

Abstract
Purpose
Two phase II studies assessed the efficacy of vismodegib, a sonic hedgehog (SHH) pathway inhibitor that binds smoothened (SMO), in pediatric and adult recurrent medulloblastoma (MB).
Patients and Methods
Adult patients enrolled onto PBTC-025B and pediatric patients enrolled onto PBTC-032 were treated with vismodegib (150 to 300 mg/d). Protocol-defined response, which had to be sustained for 8 weeks, was confirmed by central neuroimaging review. Molecular tests to identify patterns of response and insensitivity were performed when tissue was available.
Results
A total of 31 patients were enrolled onto PBTC-025B, and 12 were enrolled onto PBTC-032. Three patients in PBTC-025B and one in PBTC-032, all with SHH-subgroup MB (SHH-MB), exhibited protocol-defined responses. Progression-free survival (PFS) was longer in those with SHH-MB than in those with non-SHH–MB, and prolonged disease stabilization occurred in 41% of patient cases of SHH-MB. Among those with SHH-MB, loss of heterozygosity of PTCH1 was associated with prolonged PFS, and diffuse staining of P53 was associated with reduced PFS. Whole-exome sequencing identified mutations in SHH genes downstream from SMO in four of four tissue samples from nonresponders and upstream of SMO in two of four patients with favorable responses.
Conclusion
Vismodegib exhibits activity against adult recurrent SHH-MB but not against recurrent non-SHH–MB. Inadequate accrual of pediatric patients precluded conclusions in this population. Molecular analyses support the hypothesis that SMO inhibitor activity depends on the genomic aberrations within the tumor. Such inhibitors should be advanced in SHH-MB studies; however, molecular and genomic work remains imperative to identify target populations that will truly benefit.
INTRODUCTION
Medulloblastoma (MB) is a heterogeneous disease composed of four molecular subgroups: WNT, sonic hedgehog (SHH), and groups three and four.1 SHH-subgroup MB (SHH-MB) accounts for approximately 30% of MBs and most commonly affects children age < 5 years and adolescents age > 16 years through adulthood.2,3 With current therapy, 5-year overall survival (OS) of this subgroup is approximately 70%,4 but survival is frequently accompanied by severe morbidity.5,6 For patients with recurrent disease, survival is dismal. Therefore, improved therapy is needed not only to augment survival but also to prevent recurrence and decrease the morbidity associated with current therapy.
Smoothened (SMO) functions as a key component of the SHH pathway by regulating suppressor of fused (SUFU).7 SMO inhibitors block SUFU activation, thereby preventing translocation of GLI proteins into the nucleus (Fig 1). SMO inhibitors have shown efficacy in the treatment and prevention of basal cell carcinoma,8,9 and responses have been reported in recurrent MB.9–11 Preclinical research in genetically engineered mouse models has predicted that responses will occur in SHH-MB driven by mutations upstream of SMO and will be ineffective in tumors driven by mutations in SHH pathway genes downstream of SMO.12–14 Individual clinical responses have been consistent with these predictions10,15; however, analysis across a spectrum of SHH-MBs has not occured. Recent findings from genome sequencing of MB have revealed considerable heterogeneity among SHH-MBs; some have aberrations upstream of (eg, PTCH1) or within SMO, others have aberrations downstream (eg, SUFU, GLI2), and still others have aberrations in genes not known to directly influence SMO (eg, TP53, MYCN).16–19 This suggests that response to SMO inhibitors will be variable.
Fig 1.
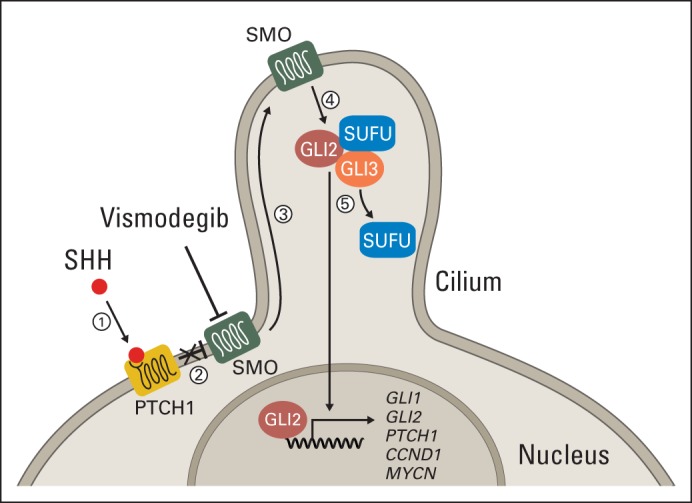
Illustration of sonic hedgehog (SHH) signaling pathway. (1) SHH ligand binds to PTCH1 transmembrane protein. (2) Binding of SHH to PTCH1 relieves inhibition of smoothened (SMO). (3) Activated SMO localizes to cilium. (4) SMO releases suppressor of fused (SUFU) inhibition of GLI proteins. (5) Activated GLI proteins translocate to nucleus and activate transcription of SHH target genes (ie, GLI1, GLI2, PTCH1, and MYCN). In SHH-subgroup medulloblastoma, disruptions to SHH pathway occur through mutation of PTCH1, SMO, or SUFU and/or amplification of GLI2 or MYCN. Vismodegib inhibits SMO.
Here we report the results of two prospective phase II Pediatric Brain Tumor Consortium (PBTC) studies, PBTC-025B and PBTC-032, which assessed the efficacy of the SMO inhibitor vismodegib in adult and pediatric patients with recurrent MB, respectively. In addition, we evaluated genomic correlates of clinical responses to vismodegib, and where possible, we described the molecular alterations in tumors that acquire resistance.
PATIENTS AND METHODS
Patients and Treatment
Patient eligibility criteria for both studies required the presence of measureable disease and a histologic diagnosis of MB that was recurrent, progressive, or refractory to standard therapy. Patients were required to have recovered from prior therapy, have stable neurologic deficits, and meet certain organ function requirements, as previously described.11 The institutional review board of each PBTC institution approved the study protocol. Patients, parents, or guardians provided written informed consent for participation.
Adults (age ≥ 22 years) with an Eastern Cooperative Oncology Group performance score of 0 to 2 were enrolled onto PBTC-025B. Measureable disease was defined as ≥ 10 mm in one dimension on imaging. Real-time prescreening immunohistochemistry (IHC) was used to group patients into strata A (non-SHH–MB), B (SHH-MB), or C (indeterminate/unknown), as previously described.11,20 Patients received oral vismodegib 150 mg once per day.
Pediatric patients (age 3 to 21 years with body-surface area of 0.67 to 2.5 m2) with a Karnofsky or Lansky score ≥ 50 were enrolled onto PBTC-032. Per the study design, patients treated at the recommended phase II dose of vismodegib during the phase I trial (PBTC-025) were counted toward the phase II accrual. Real-time prescreening IHC was used to stratify patients; however, only those in stratum B (SHH-MB) were enrolled, because stratum A (non-SHH–MB) never opened to accrual based on the results from PBTC-025 (Fig 2).
Fig 2.

Trial schematics and distribution of patients from PBTC-025 (phase I precursor study), PBTC-032 (children), and PBTC-025B (adults). In PBTC-032, strata A and C were closed to accrual before study opening, because no objective responses were seen in 13 patients with non–sonic hedgehog (SHH) medulloblastoma (MB) treated at recommended phase II dose (RP2D) of vismodegib during phase I study PBTC-025. (*) Phase I PBTC-025 patients treated at the RP2D counted toward the phase II (PBTC-032) accrual. Bottom panel shows distribution of available formalin-fixed, paraffin-embedded (FFPE) tumor samples for molecular analysis. BSA, body surface area.
Toxicity Criteria
Adverse events were graded per the Common Terminology Criteria for Adverse Events (version 4.0). Adverse events judged at least possibly attributable to the drug were recorded as toxicities.
Response Criteria
The protocol defined objective response as a complete (CR) or partial (PR) disease response that was maintained for at least 8 weeks. All reported responses by treating institutions were reviewed centrally, even if not sustained. For PBTC-025B, CR was defined as the disappearance of all target lesions, PR as a 30% reduction in the sum of the longest diameters of target lesions, progressive disease (PD) as at least a 20% increase in the sum of the longest diameters of target lesions, and stable disease (SD) as findings that did not meet the criteria for PR or PD. For PBTC-032, similar criteria were used, except that PR was defined as a 50% reduction in tumor area and PD as more than a 25% increase in tumor area.
Pharmacokinetic Analysis
Day 21 (± 7 days) bound and unbound vismodegib plasma concentrations were measured in consenting patients, as previously described.11
Molecular and Genomic Analyses
Fluorescence in situ hybridization, as previously described,20,21 was performed on all available formalin-fixed, paraffin-embedded (FFPE) tumor material to identify copy number aberrations in PTCH1, PTEN, GLI2, chromosome 17p, and MYCN. IHC for P53 was undertaken using DO-7, an anti-P53 antibody (Zeta Corporation, Sierra Madre, CA), and tumors were classified as having no staining, normal staining, or strong diffuse staining (DS). These studies were performed retrospectively by a neuropathologist (B.A.O.) blinded to the trial results.
Exome sequencing (Nextera; Illumina, San Diego, CA) was performed retrospectively on eight frozen SHH-MBs with matched germline DNA. Two MB specimens were procured during the original diagnostic surgery (patients 032-5 and 032-9); three were procured at relapse but before therapy (patients 025B-7, 025B-11, and 025B-14); three were procured after vismodegib therapy was stopped because of PD (patients 025B-1, 032-1, and 032-3; Fig 3). Paired-end sequencing reads were mapped to the human reference genome (National Center for Biotechnology Information GRCh37). Somatic and germline single-nucleotide variations/indels were called as previously described16 (Data Supplement).
Fig 3.

Clinical and molecular characteristics of patients with sonic hedgehog (SHH) –subgroup medulloblastoma (SHH-MB) enrolled onto PBTC-025B or PBTC-032. Columns arranged in descending order from longest to shortest time to disease progression (PD). Lower panels show mutations in genes (previously described as mutated in SHH-MB) in eight matched tumor and germline biospecimens. Significance assessed using Kolmogorov-Smirnov tests. FISH, fluorescence in situ hybridization; IHC, immunohistochemistry.
Statistical Analysis
The primary end point of both trials was sustained objective response. In both studies, we used identical Simon's two-stage minimax designs in each stratum with 10% type I error and 90% power to distinguish between a response rate of 5% versus 25%. The total sample size was 20 patients per stratum. Interim analysis was performed after the 13th patient, and one sustained response was adequate to expand accrual. At least three sustained responses in 20 patients were required to declare activity of vismodegib promising.
Progression-free survival (PFS) was defined as the time from the start of protocol treatment to PD or death resulting from any cause. All patients had PD or had died by the time of analysis, so no censoring was required. Kaplan-Meier estimates of PFS were plotted by pathology, genetic markers, and trial (Appendix Figs A1 and A2, online only). PFS curves were compared via log-rank tests (Mantel-Haenszel) for discrete covariates or Cox proportional hazards models for continuous covariates. For the outcome comparisons of genetic markers, where the proportion hazards assumption was clearly violated based on crossing of the survival curves, we used the Kolmogorov-Smirnov tests, which have higher power in smaller sample cases compared with weighted log-rank tests.22 P values were not corrected for multiplicity.
RESULTS
A total of 31 patients (age range, 22.4 to 51.9 years) were enrolled onto PBTC-025B (stratum A [n = 9], stratum B [n = 20], and stratum C [n = 2]; Appendix Table A1, online only). One stratum C patient (patient 025B-14) was retrospectively reassigned to stratum B, and patient 025B-32 was reassigned to stratum A after tumor material became available. Another stratum B patient (patient 025B-21) was declared ineligible after an audit because of lack of measureable disease and was not included in this analysis.
Thirteen patients with non-SHH–MB and three with SHH-MB from PBTC-025 treated at the recommended phase II dose were counted toward the accrual for PBTC-032.11 Because none of the 13 patients with non-SHH–MB experienced an objective response, enrollment onto PBTC-032 was limited to those with SHH-MB. Forty-two patients were prescreened for SHH pathway activation; 11 had SHH-MB, but two did not enroll, as a result of declining condition. Consequently, a total of 12 patients (age range, 3.9 to 20.0 years) were assigned to PBTC-032 stratum B, including the three patients from PBTC-025 (Appendix Table A2, online only).
Toxicities and adverse events for all patients revealed a low toxicity profile (Appendix Table A3, online only). No patient withdrew from therapy because of unacceptable toxicity. No drug-related bone or dental toxicity was observed in the pediatric population.
Three adults (patients 025B-4, 025B-7, and 025B-8) and one pediatric patient (patient 032-3) experienced sustained response; all had SHH-MB. No responses were observed among patients with non-SHH–MB (Appendix Table A4, online only). Radiographic responses were observed in five adults and three pediatric patients, although only four of these were maintained (Fig 4). The degree of radiographic response did not correlate with the duration of therapy (Fig 5).
Fig 4.

Magnetic resonance images showing responses in five patients with recurrent sonic hedgehog–subgroup medulloblastoma (MB) treated with vismodegib. Images were obtained at start of therapy and at 2, 4, and 6 months after. Gold arrows indicate recurrent lesions. After initial response, MB recurs locally.
Fig 5.

Time to disease progression. (A) Only patients with sonic hedgehog (SHH) - subgroup medulloblastoma (SHH-MB) enrolled onto PBTC-025B (blue shades) or PBTC-032 (light blue shades) remained progression free until second evaluation period. (B) PTCH1 loss of heterozygosity was associated with increased duration of therapy. Patients with P53 diffuse staining were all nonresponders. In all of those patients, disease progressed at or before first evaluation. (★) Patients in whom radiographic response was observed. (†) Patients in whom radiographic response was sustained beyond 8 weeks. SHH-MB patient numbers correlate with those in Figure 3. Adult non-SHH MB (gray) from PBTC-025B are numbered from 025B-22 to 025B-32. Child non-SHH patients from PBTC-025/PBTC-032 (gray) are numbered 032-13 to 032-26.
PFS for adults with SHH-MB was longer than that of adults with non-SHH–MB (P = .0279; Fig 6A). The difference in PFS between pediatric patients with SHH-MB and with non-SHH–MB was not significant (P = .2246; Fig 6B). Among all patients with SHH-MB, adults displayed a longer PFS than pediatric patients (P = .0210; Appendix Fig A1). A total of 13 (41%) of 32 patients with SHH-MB (and none of those with non-SHH–MB) reached the second evaluation period (Fig 5).
Fig 6.

Progression-free survival (PFS) of patients enrolled onto (A) PBTC-025B or (B) PBTC-032 and PBTC-025. (A) For PBTC025B, patients in stratum B (blue line) with sonic hedgehog (SHH) –activated tumors had significantly longer PFS than did those in stratum A (gray line) or C (gold line). (B) There was no significant difference between PFS of patients in PBTC032 or PBTC025 who had SHH-subgroup medulloblastoma (SHH-MB; blue line) and those with non-SHH–MB treated at recommended phase II dose of vismodegib (gold line).
Pharmacokinetic analysis of 39 patients (28 adults; 11 pediatric patients) demonstrated that the median bound or unbound vismodegib plasma concentration did not differ significantly between the trial cohorts, patients with prolonged PFS, or patients with objective responses (Appendix Table A5, online only). With a median CSF penetration of 0.53, when expressed as an area under the curve ratio of CSF vismodegib to that of unbound drug in plasma, predicted CSF vismodegib concentrations met or exceeded the free IC95 for GLI1 inhibition (0.042 to 0.068 μmol/L)23 for the majority (35 of 39) of patients.
On retrospective review, FFPE tissue was available from 35 patients (26 with SHH-MB and nine with non-SHH–MB; Fig 2). For the SHH subgroup, PTCH1 loss was evident in 10 of 26 SHH-MBs and associated with a longer PFS (P = .037; Figs 3 and 5; Appendix Fig A2). Conversely, P53 DS was associated with a reduced PFS (P = .053; Figs 3 and 5; Appendix Fig A2). No association was detected between PTEN/10q loss, 17p loss, or absence of P53 staining and treatment outcome. Although neither GLI2 amplification, MYCN amplification, nor MYCN gain were associated with a worse outcome, they overlapped with P53 DS (Appendix Fig A2). Thus, a larger cohort would be needed to determine whether these aberrations predict vismodegib insensitivity.
For the non-SHH subgroup, no response or association was detected between any molecular feature and treatment outcome. PTCH1/9q loss was not found in any of the non-SHH tumors (Appendix Table A6, online only), which is consistent with this molecular feature being enriched in the SHH subgroup.24
Exome sequencing of eight matched SHH-MB and germline samples identified mutations in SHH genes upstream of SMO in two (patients 032-1 and 032-3) of four responders (defined here as PFS until second evaluation, any PR, or CR). Three PTCH1 mutations were identified: two somatic frame-shift mutations and one concomitant germline mutation in a young patient (patient 032-1; age 2.6 years at diagnosis) with nevoid basal cell carcinoma syndrome. In both tumors, the other PTCH1/9q allele was lost (Fig 3).
Downstream activating aberrations in the SHH pathway were identified in four of four nonresponders (patients 025B-11, 025B-14, 032-5, and 032-9). All tumor samples were collected before the initiation of therapy. Three harbored somatic mutations in SUFU, with two occurring in adults (patients 025B-11 and 025B-14) and one in a young patient (patient 032-5; age 2.4 years at diagnosis) that was also accompanied by a germline mutation. Both adult samples also harbored mutations in the PI3K pathway genes (PIK3CA and PTEN, respectively), and one had a TP53 mutation. The latter tumor demonstrated P53 DS, supporting the finding that this IHC pattern correlates with the presence of a TP53 mutation.25,26 The fourth nonresponder (patient 032-9) had GLI2 amplification, which has conferred resistance to SMO inhibitors in mouse MB.14
No SMO mutations were identified in the three samples, sequenced after PD during therapy, (patients 025B-1, 032-1, and 032-3; Fig 3). These patients showed an initial favorable response to therapy, suggesting that mechanisms of resistance exist outside of the inhibitor-binding site. One patient (patient 025B-1) remained progression free during therapy for 16 months before experiencing recurrence in the lung. This metastatic lesion retained the IHC SHH phenotype and harbored mutations in BCOR, BRPF1, NRAS, and SYNE1 (Fig 3), all genes previously described as mutated in SHH-MB.17–19 The other two patients had responses but then experienced PD after 3 to 4 months of therapy. One patient (patient 032-3) had a PTEN mutation (Fig 3), which may have contributed to resistance27,28; the other (patient 032-1) had a mutation in the 3′UTR region of GNAS (Fig 3), a gene that has been implicated in a subset of aggressive SHH-MB.29 However, without molecular analysis of the tumor at diagnosis, we cannot determine if these mutations were somatically acquired during therapy or if they contributed to acquired resistance to vismodegib.
We identified numerous mutations that have previously been reported as recurrent in SHH-MB in the eight matched tumor and germline samples that underwent to exome sequencing (Fig 3; Data Supplement).16–19 The significance of these mutations remains unknown, although their association with chromatin and epigenetic regulation is noteworthy.
DISCUSSION
Vismodegib exhibited activity against adult recurrent or refractory SHH-MB and no activity against recurrent non-SHH–MB. Three of 20 patients with SHH-MB showed a sustained response, indicating that vismodegib is active in this population. For pediatric patients with recurrent or refractory SHH-MB, poor accrual limited conclusions. However, one sustained PR and two transient responses were observed, suggesting that vismodegib may have activity in this population. In addition, treatment with vismodegib resulted in a prolonged PFS in more patients with SHH-MB than just those in whom a disease response was observed, suggesting that activity is not limited to objective response.
Interestingly, favorable outcomes (radiographic responses and/or prolonged PFS) among SHH-MB were variable. Although this variability may have resulted from modest blood-brain barrier penetration of vismodegib, our pharmacokinetic data suggested adequate CSF concentrations. Hence, we hypothesized that this variability may have been attributable to genomic heterogeneity within the SHH subgroup. Our analyses revealed some key findings.
First, the position of the genomic aberration relative to SMO is predictive of SMO inhibitor activity. As would be predicted by the upstream position of PTCH1, aberrations in PTCH1 accompanied favorable outcomes. Somatic loss of heterozygosity (LOH) of PTCH1, which frequently but not exclusively accompanies PTCH1 mutations,16–19 was associated with a prolonged PFS. Moreover, loss-of-function PTCH1 mutations were found only in responders. In contrast, molecular aberrations of GLI2 or SUFU, which are downstream of SMO, were exclusively present in nonresponders.
Second, strong P53 DS in SHH-MB, which is commonly associated with dominant-negative, DNA–binding domain mutations in TP53,26 was associated with a lack of response to SMO inhibition. The reason for this is unclear, because P53 does not directly interact with SMO; however, mutations lead to chromothripsis, which may amplify SHH signaling oncogenes.30 In keeping with these observations, we found amplifications and gains of MYCN and GLI2 in four of six tumors that had P53 DS. The loss of P53 staining and hemizygous loss of 17p did not confer a similar phenotype and should not be considered markers of SMO inhibitor insensitivity. How TP53 mutations in residues outside of the DNA binding domain, which do not confer a strong DS pattern, behave relative to SMO inhibition needs to be further investigated.
Third, complete molecular profiling of all SHH-MBs is needed to identify the target population that will benefit from vismodegib. Genome sequencing and copy number analysis should be used to identify SHH pathway mutations and potential cooperating mutations; it is critical to discriminate SHH-MBs with mutations downstream of SMO that do not respond to SMO inhibition, so these can be stratified for emerging therapies. Unfortunately, clinical challenges will arise when tissue is not available or poorly preserved. As shown here, fluorescence in situ hybridization analysis for PTCH1, MYCN, and GLI2 copy number variations and P53 IHC can be performed on FFPE material. Although this approach can help predict tumor response, it is not sufficient to determine whether SMO inhibitors should be used until more robust information is made available. Response in tumors with PTCH1 LOH needs to be correlated with that in tumors with PTCH1 mutations, because LOH does not always accompany PTCH1 mutations. Methods to reliably identify mutations in genes that could confer a priori resistance, such as SMO or SUFU, from FFPE material are needed. Ways to identify TP53 mutations, in addition to those with a strong DS pattern, and their association with response will be helpful. Also, the cooperating events found in MYCN-amplified tumors should be explored before insensitivity is presumed.
Fourth, strategies to avoid or overcome resistance mechanisms are now needed. In this study, all responses to vismodegib were transient, most likely as a consequence of acquired resistance. Whereas mutations in SMO previously implicated in acquired resistance were not identified, we did uncover mutations that putatively give rise to resistance.15,31 Incorporation of SMO inhibitors into rational drug combinations aimed at preventing resistance should be considered. Aberrations in PI3K signaling genes are frequent in SHH-MB,16,18,24 and studies in preclinical models have suggested that inhibiting the PI3K pathway together with SMO may decrease resistance and recurrence.27,28 Similarly, targeting GNAS may improve the sensitivity to SMO inhibition.29 Also, preclinical modeling that pairs chromatin remodeling mutations with SHH pathway mutations may increase our understanding of this relationship and fashion drug combinations to improve outcome.
Given their therapeutic efficacy, SMO inhibitors such as vismodegib should be considered new therapeutics for patients with SHH-MB. These phase II trials and the precursor phase I trial11 suggest that vismodegib is well tolerated and safe in children and adults. However, because only a limited number of pediatric patients have been exposed to vismodegib, we recommend continued monitoring for bone and dental toxicities, given the developmental toxicity seen in preclinical models.32
The findings of these trials were limited by accrual and availability of tissue. Accrual was notably poor for adult non-SHH–MB and pediatric SHH-MB, which is no longer surprising given that various studies, published after these trials were initiated, have illustrated that SHH-MB is an uncommon subgroup in pediatric MB (age 5 to 16 years) but is widespread in adult MB.3,18,33 These observations underscore the importance of understanding the biology and distribution of the disease in all clinical trials. To this end, the best way to continue to improve understanding is through acquisition, preservation, and analysis of tissue. Even though the molecular and genomic studies performed here were limited by the quantity and type of tissue available, the association of molecular aberrations with the discrepant outcomes observed within SHH-MB serves as proof of principle that detailed molecular profiling is essential for optimal use of targeted therapy. Moreover, these associations support detailed molecular diagnostics in all specimens obtained during clinical trials to benefit all present and future patients with catastrophic diseases such as MB.
Acknowledgment
We thank Pankaj Gupta and Andrew Thrasher for their help with genomic analysis of the sequenced samples; Klo Spelhouse, Elizabeth Stevens, and Ashley Broussard from SJ Biomedical Communications for assistance with figures; Angela McArthur for editing the manuscript; and Emily Carps, who was the Operations, Biostatistics and Data Management Core/Pediatric Brain Tumor Consortium protocol coordinator for both PBTC-025B and PBTC-032.
Appendix
Table A1.
Demographic Characteristics of Patients Enrolled Onto PBTC-025B
| Characteristic | All Patients (N = 31)* |
Stratum A (n = 9)† |
Stratum B (n = 20)‡ |
Stratum C (n = 2) |
||||
|---|---|---|---|---|---|---|---|---|
| Diagnosis | Study Entry | Diagnosis | Study Entry | Diagnosis | Study Entry | Diagnosis | Study Entry | |
| Age, years | ||||||||
| Median | 23.5 | 30.3 | 16.9 | 24.0 | 26.7 | 32.0 | 20.9 | 29.1 |
| Minimum | 9.1 | 22.4 | 9.1 | 22.4 | 20.1 | 23.5 | 14.7 | 25.3 |
| Maximum | 46.9 | 51.9 | 37.4 | 40.6 | 46.9 | 51.9 | 27.1 | 32.9 |
Ineligible patient (025B-21) was excluded.
Patient 025B-32 originally assigned to stratum C was included.
Ineligible patient (025B-21) was excluded, but patient who was retrospectively assigned to sonic hedgehog group (025B-14) was included.
Table A2.
Demographic Characteristics of Patients Enrolled Onto PBTC-032
| Characteristic | All Patients (N = 25) |
Stratum A (n = 13) |
Stratum B (n = 12) |
|||
|---|---|---|---|---|---|---|
| Diagnosis | Study Entry | Diagnosis | Study Entry | Diagnosis | Study Entry | |
| Age, years | ||||||
| Median | 8.8 | 11.6 | 10.2 | 14.6 | 8.2 | 10.4 |
| Minimum | 2.4 | 3.9 | 3.8 | 5.2 | 2.4 | 3.9 |
| Maximum | 18.7 | 20.3 | 18.8 | 20.3 | 17.1 | 20.0 |
Table A3.
Toxicities and AEs in Patients Enrolled Onto PBTC-025B or PBTC-032 (n = 43)
| AE | Grade 1 |
Grade 2 |
Grade 3 |
Grade 4 |
Total |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Toxicity | AE | Toxicity | AE | Toxicity | AE | Toxicity | AE | Toxicity | AE | |
| No. of Events (No. of Patients) | No. of Events (No. of Patients) | No. of Events (No. of Patients) | No. of Events (No. of Patients) | No. of Events (No. of Patients) | No. of Events (No. of Patients) | No. of Events (No. of Patients) | No. of Events (No. of Patients) | No. of Events (No. of Patients) | No. of Events (No. of Patients) | |
| Lymphocyte count decrease | 6 (4) | 11 (7) | 15 (9) | 21 (10) | 6 (5) | 14 (11) | 1 (1) | 2 (2) | 28 (13) | 48 (18) |
| Fatigue | 13 (13) | 114 (13) | 4 (3) | 5 (4) | 11 (1) | 12 (2) | 18 (15) | 21 (17) | ||
| Hypophosphatemia | 4 (3) | 5 (4) | 8 (6) | 17 (12) | 4 (1) | 6 (2) | 16 (7) | 28 (14) | ||
| Vomiting | 14 (10) | 18 (11) | 1 (1) | 1 (1) | 1 | 2 (2) | 15 (10) | 21 (13) | ||
| Dygeusia | 11 (11) | 11 (11) | 3 (3) | 3 (3) | 14 (13) | 14 (13) | ||||
| WBC count decrease | 11 (6) | 21 (10) | 2 (2) | 6 (3) | 2 (2) | 13 (6) | 29 (11) | |||
| Musculoskeletal or connective tissue disorder | 9 (7) | 10 (8) | 4 (4) | 5 (5) | 13 (9) | 15 (10) | ||||
| Nausea | 10 (9) | 11 (10) | 2 (2) | 2 (2) | 12 (10) | 13 (11) | ||||
| Platelet count decrease | 11 (7) | 15 (9) | 11 (7) | 15 (9) | ||||||
| Diarrhea | 8 (6) | 13 (9) | 2 (2) | 3 (3) | 10 (8) | 16 (11) | ||||
| ALT increased | 8 (6) | 12 (9) | 1 (1) | 2 (2) | 9 (7) | 14 (10) | ||||
| Headache | 5 (4) | 11 (10) | 3 (2) | 8 (7) | 4 (4) | 8 (5) | 23 (17) | |||
| Anemia | 6 (5) | 10 (8) | 1 (1) | 4 (4) | 3 (1) | 7 (5) | 17 (10) | |||
| Cough | 5 (5) | 8 (7) | 1 (1) | 3 (3) | 6 (5) | 11 (9) | ||||
| Constipation | 5 (4) | 7 (5) | 1 (1) | 2 (2) | 6 (5) | 9 (7) | ||||
| Myalgia | 3 (3) | 6 (6) | 2 (2) | 2 (2) | 1 (1) | 1 (1) | 6 (5) | 9 (8) | ||
| Alopecia | 4 (4) | 5 (5) | 2 (2) | 2 (2) | 6 (5) | 7 (6) | ||||
| Hypokalemia | 5 (5) | 8 (6) | 5 (4) | 5 (5) | 13 (8) | |||||
| Hypomagnesemia | 5 (5) | 10 (9) | 1 (1) | 5 (5) | 11 (10) | |||||
| AST increased | 5 (5) | 9 (8) | 1 (1) | 5 (5) | 10 (8) | |||||
| Anorexia | 4 (4) | 5 (5) | 1 (1) | 1 (1) | 5 (5) | 6 (6) | ||||
| Hypoglycemia | 4 (4) | 12 (8) | 2 (2) | 4 (4) | 14 (9) | |||||
| Neutrophil count decreased | 1 (1) | 8 (5) | 3 (2) | 6 (4) | 4 (3) | 14 (7) | ||||
| Hypocalcemia | 4 (3) | 13 (10) | 4 (3) | 13 (10) | ||||||
| Hypoalbuminemia | 4 (3) | 9 (7) | 1 (1) | 4 (3) | 10 (7) | |||||
| Hyponatremia | 4 (3) | 8 (6) | 1 (1) | 4 (3) | 9 (7) | |||||
| Seizure | 2 (2) | 4 (4) | 1 (1) | 3 (3) | 1 (1) | 1 (1) | 4 (2) | 8 (5) | ||
| Back pain | 1 (1) | 4 (4) | 1 (1) | 1 (1) | 2 (2) | 2 (2) | 4 (3) | 7 (6) | ||
| Abdominal pain | 4 (3) | 5 (4) | 4 (3) | 5 (4) | ||||||
NOTE. Empty cells indicate no reported events. Data are sorted in decreasing frequency of total No. of toxicities. Only AEs occurring in ≥ four are listed. Data from Pediatric Brain –Tumor Consortium Operations and Biostatistics Center as of October 1, 2014. No. of courses, 162.
Table A4.
Response Rates per Stratum for PBTC-025B and PBTC-032
| Stratum | Response Rate (%) | 95% CI |
|---|---|---|
| PBTC-025B | ||
| A (non-SHH) | 0.0 | 0.0 to 20.6 |
| B (SHH) | 15.0 | 3.2 to 37.9 |
| C (unknown) | 0.0 | 0.0 to 63.2 |
| PBTC-032 | ||
| A (non-SHH) | 0.0 | 0.0 to 20 |
| B (SHH) | 8.3 | 0.2 to 38.5 |
| C (unknown) | NA | NA |
Abbreviations: NA, not available; SHH, sonic hedgehog.
Table A5.
Vismodegib Concentrations at Steady State
| Sample ID | Day 21 Total (μM)* | Unbound (μM) | Estimated CSF Penetration (unbound × 0.53; μM) |
|---|---|---|---|
| 025B-18 | 16.8 | 0.05 | 0.03 |
| 032-5 | 12.6 | 0.06 | 0.03 |
| 025B-5 | 9.93 | 0.06 | 0.03 |
| 025B-3 | 14.9 | 0.08 | 0.04 |
| 032-11 | 16.8 | 0.09 | 0.05 |
| 032-9 | 34.2 | 0.11 | 0.06 |
| 032-6 | 11.9 | 0.12 | 0.07 |
| 032-7 | 9.5 | 0.12 | 0.07 |
| 025B-2 | 19.1 | 0.14 | 0.07 |
| 025B-22 | 16.1 | 0.14 | 0.07 |
| 025B-32 | 21.2 | 0.14 | 0.07 |
| 025B-26 | 34.4 | 0.15 | 0.08 |
| 025B-28 | 27.3 | 0.16 | 0.08 |
| 025B-17 | 15.7 | 0.16 | 0.09 |
| 025B-29 | 19.2 | 0.17 | 0.09 |
| 032-10 | 28.9 | 0.18 | 0.09 |
| 025B-11 | 19 | 0.18 | 0.09 |
| 025B-27 | 20 | 0.18 | 0.09 |
| 025B-8 | 20.1 | 0.18 | 0.09 |
| 032-2 | 15.3 | 0.18 | 0.10 |
| 032-4 | 30.6 | 0.18 | 0.10 |
| 025B-14 | 20.3 | 0.21 | 0.11 |
| 025B-12 | 28.7 | 0.23 | 0.12 |
| 025B-13 | 25.5 | 0.24 | 0.13 |
| 025B-19 | 34 | 0.24 | 0.13 |
| 032-12 | 41.9 | 0.24 | 0.13 |
| 025B-6 | 30.7 | 0.25 | 0.13 |
| 025B-9 | 23.9 | 0.25 | 0.13 |
| 025B-4 | 17.5 | 0.26 | 0.14 |
| 032-1 | 18 | 0.26 | 0.14 |
| 025B-24 | 24.9 | 0.29 | 0.16 |
| 025B-1 | 17.3 | 0.30 | 0.16 |
| 025B-31 | 46.6 | 0.33 | 0.18 |
| 025B-15 | 34.3 | 0.34 | 0.18 |
| 025B-7 | 31.6 | 0.41 | 0.22 |
| 025B-23 | 29.8 | 0.46 | 0.24 |
| 025B-25 | 60.4 | 0.50 | 0.26 |
| 025B-30 | 34.2 | 0.53 | 0.28 |
| 032-3 | 45.3 | 0.53 | 0.28 |
| 025B-10 | NA | NA | NA |
| 025B-16 | NA | NA | NA |
| 025B-20 | NA | NA | NA |
| 032-8 | NA | NA | NA |
Abbreviation: NA, not available.
Or ± 7 days from day 21.
Table A6.
Molecular Analysis
| Sample ID | Trial | Central Pathology Review | Subgroup IHC | MYCN | GLI2 | P53 IHC | PTCH/9q | PTEN/10q | 17p | Molecular Analysis | Sequenced |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 025B-1 | PBTC25B | MB-CL | SHH | Poly | Poly | Lost | Lost | Poly | Poly | Yes | Yes; post-therapy lung lesion |
| 025B-2 | PBTC25B | MB-AN | SHH | Poly | Poly | Lost | Lost | Lost | Lost | Yes | No |
| 025B-3 | PBTC25B | MB-ND | SHH | NSA | NSA | NSA | Lost | NSA | NSA | Yes | No |
| 025B-4 | PBTC25B | MB-ND | SHH | Poly | Poly | NSA | NSA | Lost | Lost | Yes | No |
| 025B-5 | PBTC25B | MB-NOS | SHH | No | No | ||||||
| 025B-6 | PBTC25B | MB-ND | SHH | NSA | NSA | Lost | Lost | Lost | NSA | Yes | No |
| 025B-7 | PBTC25B | MB-NOS | SHH | Poly | NSA | NSA | NSA | NSA | NSA | Yes | Yes; pretherapy |
| 025B-8 | PBTC25B | MB-CL | SHH | NSA | Yes | No | |||||
| 025B-9 | PBTC25B | MB-CL | SHH | No | No | ||||||
| 025B-10 | PBTC25B | MB-CL | SHH | AMP | Poly | NSA | Poly | Lost | Lost | Yes | No |
| 025B-11 | PBTC25B | MB-AN | SHH | Poly | Poly | NSA | Poly | Poly | Poly | Yes | Yes; pretherapy |
| 025B-12 | PBTC25B | MB-NOS | SHH | Poly | Poly | NSA | NSA | Poly | Lost | Yes | No |
| 025B-13 | PBTC25B | MB-ND | SHH | AMP | AMP | DS | Poly | Lost | Lost | Yes | No |
| 025B-14 | PBTC25B | Inadequate material | Unknown/SHH | Poly | Poly | DS | Poly | Lost | Poly | Yes | Yes; pretherapy |
| 025B-15 | PBTC25B | MB-NOS | SHH | Gain | Poly | DS | Poly | Lost | Lost | Yes | No |
| 025B-16 | PBTC25B | MB-NOS | SHH | NSA | NSA | NSA | NSA | NSA | NSA | Yes | No |
| 025B-17 | PBTC25B | MB-ND | SHH | NSA | NSA | Lost | NSA | NSA | NSA | Yes | No |
| 025B-18 | PBTC25B | MB-ND | SHH | Poly | Poly | NSA | Lost | Poly | Lost | Yes | No |
| 025B-19 | PBTC25B | MB-CL | SHH | Poly | Poly | NSA | NSA | NSA | NSA | Yes | No |
| 025B-20 | PBTC25B | MB-ND | SHH | NSA | NSA | NSA | Lost | NSA | NSA | Yes | No |
| 025B-21 | PBTC25B | MB-ND | SHH | No | No | ||||||
| 025B-22 | PBTC25B | MB-CL | NWNS | NSA | NSA | NSA | NSA | NSA | Lost | Yes | No |
| 025B-23 | PBTC25B | MB-CL | NWNS | Poly | Poly | Lost | Poly | Poly | Lost | Yes | No |
| 025B-24 | PBTC25B | MB-CL | NWNS | Poly | Poly | NSA | Poly | Poly | Lost | Yes | No |
| 025B-25 | PBTC25B | MB-CL | NWNS | Poly | Poly | NSA | Poly | NSA | Lost | Yes | No |
| 025B-26 | PBTC25B | MB-AN | NWNS | NSA | Poly | DS | Poly | NSA | Poly | Yes | No |
| 025B-27 | PBTC25B | MB-CL | NWNS | NSA | NSA | NSA | NSA | NSA | Lost | Yes | No |
| 025B-28 | PBTC25B | MB-CL | NWNS | NSA | NSA | Lost | NSA | NSA | Lost | Yes | No |
| 025B-29 | PBTC25B | MB-CL | NWNS | NSA | NSA | Lost | NSA | NSA | Lost | Yes | No |
| 025B-30 | PBTC25B | MB-NOS | Unknown | No | No | ||||||
| 025B-31 | PBTC25B | Inadequate material | Unknown | No | No | ||||||
| 025B-32 | PBTC25B | MB-CL | Unknown/NWNS | NSA | NSA | NSA | NSA | Lost | NSA | Yes | No |
| 032-1 | PBTC032 | MB-ND | SHH | NSA | NSA | NSA | Lost | NSA | NSA | Yes | Yes; post-therapy |
| 032-2 | PBTC032/PBTC025 | MB-ND | SHH | NSA | NSA | Lost | Lost | NSA | NSA | Yes | No |
| 032-3 | PBTC032 | MB-CL | SHH | NSA | NSA | NSA | Lost | NSA | NSA | Yes | Yes; post-therapy |
| 032-4 | PBTC032 | MB-AN | SHH | AMP | AMP | Lost | NSA | Lost | Lost | Yes | No |
| 032-5 | PBTC032 | MB-ND | SHH | No | Yes; pretherapy | ||||||
| 032-6 | PBTC032 | MB-ND | SHH | Poly | Poly | DS | Poly | Poly | NSA | Yes | No |
| 032-7 | PBTC032 | MB-AN | SHH | No | No | ||||||
| 032-8 | PBTC032 | MB-ND | SHH | No | No | ||||||
| 032-9 | PBTC032 | MB-AN | SHH | AMP | Lost | Yes | Yes; pretherapy | ||||
| 032-10 | PBTC032 | MB-CL | SHH | No | No | ||||||
| 032-11 | PBTC032/PBTC025 | MB-CL | SHH | Gain | NSA | DS | Lost | Lost | Lost | Yes | No |
| 032-12 | PBTC032/PBTC025 | MB-AN | SHH | Gain | Poly | DS | Poly | NSA | Lost | Yes | No |
Abbreviations: AMP, amplified; AN, anaplastic histology; CL, classic histology; DS, diffuse staining; IHC, immunohistochemistry; MB, medulloblastoma; ND, nodular desmoplastic histology; NSA, no significant abnormality; NWNS, non-WNT non-SHH; poly, polysomy; SHH, sonic hedgehog.
Fig. A1.
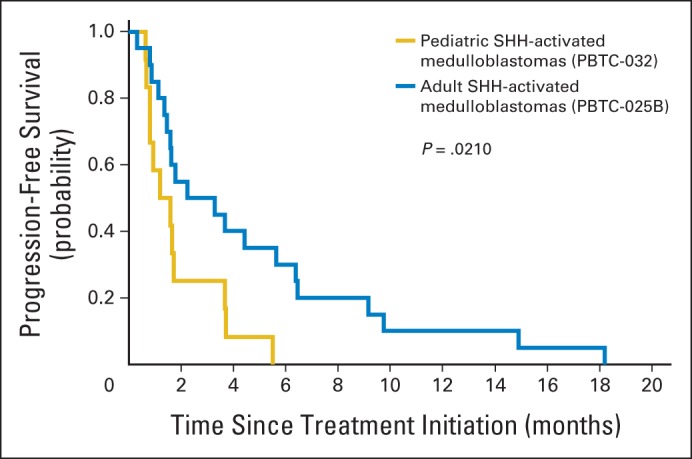
Progression-free survival of adults with sonic hedgehog (SHH) –subgroup medulloblastoma (SHH-MB; blue line) versus pediatric patients with SHH-MB (gold line). Significance assessed using log-rank test.
Fig. A2.

Progression-free survival of adult and pediatric patients with sonic hedgehog–subgroup medulloblastoma grouped based on status of following molecular characteristics: (A) PTCH1, (B) PTEN, (C) MYCN, (D) GLI2, (E) P53, and (F) 17p. Significance assessed using Kolmogorov-Smirnov tests. AMP, amplification; DS, diffuse staining; gain, imbalanced gain; loss, imbalanced loss; NSA, no significant abnormality; poly, balanced polysomy.
Footnotes
See accompanying article on page 2692
Supported in part by National Institutes of Health Grant No. U01 CA81457 to the Pediatric Brain Tumor Consortium, Cancer Center Core Grant No. CA 21765, the Noyes Brain Tumor Foundation, Musicians Against Childhood Cancer, and the American Lebanese Syrian Associated Charities.
Presented in part at the 49th Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, May 31-June 4, 2013.
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
Clinical trial information: NCT00939484, NCT01239316.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: Giles W. Robinson, Sridharan Gururangan, Roger J. Packer, Stewart Goldman, Naoko Takebe, Clinton F. Stewart, Maryam Fouladi, James M. Boyett, Richard J. Gilbertson, Tom Curran, David W. Ellison, Amar Gajjar
Administrative support: Naoko Takebe
Provision of study materials or patients: Brent A. Orr, Sridharan Gururangan, Roger J. Packer, Stewart Goldman, Michael D. Prados, Annick Desjardins, Clinton F. Stewart, Maryam Fouladi, Amar Gajjar
Collection and assembly of data: Giles W. Robinson, Brent A. Orr, Gang Wu, Sridharan Gururangan, Tong Lin, Ibrahim Qaddoumi, Stewart Goldman, Michael D. Prados, Annick Desjardins, Murali Chintagumpala, Naoko Takebe, Sue C. Kaste, Sariah J. Allen, Arzu Onar-Thomas, Clinton F. Stewart, Maryam Fouladi, James M. Boyett, David W. Ellison, Amar Gajjar
Data analysis and interpretation: Giles W. Robinson, Brent A. Orr, Gang Wu, Tong Lin, Roger J. Packer, Naoko Takebe, Sue C. Kaste, Michael Rusch, Sariah J. Allen, Arzu Onar-Thomas, James M. Boyett, Richard J. Gilbertson, Tom Curran, David W. Ellison, Amar Gajjar
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog–Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Giles W. Robinson
No relationship to disclose
Brent A. Orr
No relationship to disclose
Gang Wu
No relationship to disclose
Sridharan Gururangan
Consulting or Advisory Role: BioMarin Pharmaceuticals
Tong Lin
No relationship to disclose
Ibrahim Qaddoumi
No relationship to disclose
Roger J. Packer
Consulting or Advisory Role: Roche, AstraZeneca
Stewart Goldman
Consulting or Advisory Role: Novartis
Michael D. Prados
Consulting or Advisory Role: Novartis
Research Funding: Pfizer (Inst), Genentech (Inst), Novartis (Inst)
Annick Desjardins
Consulting or Advisory Role: Genentech, EMD Serono, Celldex, Eli Lilly, Cavion
Research Funding: Genentech, Celldex, Tactical Therapeutics, Eli Lilly
Patents, Royalties, Other Intellectual Property: Letters of patent for oncolytic poliovirus for human tumors
Murali Chintagumpala
No relationship to disclose
Naoko Takebe
No relationship to disclose
Sue C. Kaste
No relationship to disclose
Michael Rusch
Honoraria: Illumina
Travel, Accommodations, Expenses: Illumina
Sariah J. Allen
No relationship to disclose
Arzu Onar-Thomas
No relationship to disclose
Clinton F. Stewart
No relationship to disclose
Maryam Fouladi
No relationship to disclose
James M. Boyett
No relationship to disclose
Richard J. Gilbertson
Honoraria: Genentech
Tom Curran
Consulting or Advisory Role: Redx Pharma
Patents, Royalties, Other Intellectual Property: Biosource, Life Technologies, Rockland, Nantworks, Selexagen, Invitrogen, Sigma Isreal Chemicals
Expert Testimony: Genentech
David W. Ellison
Patents, Royalties, Other Intellectual Property: Immunohistochemical test for molecular subgrouping of medulloblastoma
Amar Gajjar
Consulting or Advisory Role: Astra Zeneca, Celgene
Research Funding: Genentech (Inst)
REFERENCES
- 1.Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012;123:465–472. doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924–1931. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 3.Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29:1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shih DJ, Northcott PA, Remke M, et al. Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol. 2014;32:886–896. doi: 10.1200/JCO.2013.50.9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mulhern RK, Palmer SL, Merchant TE, et al. Neurocognitive consequences of risk-adapted therapy for childhood medulloblastoma. J Clin Oncol. 2005;23:5511–5519. doi: 10.1200/JCO.2005.00.703. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong GT, Liu Q, Yasui Y, et al. Long-term outcomes among adult survivors of childhood central nervous system malignancies in the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2009;101:946–958. doi: 10.1093/jnci/djp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Svärd J, Heby-Henricson K, Persson-Lek M, et al. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian hedgehog signaling pathway. Dev Cell. 2006;10:187–197. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 8.Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med. 2012;366:2180–2188. doi: 10.1056/NEJMoa1113538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodon J, Tawbi HA, Thomas AL, et al. A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin Cancer Res. 2014;20:1900–1909. doi: 10.1158/1078-0432.CCR-13-1710. [DOI] [PubMed] [Google Scholar]
- 10.Rudin CM, Hann CL, Laterra J, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–1178. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gajjar A, Stewart CF, Ellison DW, et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: A pediatric brain tumor consortium study. Clin Cancer Res. 2013;19:6305–6312. doi: 10.1158/1078-0432.CCR-13-1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romer JT, Kimura H, Magdaleno S, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/−)p53(−/−) mice. Cancer Cell. 2004;6:229–240. doi: 10.1016/j.ccr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 13.Lee Y, Kawagoe R, Sasai K, et al. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene. 2007;26:6442–6447. doi: 10.1038/sj.onc.1210467. [DOI] [PubMed] [Google Scholar]
- 14.Dijkgraaf GJ, Alicke B, Weinmann L, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011;71:435–444. doi: 10.1158/0008-5472.CAN-10-2876. [DOI] [PubMed] [Google Scholar]
- 15.Yauch RL, Dijkgraaf GJ, Alicke B, et al. Smoothened mutation confers resistance to a hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–574. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson G, Parker M, Kranenburg TA, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–48. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones DT, Jäger N, Kool M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kool M, Jones DT, Jäger N, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pugh TJ, Weeraratne SD, Archer TC, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ellison DW, Dalton J, Kocak M, et al. Medulloblastoma: Clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011;121:381–396. doi: 10.1007/s00401-011-0800-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ellison DW, Kocak M, Dalton J, et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol. 2011;29:1400–1407. doi: 10.1200/JCO.2010.30.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fleming TR, O'Fallon JR, O'Brien PC, et al. Modified Kolmogorov-Smirnov test procedures with application to arbitrarily right-censored data. Biometrics. 1980;36:607–625. [Google Scholar]
- 23.Wong H, Alicke B, West KA, et al. Pharmacokinetic-pharmacodynamic analysis of vismodegib in preclinical models of mutational and ligand-dependent hedgehog pattern activation. Clin Cancer Res. 2011;17:4682–4692. doi: 10.1158/1078-0432.CCR-11-0975. [DOI] [PubMed] [Google Scholar]
- 24.Northcott PA, Shih DJ, Peacock J, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhukova N, Ramaswamy V, Remke M, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol. 2013;31:2927–2935. doi: 10.1200/JCO.2012.48.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tabori U, Baskin B, Shago M, et al. Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol. 2010;28:1345–1350. doi: 10.1200/JCO.2009.23.5952. [DOI] [PubMed] [Google Scholar]
- 27.Buonamici S, Williams J, Morrissey M, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med. 2010;2:51ra70. doi: 10.1126/scitranslmed.3001599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Metcalfe C, Alicke B, Crow A, et al. PTEN loss mitigates the response of medulloblastoma to hedgehog pathway inhibition. Cancer Res. 2013;73:7034–7042. doi: 10.1158/0008-5472.CAN-13-1222. [DOI] [PubMed] [Google Scholar]
- 29.He X, Zhang L, Chen Y, et al. The G protein alpha subunit Gα is a tumor suppressor in sonic hedgehog-driven medulloblastoma. Nat Med. 2014;20:1035–1042. doi: 10.1038/nm.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rausch T, Jones DT, Zapatka M, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Metcalfe C, de Sauvage FJ. Hedgehog fights back: Mechanisms of acquired resistance against Smoothened antagonists. Cancer Res. 2011;71:5057–5061. doi: 10.1158/0008-5472.CAN-11-0923. [DOI] [PubMed] [Google Scholar]
- 32.Kimura H, Ng JM, Curran T. Transient inhibition of the hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell. 2008;13:249–260. doi: 10.1016/j.ccr.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 33.Kool M, Korshunov A, Remke M, et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, group 3, and group 4 medulloblastomas. Acta Neuropahol. 2012;123:473–484. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]