anti-retroviral therapy. AIDS. 2010;24:1991–2000. doi: 10.1097/QAD.0b013e32833c93ce. [DOI] [PubMed] [Google Scholar]
- 37.Baker JV, et al. Poor initial CD4+ recovery with antiretroviral therapy prolongs immune depletion and increases risk for AIDS and non-AIDS diseases. J Acquir Immune Defic Syndr. 2008;48:541–6. doi: 10.1097/QAI.0b013e31817bebb3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gutierrez F, et al. Patients’ characteristics and clinical implications of suboptimal CD4 T-cell gains after 1 year of successful antiretroviral therapy. Curr HIV Res. 2008;6:100–7. doi: 10.2174/157016208783885038. [DOI] [PubMed] [Google Scholar]
- 39.Nakanjako D, et al. High T-cell immune activation and immune exhaustion among individuals with suboptimal CD4 recovery after 4 years of antiretroviral therapy in an African cohort. BMC Infect Dis. 2011;11:43. doi: 10.1186/1471-2334-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lederman MM, et al. Immunologic failure despite suppressive antiretroviral therapy is related to activation and turnover of memory CD4 cells. J Infect Dis. 2011;204:1217–26. doi: 10.1093/infdis/jir507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benecke A, Gale M, Jr, Katze MG. Dynamics of innate immunity are key to chronic immune activation in AIDS. Curr Opin HIV AIDS. 2012;7:79–85. doi: 10.1097/COH.0b013e32834dde31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moller HJ, Hald K, Moestrup SK. Characterization of an enzyme-linked immunosorbent assay for soluble CD163. Scand J Clin Lab Invest. 2002;62:293–9. doi: 10.1080/003655102760145852. [DOI] [PubMed] [Google Scholar]
- 43.Winckelmann AA, et al. Administration of a toll-like receptor 9 agonist decreases the proviral reservoir in virologically suppressed HIV-infected patients. PloS One. 2013;8:e62074. doi: 10.1371/journal.pone.0062074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ballak DB, et al. IL-37 protects against obesity-induced inflammation and insulin resistance. Nature Commun. 2014;5:4711. doi: 10.1038/ncomms5711. [DOI] [PubMed] [Google Scholar]
- 45.Llibre JM, et al. Treatment intensification with raltegravir in subjects with sustained HIV-1 viraemia suppression: a randomized 48-week study. Antivir Ther. 2012;17:355–64. doi: 10.3851/IMP1917. [DOI] [PubMed] [Google Scholar]
- 46.Brenchley JM, Price DA, Douek DC. HIV disease: fallout from a mucosal catastrophe? Nat Immunol. 2006;7:235–9. doi: 10.1038/ni1316. [DOI] [PubMed] [Google Scholar]
- 47.Marchetti G, Tincati C, Silvestri G. Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin Microbiol Rev. 2013;26:2–18. doi: 10.1128/CMR.00050-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van de Veerdonk FL, et al. Protective host defense against disseminated candidiasis is impaired in mice expressing human interleukin-37. Front Microbiol. 2014;5:762. doi: 10.3389/fmicb.2014.00762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wiercinska-Drapalo A, Flisiak R, Jaroszewicz J, Prokopowicz D. Increased plasma transforming growth factor-beta1 is associated with disease progression in HIV-1-infected patients. Viral Immunol. 2004;17:109–13. doi: 10.1089/088282404322875502. [DOI] [PubMed] [Google Scholar]
- 50.Garba ML, Pilcher CD, Bingham AL, Eron J, Frelinger JA. HIV antigens can induce TGF-beta(1)-producing immunoregulatory CD8+ T cells. J Immunol. 2002;168:2247–54. doi: 10.4049/jimmunol.168.5.2247. [DOI] [PubMed] [Google Scholar]
- 51.Kelesidis T, Kendall MA, Yang OO, Hodis HN, Currier JS. Biomarkers of microbial translocation and macrophage activation: association with progression of subclinical atherosclerosis in HIV-1 infection. J Infect Dis. 2012;206:1558–67. doi: 10.1093/infdis/jis545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sandler NG, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis. 2011;203:780–90. doi: 10.1093/infdis/jiq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lien E, et al. Elevated levels of serum-soluble CD14 in human immunodeficiency virus type 1 (HIV-1) infection: correlation to disease progression and clinical events. Blood. 1998;92:2084–92. [PubMed] [Google Scholar]
- 54.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 55.Dinarello CA. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 2010;29:317–29. doi: 10.1007/s10555-010-9229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zeng Q, et al. Therapeutic potential of interleukin-37 for suppression of aortic valve inflammatory and osteogenic responses. Circulation. 2012;126:A11803. [Google Scholar]
- 57.Cassol E, et al. Persistent microbial translocation and immune activation in HIV-1-infected South Africans receiving combination antiretroviral therapy. J Infect Dis. 2010;202:723–33. doi: 10.1086/655229. [DOI] [PubMed] [Google Scholar]
- 58.Burdo TH, et al. Soluble CD163 made by monocyte/macrophages is a novel marker of HIV activity in early and chronic infection prior to and after anti-retroviral therapy. J Infect Dis. 2011;204:154–63. doi: 10.1093/infdis/jir214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moller HJ. Soluble CD163. Scand J Clin Lab Invest. 2012;72:1–13. doi: 10.3109/00365513.2011.626868. [DOI] [PubMed] [Google Scholar]
- 60.Hatano H, et al. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J Infect Dis. 2013;208:50–6. doi: 10.1093/infdis/jis630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Poizot-Martin I, et al. Lack of correlation between the size of HIV proviral DNA reservoir and the level of immune activation in HIV-infected patients with a sustained undetectable HIV viral load for 10 years. J Clin Virol. 2013;57:351–5. doi: 10.1016/j.jcv.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 62.Marchetti G, et al. Skewed T-cell maturation and function in HIV-infected patients failing CD4+ recovery upon long-term virologically suppressive HAART. AIDS. 2010;24:1455–60. doi: 10.1097/QAD.0b013e328339cf40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.