

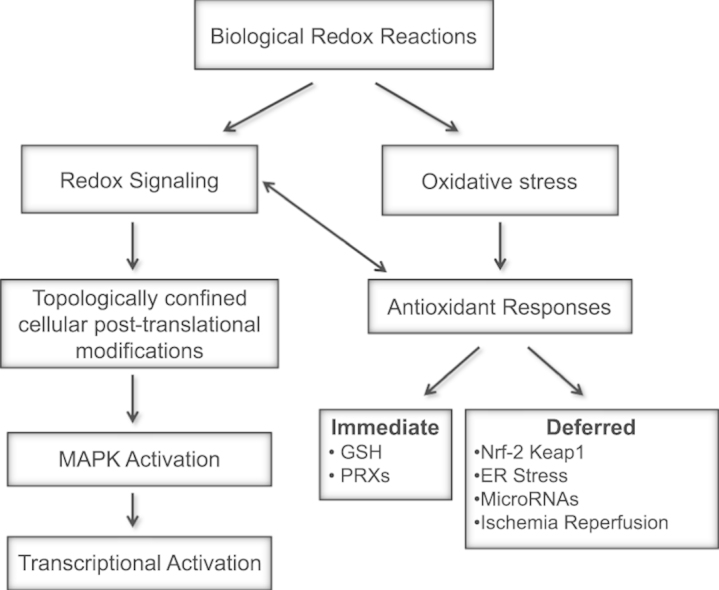
Abstract
Redox biological reactions are now accepted to bear the Janus faceted feature of promoting both physiological signaling responses and pathophysiological cues. Endogenous antioxidant molecules participate in both scenarios. This review focuses on the role of crucial cellular nucleophiles, such as glutathione, and their capacity to interact with oxidants and to establish networks with other critical enzymes such as peroxiredoxins. We discuss the importance of the Nrf2-Keap1 pathway as an example of a transcriptional antioxidant response and we summarize transcriptional routes related to redox activation. As examples of pathophysiological cellular and tissular settings where antioxidant responses are major players we highlight endoplasmic reticulum stress and ischemia reperfusion. Topologically confined redox-mediated post-translational modifications of thiols are considered important molecular mechanisms mediating many antioxidant responses, whereas redox-sensitive microRNAs have emerged as key players in the posttranscriptional regulation of redox-mediated gene expression. Understanding such mechanisms may provide the basis for antioxidant-based therapeutic interventions in redox-related diseases.
Keywords: Redox signaling, Antioxidants, Transcription factors, Ischemia–reperfusion, ER stress
Graphical abstract
Highlights
-
•
Antioxidant responses are crucial for both redox signaling and redox damage.
-
•
Glutathione-mediated reactions and Nrf2-Keap1 pathway are key antioxidant responses.
-
•
Redox-related post-translational modifications activate specific signaling pathways.
-
•
Redox-sensitive microRNAs contribute to redox-mediated gene expression regulation.
-
•
ER stress and ischemia-reperfusion are antioxidant-related pathophysiological events.
1. Introduction
Free radicals and oxidant species may behave as deleterious and toxic products, involved in cellular and tissular dysfunction. Overproduction of these species may result in DNA, lipid and protein damage. Nevertheless, low or moderate concentrations of reactive oxygen species (ROS) or reactive nitrogen species (RNS) are also involved in physiological responses as part of signaling processes and defense mechanisms against infectious agents. In fact, ROS protect the cell against ROS damage by inducing different antioxidant responses and re-establishing or maintaining redox homeostasis. For the sake of clarity in this review we will focus on endogenous antioxidant systems and the different factors involved in their regulation.
2. Glutathione and glutathione-related post-translational modifications
Antioxidant molecules are in fact nucleophilic and reductant molecules able to react with oxidants, which are generally electrophiles, giving them one or two electrons. Glutathione (GSH) is considered the most abundant molecule among endogenous antioxidants. GSH is a reduced peptide consisting of three-residues (γ-l-glutamyl-l-cysteinyl glycine) which can donate an electron with the consequence that two electron donating GSH molecules form oxidized GSSG. In humans, GSH is almost uniquely present in a quite high concentration (1–10 mM) which allows to scavenge ROS either directly or indirectly [1]. It can directly react with and some other ROS, but its indirect ROS-scavenging functions, such as revitalizing other antioxidants, are likely more important; e.g. it can reduce dehydroascorbic acid which is formed in the reconversion of α-tocopheroxyl radical to α-tocopherol, a lipophilic chain breaking antioxidant, which interacts with the polyunsaturated acyl groups of lipids, stabilizes membranes and scavenges various reactive oxygen species and lipid oxy-radicals [2,3]. As an antioxidant it reacts with ROS, RNS and radicals produced in association with electron transport, xenobiotic metabolism and inflammatory responses [4,5]. GSH is synthesized from its constituent amino acids forming a tripeptide thiol and this synthesis requires two ATP-dependent steps (Fig. 1). The first and limiting synthesis is catalyzed by γ-glutamyl-cysteine ligase (GCL) and the second step mediated by GSH synthetase (GS). GCL is a heterodimeric enzyme composed by a heavy subunit, GCLc (73 kDa) with catalytic activity and a smaller one, GCLm (33 kDa) that has a regulatory role on the other subunit [6]. GSH homeostasis in the cell is not only regulated by its de novo synthesis, but also by other factors such as utilization, recycling and cellular export. This redox cycle is known as the GSH cycle and incorporates other important antioxidant, redox-related enzymes. Aerobic respiration may result in an increase in hydrogen peroxide, which will be metabolized by glutathione peroxidase (GPx) by converting 2 GSH molecules to its oxidized form (GSSG). GSH is recycled by the action of glutathione reductase (GR, see below). Moreover, GSH is also able to react with critical Cys residues in proteins, by forming mixed disulfides (see below). This is a labile process that can be reverted by glutathione S-transferase (GST). Another cellular mechanism used to prevent redox unbalance is exporting GSSG to the extracellular medium. In addition, the biosynthetic capacity to form GSH is also regulated by allosteric mechanisms and substrate abundance. An excess of GSH in the cell produces a competitive inhibition in GCLc, while l-Cys is one of the limiting substrates [6,7].
Fig. 1.

GSH biosynthetic route and GSH cycle. GSH biosynthesis occurs in two different and ATP-dependen steps. The first and limiting step is carried out by GCL, formed by two subunits GCLc and GCLm. In this step l-Glu and l-Cys react to form γ-glutamyl-cisteine. GS is responsible of the second step, that joins l-Gly forming γ-glutamyl-cisteinyl-glicine (GSH). As well as aerobic respiration or other ROS sources increase H2O2, that should be metabolized, in this case GPx generating GSSG. This GSSG could be reduced to GSH again with the help of GR GR, creating a redox cycle, using as reducing agent NAPDH, from Penthoses Phosphate Pathway (PPP).
Alterations in the ratio of the redox pair 2GSH/GSSG towards a more oxidized status form the biochemical basis of targeting redox-sensitive cysteine residues in proteins by generating mixed disulfides between the thiol and GSH [2,8]. However, the calculation and quantification of this ratio is not easy and depends on the detection method, the tissue of interest and the abundance of recycling enzymes such as GPx1 [9]. This post-translational modification, known as S-glutathionylation, is a reversible process, which has been extensively reviewed elsewhere [10,11]. S-glutathionylation may result in the activation or inactivation of protein function [12]. Thereby S-glutathionylation is able to modulate different cellular pathways, and affect gene expression profiles by affecting different transcription factors as Nrf2 (see below), or NF-κB. Although these mechanisms were initially proposed to be part of a protective pathway against other irreversible oxidative modifications [2,8], it was recently shown that S-glutathionylation of eNOS at Cys689 and Cys908 leads to eNOS uncoupling, diminished NO production, and enhanced oxidative stress and superoxide anion production [13]. Thus, S-glutathionylation may be a double-edged sword in the sense that it may promote antioxidant or pro-oxidant responses.
2.1. GSH-related enzymatic antioxidants
Glutathione reductase (GR) is the enzyme critical for the reduction of GSSG back to GSH. GR utilizes FAD and NADPH to reduce one GSSG to two GSH (Fig. 2). The link of GR to oxidative stress was established due to its role in the ROS-releasing macrophage oxidative burst response as cells lacking GR had a more persistent response. In addition, GR deficiency has been linked to lupus erythematosus in Afro-Americans, favism and cataracts [14]. Glutathione peroxidases (GPXs; GPX1-8) couple oxidation of GSH with detoxification of H2O2. GPX1 deficiency in mice was also shown to contribute to the development of cataracts at an early age. From the GPX family members known, GPX1 is the ubiquitous enzyme that reduces cytosolic, mitochondrial and in some cells peroxisomal peroxides. GPX2 is an epithelial-specific form with highest expression in intestine [15]. GPX3 is a secreted form mainly expressed in lung and kidney; it is measurable in plasma and seems to be involved in the protection against external peroxides [15]. There are three different isoforms of GPX4 (also known as phospholipid–hydroperoxide glutathione peroxidase) derived from a single gene: cytosolic (c-GPX4), mitochondrial (m-GPX4), and nuclear (n-GPX4) [16]. Unlike other glutathione peroxidases, GPX4 has a broad range of substrates; in addition to H2O2, these include derivatives from cholesterol and cholesteroyl esters and thymine hydroperoxide [16]. Importantly, the catalytic sites of various GPX enzymes and thioredoxin reductases (TrxR) contain selenium in the form of selenocysteine. Selenium is a trace element, and its appearance in the catalytic centers of antioxidant enzymes underlines even more its essential biological roles [17]. In addition to GPX and TrxR, selenoprotein P has also been reported to have antioxidant properties [17] because selenium-containing compounds scavenged ROS in vitro and in various animal studies [18–20].
Fig. 2.

Interconnection of glutaredoxin, peroxiredoxin, thioredoxin, and glutathione containing antioxidant systems. Hydrogen peroxide (H2O2) can be reduced by peroxiredoxins (Prx) or glutathione peroxidases (GPX), which couple reduction of H2O2 with oxidation of glutathione (GSH). Oxidized Prx can be reduced by thioredoxins (Trx). Subsequently, oxidized Trx become reduced by thioredoxin reductase (TrxR) in a NADPH-dependent manner. Oxidized glutathione disulfide (GSSG) is reduced by glutathione reductase (GR) in the presense of NADPH. Further, glutaredoxins (Grx) can reduce disulfide (S–S) bonds in proteins (Pr), and glutathione S transferase (GST) is using GSH to conjugate and thus to detoxify reactive electrophilic compounds (R).
Glutathione is also a cofactor and substrate for multiple glutaredoxins (Grx), which catalyze disulfide reductions in the presence of NADPH and GR [21]. Another important enzyme family consists of glutathione-S-transferases (GSTs). In humans, 22 family members known which are classified in classes based upon their structure. GSTs are dimeric, with both subunits being from the same class of GSTs. The activity of GSTs is also dependent upon the presence of GSH, again stressing its importance as an antioxidant molecule. The primary role of GSTs is the detoxification of reactive electrophilic compounds, including environmental toxins, and products of oxidative stress, by conjugation with GSH. Due to the action of specific transporters GSH conjugates are removed from the cell, thereby preventing crucial cellular proteins and nucleic acids from the action of reactive electrophilic compounds.
A peroxidase activity is not only found in GPX but is also characteristic of the peroxiredoxin (PRX) family (Fig. 3). Peroxiredoxins are thiol-specific proteins that react with H2O2 at a very high rate [22]. The activity of peroxiredoxins is crucially dependent on a conserved, so called ’peroxidatic’, cysteine residue in the active site. Importantly, the catalytic cysteine can become hyperoxidized and catalytically inactive but still be able to participate in ROS signaling. Hyperoxidized peroxiredoxin can be recycled by sulphiredoxin [23–25]. It is important to note that PRXs can also reduce and detoxify peroxynitrite and a wide range of organic hydroperoxides. Prx1, 2 and 4 are found primarily in the cytoplasm but are also expressed in nuclei. In addition, Prx1 is also present in mitochondria and peroxisomes while Prx4 is found in lysosomes [26,27]. While Prx3 is localized in the mitochondria [26], Prx5 is found in mitochondria, cytoplasm, nuclei and peroxisomes [28]. It has been suggested that Prx2, due to its high reaction rate and abundance, traps almost all H2O2 in vivo [29–31]. Oxidized cysteine residues of Prx are specifically reduced by thioredoxins (Trx), thiol-disulfide oxidoreductases that can be oxidized upon oxidative stress caused by a variety of stimuli [32]. Oxidized Trx (as well as other oxidized cellular proteins) can be reversibly reduced by thioredoxin reductase (TrxR) in a NADPH-dependent manner [32]. Interestingly, a compartment-specific specialization can also be found here: Trx1 and TrxR1 are in the cytoplasm and nucleus whereas Trx2 and TrxR2 show mitochondrial localization. Overall, they appear to have a protective function against oxidative stress [26,33]; e.g. the Trx/TrxR system supports the activity of ribonucleotide reductase and inhibits apoptosis signal-regulated kinase-1 [33].
Fig. 3.

Two-step mechanism reaction of a peroxiredoxin dimer with hydrogen peroxide. (i) the sulfhydryl group at the peroxidatic cysteine of one peroxiredoxin (Prx) subunit is oxidized to sulfenic acid (–SOH); (ii) the sulfenic acid condenses with the –SH group at the resolving cysteine from the other subunit to form an intersubunit disulfide bond. The disulfide bond can be reduced by thioredoxin or another reductase (red) depending on the species. Continuous peroxide signaling leads to irreversible hyperoxidation and formation of sulfinic acid (–SOOH) at the peroxidatic cysteine to (shown as the “Hyperoxidation”). The species peroxynitrite (ONOO–) has been included in the cartoon because it is susceptible of reduction by peroxiredoxins.
Among the antioxidant enzymes SODs catalyze the dismutation of to H2O2, and catalase stops formation of •OH by converting H2O2 to oxygen and water [34]. Whereas catalase is considered to be a rather peroxisome-specific antioxidant enzyme and will not be covered in this review, SODs are topologically confined. The Cu- and Zn-containing Cu/ZnSOD (SOD1) is located mainly in the cytosol and peroxisomes. In contrast, a Mn-dependent MnSOD (SOD2) is found in the mitochondrial matrix. A third extracellular variant, EcSOD or SOD3, is secreted and forms a glycosylated heterotetramer bound to the extracellular matrix [35] but again the enzyme activity is Cu-, Zn-dependent. Importantly, changes in the cytosolic Cu/ZnSOD (SOD1), mitochondrial MnSOD (SOD2) or extracellular EcSOD (SOD3) activity or expression correlate with ROS levels [36].
Dysfunction or failure in the dismutation process is linked to the pathogenesis of a number of diseases [36]. Thereby the impact and tissue affected by the different SODs seems to vary; SOD1 has been clearly linked to amyotrophic lateral sclerosis, the extracellular superoxide dismutase (SOD3, ecSOD) seems to be involved in hypertension, acute respiratory distress syndrome, or chronic obstructive pulmonary disease [37] and SOD2 has been linked with carcinogenesis [36]. The removal of superoxide anion by superoxide dismutase(s) appears to be important for a number of cellular processes, among them mitochondrial and ER function (see below).
3. Redox-dependent transcriptional and post-transcriptional regulation of antioxidants: main examples
Multiple H2O2 sensors and pathways are triggered converging in the regulation of transcription factors, including AP-1, Nrf2, CREB, HSF1, HIF-1, TP53, NF-κB, Notch, SP1 and CREB-1, which induce the expression of a number of genes, including those required for the detoxification of oxidizing molecules and for the repair and maintenance of cellular homeostasis, controlling multiple cellular functions like cell proliferation, differentiation and apoptosis. In addition, the family of FoxO-related transcription factors plays an important role in redox responses and is the object of a separate review within this thematic series. H2O2-mediated regulation of transcription factors can take place at different levels: synthesis/degradation, cytoplasm-nuclear trafficking, DNA binding and transactivation [31]. In this section we focus on the most important transcription-related pathways involved in antioxidant responses.
3.1. The Nrf2-Keap1 pathway
The Nrf2 (nuclear factor erythroid 2 [NF-E2]-related factor 2)-Keap1 (Kelch-like ECH-associated protein 1) pathway is the major regulator of cytoprotective responses to oxidative and electrophilic stress [38,39]. Nrf2 is a basic region-leucine zipper-type transcription factor that belongs to the Cap ‘n’ Collar (CNC) family of regulatory proteins that also includes NF-E2, Nrf1, Nrf3, Bach1 and Bach2 [40]. The protein contains seven functional domains, known as Neh1–Neh7. The major regulatory domain is Neh2, which is located in the N terminus of Nrf2. The Neh2 domain contains seven lysine residues critically involved in ubiquitin conjugation [41] as well as two binding sites, known as ETGE and DLG motifs, that help to regulate Nrf2 stability [42,43].
The Nrf2 protein is rapidly turned over; it has a short half-life under basal conditions (7–15 min) which is increased to 30–100 min in the presence of inducers [44,45]. The Nrf2 protein stability is primarily regulated by Keap1 [46] (Fig. 4). The ETGE and DLG motifs within Nrf2 interact with Keap1. Keap1 is a substrate adaptor protein for a Cullin 3 (Cul3)-dependent E3 ubiquitin ligase complex; hence binding of Keap1 with Nrf2 mediates ubiquitination and subsequent proteasomal degradation of Nrf2 [41,46–50].
Fig. 4.

Keap1 dependent regulation of Nrf2. Under basal conditions, Nrf2 is sequestered in the cytosol by a Keap1 homodimer, which facilitates the ubiquitination and proteasomal degradation of Nrf2. Inducers react with specific cysteine residues in Keap1, leading to the release of Nrf2 and allowing its nuclear translocation. In the nucleus, Nrf2 heterodimerizes with small Maf proteins and binds to the antioxidant response element (ARE), activating the expression of a battery of cytoprotective genes.
Sulfhydryl groups within Keap1 act as sensors for electrophiles and oxidants [51]. In the presence of reactive oxygen species, critical cysteine residues in Keap1 become oxidized leading to a conformational change of Keap1, which prevents its binding to Nrf2. As a consequence, Nrf2 ubiquitination and degradation is stopped, and its nuclear translocation promoted.
Among the 27 cysteine residues within Keap1, Cys151 seems to be the most critical sensor residue which mediates the formation of an intermolecular disulfide – Cys151–Cys151 – between two Keap1 molecules. Thus, in response to a diverse array of stimuli, redox-sensitive residues, especially Cys151 within Keap1, can be covalently modified, allowing Nrf2 to evade Keap1-mediated ubiquitination [52,53]. As a result, Nrf2 accumulates in the nucleus and heterodimerizes with a small Maf protein. The Nrf2-small Maf heterodimer binds to regulatory gene regions known as ARE sites with a consensus sequence 5′-TGACnnnGC-3′, also called electrophile response elements (EpRE) [54]. Substances able to induce a response at the ARE are derived from a variety of sources, including phytochemicals and derivatives such as genistein, quercetin, curcumin and sulforaphane; therapeutics such as oltipraz, auranofin and acetaminophen; environmental agents like paraquat and metals and endogenous inducers such as nitric oxide, nitro-fatty acids, hydrogen peroxide and 4-hydroxynonenal [55]. Nrf2 target genes mainly include enzymes involved in the antioxidant response such as NAD(P)H:quinone oxidoreductase 1 (NQO1), glutathione S-transferases (GSTs), cysteine–glutamate exchange transporter, and multidrug resistance-associated protein.
In addition to cysteine-thiol modification, several other mechanisms have been described to regulate Nrf2 signaling. For example, some proteins, such as autophagy substrate p62 and p53-regulated p21, interfere with the Keap1-dependent ubiquitination of Nrf2 by disrupting Nrf2-Keap1 binding causing persistent activation of Nrf2 [56,57]. ROS also regulate Nrf2 by promoting the Ser209 phosphorylation of eIF4E, the initiation factor 4F complex which recruits the 40S ribosome to the 5′-UTR of the Nrf2 mRNA. An additional level of regulation is provided by an Internal Ribosome Entry Site (IRES) sequence in the 5′-UTR in the Nrf2 mRNA. IRES activity is regulated by IRES trans-acting factors (ITAFs), which recruit eIFs and ribosomes to initiate translation. One of such ITAFs, La Autoantigen, is activated by ROS promoting dephosphorylation of Ser366 or phosphorylation of Thr301 by AKT. Concerning Nrf2 export from the nucleus, oxidation of Cys183 in Nrf2 inhibits its binding to the nuclear exporting protein CRM1, while the phosphorylation of Tyr568 by FYN, a kinase activated by GSK-3β strengthens this interaction [58].
Moreover, Nrf2 can bind to the E3 ubiquitin ligase β-TrCP (β-transducing repeat-containing protein) via its Neh6 domain, which can be phosphorylated by GSK-3β (glycogen synthase kinase 3β), leading to Cullin 1 (Cul1)-dependent ubiquitination and degradation of Nrf2 in a Keap1-independent manner [44,58]. GSK-3β is inhibited by phosphorylation, which can be catalyzed by AKT, ERK, p38 MAPK and PKC kinases, all known to be activated by H2O2. Also, Nrf2 cross-interacts with several other signaling pathways such as NF-κB, AhR, p53, and homeodomain transcription factors, which also expands the scope of Nrf2-regulated genes [59–62]. Additionally, Nrf2 phosphorylation by protein kinase RNA (PKR)-like ER kinase (PERK) or PKC-δ, both activated by ROS, causes dissociation of the Nrf2/Keap1 complex.
Post-translational modifications at the level of Keap1 that prevent its interaction with Nrf2 are another mechanism leading to Nrf2 activation. Indeed, Keap1 phosphorylation at Tyr141 renders the protein highly stable and its dephosphorylation induced by hydrogen peroxide results in rapid Keap1 degradation and Nrf2 activation [63]. Interestingly, Keap1 knockout mice die short after birth, but the lethal phenotype can be reversed by the double knockout of Keap1 and Nrf2 [64], which suggests that either excessive or insufficient Nrf2 activity may be detrimental for normal physiology. In fact, the constitutive activation of the Nrf2 pathway can be beneficial for tumor survival [65].
3.2. NF-κB
The NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells)/REL family of transcription factors consist of homo- and heterodimers of five distinct proteins, the REL subfamily proteins (p65/RELA, RELB, and c-REL) and the NF-κB subfamily proteins (p50, and p52, and its precursors p105 and p100, respectively). All NF-κB/REL proteins contain a Ref-1-homology domain (RHD), which is responsible for dimerization, recognition and binding to DNA as well as interaction with the inhibitory κB (IκB) proteins. The IκB family is composed of IκB-α, IκB-β, IκB-ϵ, IκB-γ, and BCL-3, all of which possess typical ankyrin repeats that bind to the RHD of NF-κB/REL proteins to interfere with their function. The IκB-kinase complex (IKK complex) catalyzes phosphorylation of IκBs with the result that IκBs are targeted for degradation by the 26S proteasome thereby freeing NF-κB. The latter then binds to the DNA consensus sequence 5′-GGGRNNYYCC-3′ (R is a purine, Y is a pyrimidine, and N is any base) in the promoter/enhancer regions of target genes [66].
Activation of IKK occurs also by phosphorylation and is catalyzed by an IKK kinase, including TGF-β-activated kinase (TAK1), AKT, or NF-κB inducing kinase-1 (NIK1), all of which can be regulated by H2O2 [67]. Alternatively, the reduced form of the dynein light chain protein LC8 binds to IκB and inhibits its phosphorylation by IKKs. H2O2 induces dimerization of LC8 by a disulfide bond, promoting dissociation from IκB, and NF-κB activation [68]. On the other hand, inhibition of activation of NF-κB by H2O2 can be mediated by Keap1-dependent degradation of IKKβ [69].
NF-κB target genes mainly include enzymes involved in the antioxidant response such as ferritin heavy chain [70] and SOD2 [71]. Another NF-κB target gene that contributes to both survival and innate immune functions is the HIF-1α gene, encoding the oxygen-regulated subunit of the hypoxia responsive transcription factor HIF-1 [72]. On the other hand, hydroquinone and tert-butyl hydroquinone, prototypes of phenolic antioxidants, block lipopolysaccharide (LPS)-induced transcription of TNFα and NF-κB as they block the formation of NF-κB/DNA binding complexes [73].
3.3. AP-1
AP-1 (activator protein 1) is a family of dimeric bZIP transcription factors including Jun, Fos, ATF/CREB, JDP and MAF that usually form heterodimers that bind to a TPA-responsive element (TRE, 5′-TGAG/CTCA-3′) or cAMP response elements (CRE, 5′-TGACGTCA-3′). They regulate several cellular processes, including cell proliferation, apoptosis, survival, and differentiation. AP-1, when upregulated, concentrates in the nucleus to activate gene expression [74]. H2O2 was shown to induce transcription of both c-JUN and c-FOS via activating the JNK, p38 MAPK and ERK signaling cascades [75], while antioxidants like butylated hydroxyanisole (BHA) and pyrrolidine dithiocarbamate (PDTC) induce AP-1 binding activity and AP-1-dependent gene expression including glutathione S-transferase [76].
Although several other pathways can be regulated by ROS (see above), the primary responses against different intensities of intermediate oxidative stress are mainly modulated by the cooperation of the three pathways: Nf-κB, AP1 and MAP kinases. However, with increasing oxidative stress, the Nrf2/ Keap pathway (see above) is also needed to induce antioxidant defenses and to minimize oxidative damage. When these defenses are not strong enough to contend against oxidative stress, the cell induces formation/opening of the mitochondrial permeability transition pore, as an efficient way to decrease ROS production, by decreasing the mitochondrial membrane mitochondrial potential. However, this increases the oxidative state in the cell [77,78], resulting in a homeostatic disruption of the redox balance, cell damage, and apoptosis [79].
3.4. Role of microRNAs in antioxidant responses
MicroRNAs are endogenous non-coding RNAs. They are small single-stranded RNAs (22–26 nt) and are now considered essential post-transcriptional regulators of gene expression. They bind to the 3′-UTR region of mRNAs by Watson-Crick complementarity and through different molecular interactions, they are able to block their translation into proteins. In 1998, Craig Mello and Andrew Fire showed how the formation of double stranded RNA in C. elegans produced transcriptional silencing and they described the first microRNA named Lin-4 in this same model [80,81]. During the last decades knowledge in this field has grown exponentially in such a way that a recent update of the main miRNA database (miRbase 21, June 2014) [82], listed 28,645 miRNAs in more than 142 species. The vast majority of these microRNA are highly conserved across species, including non-vertebrate ones as Drosophila or C. elegans.
MicroRNA transcription depends on its localization across the genome. These small RNAs can be encoded by intronic regions (about 80%) [83], intergenic regions, non-coding genes or even exons [84]. Transcriptional regulation of microRNAs is a complex process and has been reviewed in depth elsewhere [85,86]. miRNAs are also processed sequentially in the nucleus and cytoplasm by specific proteins finally leading to the generation of a mature miRNA (for review see [85,87]).
In the last years miRNAs have been found to regulate stress responses including redox balance, to the extent that the term “redoximiRs” has been coined to define a subset of miRNAs that either regulate redox pathways or are themselves regulated by the redox status [88]. As an example, a significant number of publications describe miRNAs that might target the Nrf2 pathway or that of its co-regulators Keap1 and Bach1 (Table 1) [88–103]. Also, we have recently described one miRNA, miR-433, which targets GSH biosynthetic enzymes in an Nrf2-independent manner, resulting in decreased GSH levels and antioxidant capacity with important implications for organ fibrosis [104]. Of interest Yang et al. have described Nrf2 and GSH regulation in hepatic fibrosis produced by miR-27b, a microRNA modulated by c-Myc [96].
Table 1.
.
| microRNA ID | Target | Relevance/disease | Reference |
| RedoximiRs | |||
| miR-144 | Nrf2 | Sickle cell disease | [94] |
| miR-28 | Nrf2 | Breast cancer | [95] |
| miR-27b | Nrf2 | Liver fibrosis | [96] |
| miR-34a | Nrf2 | Liver aging | [97–99] |
| miR-132 | Nrf2 | Nephrotoxicity | [89] |
| miR-200a | Keap1 | Breast cancer/fibrosis | [92,100] |
| miR-155 | Bach1 | Cardiovascular diseases, carcinomas | [101–103] |
| MitomiRs | |||
| miR-27 | MFF | Mitochondrial fission | [113] |
| miR-34a | Txnrd2 | Renal senescence | [114] |
| miR-145 | Bnip3 | [115] | |
| miR181a | Bcl-2 | Heart disease | [116] |
| miR-210 | ISCU/ AIFM3 | Hypoxia/cancer | [117] |
| miR-335 | SOD2 | Renal senescence | [114] |
| miR-494 | Foxj3/ mtF2 | Skeletal muscle differentiation | [118] |
| HipoxymiRs | |||
| miR-155 | HK2 | Hypertension | [122] |
| miR-210 | GPD1L/ISCU/COX10 | [123] | |
| miR-200 | PHD2 | Ischemia/precondition | [124] |
| miR-1/133 | Hsp60/ Hsp70/ IGF-1/ Bcl-2 | Ischemia/precondition | [124] |
| miR-34a | Sirt1 | Cardiovascular disease/myocardial infartation | [125] |
In the case of Dicer, one of the endonucleases involved in miRNA maturation, several publications have reported its downregulation in aging, senescence and stress models, hence altering the abundance of mature miRNA number [105,106]. Dicer downregulation can be prevented by the use of exogenous molecules with antioxidant potential, such as resveratrol and sulforaphane, both known to enhance Nrf2 activation. This is consistent with the fact that Dicer bears ARE sequences in its promoter region, which are conserved in human and murine species [105]. The NF-κB family of transcription factors (see above) is also able to regulate acute stress responses during inflammation through their interaction with miRNAs, specifically by upregulating the expression of miR-9, miR-155 and miR-146. Interestingly, some of the validated target genes for these three miRNAs are also pro-inflammatory molecules themselves [107]. Thus, these miRNAs may contribute to limit the duration of acute stress responses and reduce their related damage [108].
MicroRNAs are also susceptible of suffering epigenetic modifications that modulate their expression. Reciprocally, epigenetic enzymes like histone deacetylases (HDAC) and DNA-methyl transferases are also targets of oxidative stress and hence changes in their activities may result in changes in miRNA expression [109,110].
The study of miRNAs in pathological contexts has been useful to link them with new targets including enzymes professionally dedicated to ROS generation such as NOXs. For example, NOX4 has been reported to be regulated by miR-25, miR-146a and miR-23a [111]. Moreover other miRNAs, as miR-210 and miR-128a, have been identified in mitochondria as capable of regulating their metabolic activity [112]. These mitomiRs may play a role in the initiation and progression of cardiovascular disease (Table 1) [113–118]. Regarding other stress conditions, the unfolded protein response or endoplasmic reticulum (ER) stress (see below) includes an adaptive response whereby some miRNAs are modulated or regulate ER signaling. For example ER stress induces changes in the localization of AGO proteins, one of the essential set of proteins in the RISC complex, suggesting a miRNA repression role during ER stress [119]. However, other miRNAs are upregulated in these conditions [120]. In addition, other environmental stresses such as hypoxia also induce a specific subset of miRNAs (hypoximiRs), some of them involved in antioxidant responses [121–125]. Among these, miR-155, miR-210, miR-200 or miR-24 are related to the archetypal hypoxia-regulated transcription factor Hif-1α. In the context of ischemia–reperfusion (see below) miR-1 and miR-133 have been described in the heart in the setting of arrytmhia [124,126] while the miR-34a/Sirt1 pathway has been reported to be protective [127] ( Table 1).
4. Antioxidant responses in specific pathophysiological contexts
Obviously, antioxidant responses are involved in an enormous amount of cellular and tissular processes in relation to both physiology and pathosphysiology. Excellent reviews covering these issues are already available [8,128–130]. For the sake of clarity we now discuss in depth two examples, one related to a cellular biochemical process, ER stress and the other one in the context of a relevant pathophysiological common event, ischemia–reperfusion.
4.1. ER stress
The endoplasmic reticulum (ER) has two principal major functions. First, it is the primary site of protein synthesis and packaging, second, it plays a central role in metabolism and various signaling processes. The synthesis and the packing of proteins within the ER is strongly coupled to native disulfide bond formation. Correct disulfide bond formation is coupled to oxidation, reduction and isomerisation of disulfide bonds within the respective proteins. Indeed, there is growing evidence that a large number of different gene families and redox carriers play a role in the supply of redox equivalents to achieve a correct protein disulfide bond formation.
The rate-limiting step for disulfide bond formation is late-stage isomerization, which is necessary to achieve a substantial regular secondary protein structure. Since protein folding is not error-free, correction of incorrect disulfide bonds requires a partial unfolding of the already formed structures. Remarkably, proper folding and disulfide bond formation of proteins appears to be dependent on the redox status within the ER. Although reducing conditions prevail in the cytosol, the lumen of the ER represents a more oxidizing environment, with a higher ratio of oxidized to reduced glutathione [GSSG/GSH] [131]. Especially, reduced glutathione (GSH) or protein thiols react with ROS and thus contribute to cellular redox homeostasis. In particular, GSH is considered as the major thiol-disulfide redox buffer of cells and often the ratio between GSSG and GSH is used to determine the redox state. In the cytoplasm, the ratio of reduced glutathione to oxidized glutathione is >50:1 [132]. By contrast, this ratio is 1:1 to 3:1 in the ER. It appears that the oxidizing conditions are mainly important for de-novo disulfide bond formation of most proteins synthesized in the ER. However, disulfides can also be reduced in the ER, and hence redox homeostasis must be maintained.
In general the factors catalyzing disulfide bond formation become reduced and in order to fulfill further catalytic cycles these factors need to be re-oxidized [133]. In eukaryotic cells, in particular mammalian cells, it is believed that molecular oxygen functions as the final electron acceptor. Within that system, the ER resident oxidoreductase Ero1 and protein disulfide isomerase (PDI) appear to play key roles [134,135]. Overall, this implies that disturbances in the nutrient and metabolic flux are important effectors of Ero1 and PDI function and could thereby contribute to the generation of ROS and the build-up of an ER stress response.
The ER stress or unfolded protein response (UPR) is in general triggered when the ER function is diminished, e.g. when the entry of newly synthesized proteins exceeds the folding capacity. The accumulation of unfolded proteins within the ER leads to an activation of signaling events with the aim to increase the folding capacity and to reduce folding load. The folding capacity is enhanced after ER enlargement [136] and synthesis of ER resident molecular chaperones and foldases [137]. Decreasing expression of genes encoding secretory proteins [138] as well as increasing the ER-associated degradation (ERAD) of slowly folding or misfolded proteins [139] reduces the folding load. Three distinct branches of the ER stress response are in principle distinguished; the double-stranded RNA-activated protein kinase (PKR)-like endoplasmic reticulum kinase (PERK) branch, the inositol requiring kinase 1 (IRE1) branch, and the activating transcription factor 6 (ATF6) branch. All three components associate with the ER luminal chaperon BiP (also known as glucose-regulated protein 78, GRP78; or Kar2p in yeast). Once unfolded proteins accumulate in the ER, BiP preferentially associates with the unfolded proteins instead of PERK, ATF6, and IRE1, resulting in activation of their downstream signalling molecules [133]. So far, the cross-talk between ROS and ER stress appears to be most effective for the PERK branch. Activation of PERK promotes phosphorylation of the α-subunit of eukaryotic translation initiation factor 2 (eIF2α) [140] with the result of a block in protein translation-initiation except that of ATF4. ATF4 in turn induces expression of genes like growth arrest and DNA damage-34 (GADD34), and CAAT/Enhancer binding protein (C/EBP) homologous protein (CHOP) [141]. In addition, another direct PERK target appears to be Nrf2 (see above). Interestingly, Nrf2-/- cells are very sensitive to ER stress [142] implying that ER stress via PERK phosphorylates Nrf2, resulting in its dissociation from Keap1, nuclear localization of Nrf2, which, after heterodimerically partnering with other transcription factors, activates transcription through ARE binding as detailed above.
Although a redox unbalance can be caused by affecting the PERK branch, most ROS in the ER appear to be generated via Ero1 [143,144]. In humans, there are two ERO1 isoforms, hERO1-La and hERO1-Lb [145–147], which contain flavin adenine dinucleotide (FAD). Indeed, overexpression of both Ero1 proteins shifts the redox state of PDI towards the oxidized form (Fig. 5), which thus also influences oxidation of PDI substrates [148,149]. The isoform Ero1α was also localized in the mitochondria-associated membrane (MAM), and this localization depends on the oxidizing conditions within the ER [150]. During disulfide bond formation electrons pass several thiol-disulfide exchange reactions, the thiols of the substrate, PDI and Ero1 from where they finally reach molecular oxygen [143]. In a simplistic view, Ero1 usually donates a disulfide bond to PDI and at the same time the receiving electrons are passed on via Ero1´s FAD moiety to reduce oxygen and to generate hydrogen peroxide (Fig. 5) [151,152]. Interestingly, the hydrogen peroxide produced can be used by the ER-localized peroxiredoxin 4 to oxidize PDI and some PDI family members. This process does not only increase the efficiency of the Ero1-driven disulfide bond formation but also allows disulfide bond formation via alternative sources of H2O2 [153,154]. Although these findings underline the importance of hydrogen peroxide for proper disulfide bond formation, the incomplete reduction of oxygen during this process results in the formation of superoxide anion radicals, which can then be dismutated to H2O2 or converted to other ROS.
Fig. 5.
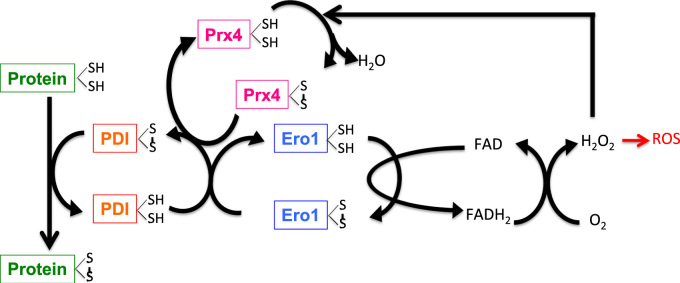
Protein folding in the endoplasmic reticulum is coupled to ROS formation. The ER enzymes PDI and ERO1 are crucial for protein folding. PDI oxidizes thiol (SH) groups, i.e. generates disulfide bonds, in the folding substrates, thereby it accepts electrons (e−) and becomes reduced. Ero1 re-oxidizes PDI by using its FAD moiety to transfer electrons from PDI to molecular oxygen (O2) to form hydrogen peroxide (H2O2), which may give rise to further formation of ROS. Hydrogen peroxide may also be used by peroxiredoxin 4 (Prx4) to oxidize PDI, thereby increasing the efficiency of Ero1-dependent disulfide bond formation.
In addition, removal of superoxide by SODs appears to be important for ER function. This is supported by findings from mice transgenic for a non-functional Cu/Zn-superoxide dismutase mutant. These mice show protein aggregate formation and extensive dilation of the ER along with degeneration of motor neurons of the spinal cord, features also seen in patients with amyotrophic lateral sclerosis [155–157]. Whether the ER dilation observed in these mice is the direct effect of ROS on protein folding or the more indirect result via activation of signalling events is not known yet. However, the Cu/Zn-superoxide dismutase-induced degeneration of neurons could be blocked by overexpression of the cytosolic chaperone heat-shock-protein 70 (HSP70) [158,159] suggesting that cytosolic ROS may affect protein folding in the ER. This is further supported by the finding that mutant CuZn-superoxide dismutase induces BiP (GRP78) levels [156].
The connection between oxygen, ROS, energy metabolism and ER resident proteins is also underlined by the fact that some of the latter, known as glucose-regulated proteins (GRPs) [160], are induced by hypoxia [155,161,162]. In line with this, severe hypoxia or anoxia can also activate ATF4 [163,164]. Although ER stress and anoxia induce ATF4, the latter was shown to be independent of hypoxia-inducible transcription factors (HIFs). However, the link between ER function and regulation of the O2-sensitive HIF-1α subunit seems to be independent of ATF4 since HIF-1α localizes to the perinuclear ER membrane under normoxia, where ROS such as hydroxyl radicals are generated in an iron-dependent Fenton reaction [165]. Moreover, the so called transmembrane proline 4 hydroxylase (P4HTM), one member of the four HIF-proline hydroxylases, which are critically involved in the regulation of HIF protein stability was found to be an ER resident enzyme [166].
Interestingly, from the two mammalian Ero members only Ero1Lα but not Ero1Lβ appears to be under transcriptional control from HIF-1 [167]. Thus, ROS-dependent HIF-1α regulation may be part of a feedback cycle contributing to thiol-oxidation in the ER via adaptation of Ero1Lα transcription to the overall cellular redox status.
However, ER-derived ROS, e.g. due to the action of Ero1, may not only have an intricate role in disulfide bond formation but also for the regulation of cell death and life span. This is in line with data showing that ER stress induces accumulation of ROS in PERK or ATF-4 knockout cells to higher levels than in wild-type cells [144] and that this contributes to apoptosis and a reduced life span in C. elegans. Interestingly, repression of Ero-1 restored normal life span in C. elegans with repressed PERK expression [144]. Furthermore, the cytokine TNFα caused cell death in a ROS- and ER stress dependent fashion in murine fibrosarcoma L929 cells [168]. However, pre-conditioning these cells with the ER stressor tunicamycin substantially inhibited TNFα-induced ROS accumulation and cell death. Within that scenario, the tumor necrosis factor receptor-associated factor 2 (TRAF2) was found to interact with the cytosolic domain of IREα [169] and with apoptosis signal-regulating kinase 1 (ASK1) [170,171]. This trimeric complex can be activated by ROS involving thioredoxin, another interacting partner of ASK1 [172–174]. Oxidative stress disrupts the ASK1–thioredoxin complex by oxidation of thioredoxin and thereby activating ASK1 [173] and subsequently JNK, p38 and cell death [175]. Interestingly, the Jun activation domain-binding protein (JAB-1, also known as COP9 signalosome subunit 5) may be a negative feed-back regulator since it was shown that it interacts with IRE1α [176]. Thus, ER stress and oxidative stress may induce cell death by using the same molecular complex consisting of IREα/TRAF2/ASK1/thioredoxin with JAB1 acting as negative regulator.
4.2. Ischemia-reperfusion
The process of ischemia followed by reperfusion is often accompanied by the activation of an injurious cascade. Although the pathogenesis of ischemia–reperfusion (IR) is not completely understood, there is considerable evidence implicating ROS as an initial cause of the injury. Ischemia results in a build-up of NADH that, upon reperfusion, generates a burst of free radicals from the electron transport chain (ETC). This can adversely alter proteins and DNA, and further propagate lipid radical reactions that produce electrophile species.
4.2.1. Post-translational modifications of mitochondrial proteins in ischemia–reperfusion
The specific modifications of mitochondrial proteins leading to improved mitochondrial function have the potential to modulate cardiac IR damage. The metabolism of (NO) can lead to the S-nitrosylation of proteins containing thiol groups, including mitochondrial proteins, thereby reversibly regulating their activity. In particular, complex I, the entry point for electrons from NADH into the respiratory chain, can be S-nitrosylated [177–179], which may alter its activity and thereby protect against damage during IR injury [180,181]. In agreement with this, mitochondria-targeted S-nitrosothiols (MitoSNO), which induce functional modification of mitochondrial proteins following S-nitrosylation, protect against heart IR injury [182–184]. This modification of complex I appears to increase superoxide production from the complex [178,185]. However, in the context of IR, reversible S-nitrosylation of complex I, in particular, a specific Cys residue (Cys 39) within ND3 (Fig. 6), slows the reactivation of mitochondria during the first minutes of reperfusion, thereby decreasing ROS production, oxidative damage and tissue necrosis [184]. In addition, consistent with their potential role in cardioprotection, S-nitrosothiols were detected in mitochondria isolated from perfused rat hearts subjected to ischemic preconditioning [177,186].
Fig. 6.

S-Nitrosylation of ND3 Cys39 protects from IR injury. The low activity of complex I after ischemia causes the ND3 Cys39 to become exposed. Reperfusion of ischemic tissue rapidly reactivates complex I and generates superoxide leading to oxidative damage and cell death. The presence of MitoSNO or other S-nitrosylating agents at reperfusion, causes the exposed ND3 cysteine to become S-nitrosylated, holding complex I in a low activity state and decreasing ROS production. Complex I is gradually reactivated through the reduction of the S-nitrosothiol by glutathione and thioredoxin.
Besides S-nitrosylation, deacetylation of mitochondrial proteins has been reported to attenuate IR damage. The sirtuin family are NAD+-dependent protein deacetylases that mediate the effects of caloric restriction [187]. It has been shown that caloric restriction primes cardiac mitochondria for ischemic stress by deacetylating specific mitochondrial proteins of the ETC [188], suggesting that deacetylation of specific mitochondrial proteins by sirtuin preserves mitochondrial function and attenuates myocardial damage during IR.
4.2.2. The Nrf2 pathway in ischemia–reperfusion
Since the generation of reactive species has been involved in the pathogenesis of IR, reducing oxidative stress is a potential therapeutic approach to prevent IR injury. Nrf2 accumulates in the nucleus after IR in cardiac [189] and renal tissues [190]. Moreover, several compounds that activate the Nrf2 pathway are protective against IR damage. Thus, curcumin, a natural compound in turmeric with antioxidant activity via the Nrf2 pathway, protects neurons from death caused by oxygen and glucose deprivation in an in vitro model of IR [191], and exerts beneficial effects against cardiac [192] and renal [193] IR injury. Likewise, mono-carbonyl analogues of curcumin such as 14p have been reported to decrease oxidative stress and limit myocardial IR injury via Nrf2 activation [194]. In addition, sulforaphane, a compound obtained from cruciferous vegetables, which induces Nrf2-dependent gene expression, attenuates liver injury induced by intestinal IR in rats [195], protects neurons and astrocytes from delayed cell death induced by oxygen and glucose deprivation [196,197], reduces the infarct size of isolated perfused rat hearts subjected to IR [198], and prevents renal injury caused by IR in vivo and in vitro [199]. This compound also protects rat hearts from early injury after experimental transplantation [200]. Taken together, these observations indicate that the activation of the Nrf2 pathway protects against IR injury and is a potential target to ameliorate this type of damage.
4.2.3 Lipid peroxidation products in cardioprotection against ischemia–reperfusion
As a result of oxidative damage, lipid peroxidation products accumulate in the heart. Mitochondria are both a primary source and target of lipid peroxidation products [201]. Reactive aldehydes such as 4-hydroxy-2-nonenal (4-HNE) are formed during cardiac IR. High concentrations of aldehydes are responsible for much of the damage produced during IR. In fact, aldehyde dehydrogenase 2 (ALDH2) has been reported to protect myocardium against IR injury through detoxification of aldehydes [202]. By contrast, low concentrations of aldehydes can stimulate stress resistance pathways to confer cardioprotection. Thus, lipid peroxidation products are involved in the transcriptional regulation of endogenous antioxidant systems. Recent evidence demonstrates that transient increases in lipid peroxidation products may be beneficial in cardioprotection by contributing to mitohormesis (i.e. induction of antioxidant systems) in cardiomyocytes. Thus, 4-HNE activates Nrf2 in the heart and increases the intramyocardial GSH content, improving the functional recovery of the left ventricle following IR in isolated perfused hearts [203]. 4-HNE also induces mitochondrial uncoupling protein 3 (UCP3) via Nrf2 in cardiomyocytes, promoting cell survival [204].
5. Signal transduction in the context of antioxidant responses: some key players
Historically considered as cellular damaging agents, ROS have emerged as important modulators of intracellular transduction signaling. ROS interact with redox-sensitive signaling molecules including transcription factors, protein tyrosine phosphatases, protein kinases and ion channels, that contain cysteine residues whose SH groups are oxidized, causing a change in their biological activity, regulating cellular processes like growth factor signaling, hypoxic signal transduction, autophagy, immune responses, and stem cell proliferation and differentiation [129,205].
These redox changes include activation/deactivation cycles of redox metabolism and expression of antioxidant enzymes expression that are essential to maintain nucleofilic tone and oxidant/antioxidant balance in the cells [130]. In the following section a brief overview of some examples of redox-sensitive cytosolic and mitochondrial proteins is given.
5.1. Cytosolic regulation
5.1.1. Protein tyrosine phosphatases
One of the most studied direct targets of ROS signaling is the family of protein tyrosine phosphatases. All tyrosine phosphatases have a conserved 230-amino acid domain that contains a reactive and redox-regulated cysteine, which catalyzes the hydrolysis of protein phosphotyrosine residues by the formation of a cysteinyl-phosphate intermediate, that later is hydrolyzed by an activated water molecule. Because of the unique environment of the tyrosine phosphatase active site, the catalytic cysteine presents an unusually low pKa value and is therefore deprotonated at physiological pH, existing as a thiolate anion (Cys–S−). Oxidation of this residue to sulfenic acid by H2O2 renders the tyrosine phosphatases inactive. This oxidation of cysteine to sulfenic acid is reversible, while oxidation by the addition of two (sulfinic acid) or three (sulfonic acid) oxygens to the active site cysteine is irreversible. These modifications can be further stabilized by the formation of inter or intramolecular disulfide (S–S) or sulfenyl–amide bonds. Thus ROS significantly inhibit activity of tyrosine phosphatases, resulting in increased tyrosine phosphorylation [206]. For instance, redox regulation of protein tyrosine phosphatase 1B involves a sulfenyl–amide intermediate, also accompanied by large conformational changes in the catalytic site that inhibit substrate binding. This is an unusual protein modification that protects the active-site cysteine residue of PTP1B from irreversible oxidation [207,208]. Interestingly, S-glutathionylation of the sulfenic acid form of these proteins is proposed as a mechanism to protect against irreversible oxidation [209].
5.1.2. Protein Kinases
Protein kinases are a group of enzymes that phosphorylate tyrosine, threonine, and/or serine residues of target proteins altering their function. Susceptible cysteines in some serine/theonine protein kinases are directly modified by ROS. Protein kinase C (PKC) contains a cysteine rich domain susceptible to oxidation, while oxidation of Cys-245 and Cys-487 in the kinase domain of the nonreceptor tyrosine kinase Src results in the activation of the protein [210]. Mitogen-activated protein kinases (MAPKs) are a family of serine/threonine kinases that play a central role in coupling various extracellular signals to a variety of biological processes, such as gene expression, cell proliferation, differentiation, and cell death. The activity of MAP kinases (ERK, c-Jun and p38) is regulated by phosphorylation cascades: MAPKs activation is induced through the phosphorylation of their threonyl and tyrosyl residues within a tripeptide motif TXY by a dual specificity kinase termed MAP kinase kinase (MKK), which in turn is phosphorylated and activated by an upstream kinase called MAPK kinase kinase (MAPKKK) [211]. However, MAPKs can also be activated by ROS.
In vascular smooth muscle cells, intracellular ROS are critical for AII-induced activation of p38MAPK, JNK and ERK5, whereas phosphorylation of ERK1/2 activation in smooth muscle cells is redox sensitive, in contrast to fibroblasts, suggesting that redox-regulation of MAP kinases may be ligand- and cell-specific [212].
Many growth factor and cytokine receptors bearing cysteine-rich motifs can be targets of oxidative stress. Thus, insulin-like growth factor-I (IGF-I) activates ERK pathway through ROS-mediated activation of EGF receptor, which by mediation of Ras-GTPase activates Raf and MEK, phosphorylating ERK [213]. ROS may also activate MAPK pathways through the oxidative modification of the intracellular kinases. ASK-1, a member of the MAP3K superfamily for JNK and p38, binds to reduced thioredoxin in nonstressed cells. Upon oxidative stress, thioredoxin becomes oxidized and dissociates from ASK-1, leading to activation of JNK and p38 pathways through oligomerization of ASK-1 [214].
Another mechanism for MAPK activation by ROS includes the inactivation and degradation of the MAPK phosphatases (MPKs). Kamata et al. reported that H2O2 inactivates MKPs by oxidation of their catalytic cysteine, which leads to sustained activation of the MAPK pathway [71]. However, Zhou et al. found that upregulation of MKP-1 expression by H2O2 correlates with inactivation of JNK and p38 activity [215]. This paradox in the roles of ROS as “inducers” in the regulation of MKP-1 expression and as “inhibitors” may be, at least in part, related to differences in the concentrations of ROS and of the redox sensitivity of Cys residues within MAPK phosphatases.
Some antioxidant responses are also regulated by MAPKs signaling. Activation of mitogen activated protein kinase pathways induces antioxidant response element-mediated gene expression via a Nrf2-dependent mechanism [216]. Prx I is an antioxidant that is up-regulated in a ROS/p38 MAPK-dependent manner [217]. In addition, MAPK regulates NK-κB activation by modulating the phosphorylation of IκB-alpha and p65 NF-κB, stimulating iNOS and AP1 gene expression (Fig. 7) [218]. It also has been reported that mitogen-activated protein kinases (MAPKs) are involved in some polyphenol-mediated regulation Phase II enzymes gene expression and induction of apoptosis [219].
Fig. 7.

Induction of antioxidant responses by ROS-mediated activation of cytosolic cell signaling pathways. ROS drive activation of MAPKs. ERK and JNK are involved in recruiting c-Fos and c-Jun to the nucleus where they activate the transcription factor AP-1, whereas activation of p38 and IKK is important in the transcriptional activation of NF-ĸB. Both of these factors are important in regulating diverse genes, including antioxidants (p53, NQO1, GSTs, SOD2, Ap-1,p53, and Prx I).
5.2. Mitochondrial regulation
Mitochondria are regulated in an intricate manner by oxidants and electrophiles. The activity of several complexes of the electron transport chain is modulated by post-translational modifications such as S-nitrosylation, S-glutathionylation or electrophile additions (Fig. 6). As mentioned above, complex I (NADH ubiquinone oxidoreductase) is modified by nitric oxide or its derivatives [177,178,185,220,221] and glutathione [222–225]. The S-nitrosylation of complex I correlates with a significant loss of activity that is reversed by thiol reductants. S-nitrosylation was also associated with increased superoxide production from complex I [178,185]. The fact that mitochondrial superoxide formation can be regulated by S-nitrosylation of complex I may play an important role in mitochondrial redox signalling [226]. Complex I has two transitional states, the active (A) and the deactive (D) states, and the complex is S-nitrosylated in the D state [179]. The A to D transition may take place during hypoxia and this might be important in the setting of ischemia–reperfusion. Other studies have reported nitrotyrosine modification on the complex [227,228]. In addition, reversible glutathionylation of complex I increases mitochondrial superoxide production [222]. Other electron transport complexes, in particular, complex II (succinate dehydrogenase) and complex V (ATP synthase), have been shown to be modified by reactive species [229,230]. Likewise, mitochondrial matrix proteins such as NADP+-isocitrate dehydrogenase [231,232], α-ketoglutarate dehydrogenase [233,234] and aconitase [230,235], as well as proteins of the intermembrane space such as creatine kinase [236] and cytochrome c [237] can also be modified by reactive species, affecting in most cases their catalytic activity.
In response to stress or to an increased demand in energy, mitochondria produce a limited amount of ROS that act as signaling molecules, initiating a molecular stress response that leads to transcriptional changes in the nucleus. A transient ROS signal generates an endogenous response that allows detoxification of ROS by inducing defence enzymes such as superoxide dismutase or catalase, as well as other stress defence pathways [238]. This process is called retrograde response [239,240]. The release of ROS from mitochondria can function in numerous signalling pathways [241]. Some examples are the regulation of cytosolic stress kinases [71], modulation of hypoxic signalling [242], and activation of macroautophagy [243].
In contrast to this function of ROS as signalling molecules, sustained high levels of ROS can cause intracellular damage. This nonlinear response to mitochondrial ROS is known as mitochondrial hormesis or mitohormesis [240]. This concept considers ROS as essential signalling molecules, and not just harmful and damaging by-products of mitochondrial metabolism.
5.2.1. Targeting mitochondria to eliminate ROS production
Antioxidants can be targeted selectively to mitochondria to reduce oxidative stress. Mitochondria-targeted antioxidants comprise a triphenylphophonium cation (TPP+) covalently attached to an antioxidant, which enables mitochondrial delivery of the compound. The most extensively studied is MitoQ, which contains the antioxidant ubiquinol covalently attached to TPP+. MitoQ decreases cardiac [244] and renal [245] IR injury and it is therefore a potential therapeutic agent against this type of damage. Ongoing clinical trial are evaluating the efficacy of Bendavia, Szeto-Schiller peptides that also improve post-infarction cardiac function and post-IR kidney injury post-IR [246–248]. Another strategy to eliminate harmful ROS generation is the removal of damaged mitochondria, which can produce excessive ROS. Recent studies have focused on the inhibition of Drp1 (dynamin-related protein 1), a large GTPase involved in mitochondrial fission. In particular, the inhibitor mdivi-1 reduces myocardial infarct size in mice undergoing IR, and reduces excessive fragmentation in adult rat cardiomyocytes exposed to simulated IR injury [249]. In addition, a small separation of function peptide, P110, which specifically affects the interaction between Drp1 and fission 1, diminishes excessive mitochondrial fission and reduces neurotoxicity in cells derived from patients with Parkinson disease [250]. Another mechanism to prevent pathological ROS production consists of eliminating damaged mitochondria. After cellular insults such as oxidative stress caused by IR, the elimination of damaged or dysfunctional mitochondria through autophagy (mitophagy) is upregulated by diverse molecular mechanisms [251,252], contributing to the maintenance of mitochondrial quality control. Finally, other mechanisms that maintain mitochondrial quality control are the unfolded protein response and the proteasome machinery, which are also potential therapeutic targets.
6. Conclusion
Since the discovery of ROS there has been increased interest and research into the processes of ROS generation, ROS-mediated effects and functional consequences of ROS driven processes. We now have a relatively comprehensive view of how ROS can be generated, converted into each other and how their action affects lipids, proteins and DNA. A concept that has gained momentum in redox biology and is probably to remain for a long time is the major importance of hydrogen peroxide as the paradigmatic signaling molecule [253]. However, we still need more knowledge about how this signaling proceeds, the antioxidant responses involved and their thresholds to redox perturbations. Moreover, mechanisms of how ROS may be transported or interconverted inside or outside cellular compartments and how these conversions contribute to cellular homeostasis are lacking. The next few years may well provide further examples or may uncover new or alternate mechanisms contributing to redox balance, the antioxidant defense and ROS/peroxide signaling.
References
- 1.Hwang C., Sinskey A.J., Lodish H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257(5076):1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 2.Pastore A., Piemonte F. S-glutathionylation signaling in cell biology: progress and prospects. Eur. J. Pharm. Sci. 2012;46(5):279–292. doi: 10.1016/j.ejps.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 3.Ray P.D., Huang B.W., Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24(5):981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rahman I. Glutathione, stress responses, and redox signaling in lung inflammation. Antioxid. Redox Signal. 2005;7(1–2):42–59. doi: 10.1089/ars.2005.7.42. [DOI] [PubMed] [Google Scholar]
- 5.Haddad J.J., Harb H.L. L-gamma-glutamyl-l-cysteinyl-glycine (glutathione; GSH) and GSH-related enzymes in the regulation of pro- and anti-inflammatory cytokines: a signaling transcriptional scenario for redox(y) immunologic sensor(s)? Mol. Immunol. 2005;42(9):987–1014. doi: 10.1016/j.molimm.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 6.Lu S.C. Glutathione synthesis. Biochim. Biophys. Acta. 2013;1830(5):3143–3153. doi: 10.1016/j.bbagen.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu S.C. Regulation of glutathione synthesis. Mol. Aspects Med. 2009;30(1–2):42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forman H.J., Davies K.J., Ursini F. How do nutritional antioxidants really work: nucleophilic tone and para-hormesis versus free radical scavenging in vivo. Free Radic. Biol. Med. 2014;66:24–35. doi: 10.1016/j.freeradbiomed.2013.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oelze M. Glutathione peroxidase-1 deficiency potentiates dysregulatory modifications of endothelial nitric oxide synthase and vascular dysfunction in aging. Hypertension. 2014;63(2):390–396. doi: 10.1161/HYPERTENSIONAHA.113.01602. [DOI] [PubMed] [Google Scholar]
- 10.Gallogly M.M., Mieyal J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007;7(4):381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Martinez-Ruiz A., Lamas S. Signalling by NO-induced protein S-nitrosylation and S-glutathionylation: convergences and divergences. Cardiovasc. Res. 2007;75(2):220–228. doi: 10.1016/j.cardiores.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 12.Grek C.L. Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem. 2013;288(37):26497–26504. doi: 10.1074/jbc.R113.461368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen C.A. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468(7327):1115–1118. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Zwieten R., Verhoeven A.J., Roos D. Inborn defects in the antioxidant systems of human red blood cells. Free Radic. Biol. Med. 2014;67:377–386. doi: 10.1016/j.freeradbiomed.2013.11.022. [DOI] [PubMed] [Google Scholar]
- 15.Lubos E., Loscalzo J., Handy D.E. Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011;15(7):1957–1997. doi: 10.1089/ars.2010.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imai H., Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic. Biol. Med. 2003;34(2):145–169. doi: 10.1016/s0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- 17.Rayman M.P. Selenium and human health. Lancet. 2012;379(9822):1256–1268. doi: 10.1016/S0140-6736(11)61452-9. [DOI] [PubMed] [Google Scholar]
- 18.Chen H., Tappel A.L. Protection of vitamin E, selenium, trolox C, ascorbic acid palmitate, acetylcysteine, coenzyme Q0, coenzyme Q10, beta-carotene, canthaxanthin, and (+)-catechin against oxidative damage to rat blood and tissues in vivo. Free Radic. Biol. Med. 1995;18(5):949–953. doi: 10.1016/0891-5849(94)00238-f. [DOI] [PubMed] [Google Scholar]
- 19.Fiala E.S. Inhibition of 2-nitropropane-induced rat liver DNA and RNA damage by benzyl selenocyanate. Carcinogenesis. 1997;18(9):1809–1815. doi: 10.1093/carcin/18.9.1809. [DOI] [PubMed] [Google Scholar]
- 20.Leibovitz B., Hu M.L., Tappel A.L. Dietary supplements of vitamin E, beta-carotene, coenzyme Q10 and selenium protect tissues against lipid peroxidation in rat tissue slices. J. Nutr. 1990;120(1):97–104. doi: 10.1093/jn/120.1.97. [DOI] [PubMed] [Google Scholar]
- 21.Fernandes A.P., Holmgren A. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid. Redox Signal. 2004;6(1):63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- 22.Wood Z.A. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003;28(1):32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 23.Wood Z.A., Poole L.B., Karplus P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300(5619):650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 24.Hofmann B., Hecht H.J., Flohe L. Peroxiredoxins. Biol. Chem. 2002;383(3–4):347–364. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- 25.Fujii J., Ikeda Y. Advances in our understanding of peroxiredoxin, a multifunctional, mammalian redox protein. Redox Rep. 2002;7(3):123–130. doi: 10.1179/135100002125000352. [DOI] [PubMed] [Google Scholar]
- 26.Oberley T.D. Localization of the thioredoxin system in normal rat kidney. Free Radic. Biol. Med. 2001;30(4):412–424. doi: 10.1016/s0891-5849(00)00486-x. [DOI] [PubMed] [Google Scholar]
- 27.Immenschuh S. Differential cellular and subcellular localization of heme-binding protein 23/peroxiredoxin I and heme oxygenase-1 in rat liver. J. Histochem. Cytochem. 2003;51(12):1621–1631. doi: 10.1177/002215540305101206. [DOI] [PubMed] [Google Scholar]
- 28.Seo M.S. Identification of a new type of mammalian peroxiredoxin that forms an intramolecular disulfide as a reaction intermediate. J. Biol. Chem. 2000;275(27):20346–20354. doi: 10.1074/jbc.M001943200. [DOI] [PubMed] [Google Scholar]
- 29.Winterbourn C.C., Metodiewa D. The reaction of superoxide with reduced glutathione. Arch. Biochem. Biophys. 1994;314(2):284–290. doi: 10.1006/abbi.1994.1444. [DOI] [PubMed] [Google Scholar]
- 30.Winterbourn C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008;4(5):278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 31.Marinho H.S. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014;2:535–562. doi: 10.1016/j.redox.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holmgren A. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem. Soc. Trans. 2005;33(6):1375–1377. doi: 10.1042/BST0331375. [DOI] [PubMed] [Google Scholar]
- 33.Go Y.M., Jones D.P. Redox control systems in the nucleus: mechanisms and functions. Antioxid. Redox Signal. 2010;13(4):489–509. doi: 10.1089/ars.2009.3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCord J.M., Fridovich I. Superoxide dismutases: you’ve come a long way, baby. Antioxid. Redox Signal. 2014;20(10):1548–1549. doi: 10.1089/ars.2013.5547. [DOI] [PubMed] [Google Scholar]
- 35.Hileman E.A., Achanta G., Huang P. Superoxide dismutase: an emerging target for cancer therapeutics. Expert Opin. Ther. Targets. 2001;5(6):697–710. doi: 10.1517/14728222.5.6.697. [DOI] [PubMed] [Google Scholar]
- 36.Konzack A., Kietzmann T. Manganese superoxide dismutase in carcinogenesis: friend or foe? Biochem. Soc. Trans. 2014;42(4):1012–1016. doi: 10.1042/BST20140076. [DOI] [PubMed] [Google Scholar]
- 37.Gongora M.C. Role of extracellular superoxide dismutase in hypertension. Hypertension. 2006;48(3):473–481. doi: 10.1161/01.HYP.0000235682.47673.ab. [DOI] [PubMed] [Google Scholar]
- 38.Motohashi H., Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004;10(11):549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 39.Baird L., Dinkova-Kostova A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011;85(4):241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- 40.Motohashi H. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene. 2002;294(1–2):1–12. doi: 10.1016/s0378-1119(02)00788-6. [DOI] [PubMed] [Google Scholar]
- 41.Zhang D.D. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004;24(24):10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McMahon M. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 2006;281(34):24756–24768. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- 43.Tong K.I. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol. Chem. 2006;387(10–11):1311–1320. doi: 10.1515/BC.2006.164. [DOI] [PubMed] [Google Scholar]
- 44.McMahon M. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004;279(30):31556–31567. doi: 10.1074/jbc.M403061200. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen T. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26S proteasome. J. Biol. Chem. 2003;278(7):4536–4541. doi: 10.1074/jbc.M207293200. [DOI] [PubMed] [Google Scholar]
- 46.Sun Z. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol. Cell. Biol. 2007;27(18):6334–6349. doi: 10.1128/MCB.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kobayashi A. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004;24(16):7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cullinan S.B. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004;24(19):8477–8486. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Furukawa M., Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 2005;25(1):162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kobayashi A. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006;26(1):221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dinkova-Kostova A.T. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA. 2002;99(18):11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fourquet S. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J. Biol. Chem. 2010;285(11):8463–8471. doi: 10.1074/jbc.M109.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yamamoto T. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol. 2008;28(8):2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rushmore T.H., Morton M.R., Pickett C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991;266(18):11632–11639. [PubMed] [Google Scholar]
- 55.Ma Q., He X. Molecular basis of electrophilic and oxidative defense: promises and perils of Nrf2. Pharmacol. Rev. 2012;64(4):1055–1081. doi: 10.1124/pr.110.004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen W. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell. 2009;34(6):663–673. doi: 10.1016/j.molcel.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Komatsu M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010;12(3):213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 58.Chowdhry S. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene. 2013;32(32):3765–3781. doi: 10.1038/onc.2012.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Malhotra D. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010;38(17):5718–5734. doi: 10.1093/nar/gkq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma Q. Induction of murine NAD(P)H:quinone oxidoreductase by 2,3,7,8-tetrachlorodibenzo-p-dioxin requires the CNC (cap ‘n’ collar) basic leucine zipper transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2): cross-interaction between AhR (aryl hydrocarbon receptor) and Nrf2 signal transduction. Biochem. J. 2004;377(1):205–213. doi: 10.1042/BJ20031123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iida K. Nrf2 and p53 cooperatively protect against BBN-induced urinary bladder carcinogenesis. Carcinogenesis. 2007;28(11):2398–2403. doi: 10.1093/carcin/bgm146. [DOI] [PubMed] [Google Scholar]
- 62.Li W. Activation of Nrf2-antioxidant signaling attenuates NFkappaB-inflammatory response and elicits apoptosis. Biochem. Pharmacol. 2008;76(11):1485–1489. doi: 10.1016/j.bcp.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jain A.K., Mahajan S., Jaiswal A.K. Phosphorylation and dephosphorylation of tyrosine 141 regulate stability and degradation of INrf2: a novel mechanism in Nrf2 activation. J. Biol. Chem. 2008;283(25):17712–17720. doi: 10.1074/jbc.M709854200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Wakabayashi N. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003;35(3):238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 65.Jiang T. High levels of Nrf2 determine chemoresistance in type II endometrial cancer. Cancer Res. 2010;70(13):5486–5496. doi: 10.1158/0008-5472.CAN-10-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ghosh S., May M.J., Kopp E.B. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 67.Oliveira-Marques V. Role of hydrogen peroxide in NF-kappaB activation: from inducer to modulator. Antioxid. Redox Signal. 2009;11(9):2223–2243. doi: 10.1089/ars.2009.2601. [DOI] [PubMed] [Google Scholar]
- 68.Jung Y. Dynein light chain LC8 negatively regulates NF-kappaB through the redox-dependent interaction with IkappaBalpha. J. Biol. Chem. 2008;283(35):23863–23871. doi: 10.1074/jbc.M803072200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee D.F. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol. Cell. 2009;36(1):131–140. doi: 10.1016/j.molcel.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pham C.G. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119(4):529–542. doi: 10.1016/j.cell.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 71.Kamata H. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120(5):649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 72.Rius J. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453(7196):807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ma Q. Inhibition of nuclear factor kappaB by phenolic antioxidants: interplay between antioxidant signaling and inflammatory cytokine expression. Mol. Pharmacol. 2003;64(2):211–219. doi: 10.1124/mol.64.2.211. [DOI] [PubMed] [Google Scholar]
- 74.Karin M. Oxidative stress and gene expression: the AP-1 and NF-kappaB connections. Biofactors. 2001;15(2–4):87–89. doi: 10.1002/biof.5520150207. [DOI] [PubMed] [Google Scholar]
- 75.Whitmarsh A.J., Davis R.J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. (Berl) 1996;74(10):589–607. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- 76.Pinkus R., Weiner L.M., Daniel V. Role of oxidants and antioxidants in the induction of AP-1, NF-kappaB, and glutathione S-transferase gene expression. J. Biol. Chem. 1996;271(23):13422–13429. doi: 10.1074/jbc.271.23.13422. [DOI] [PubMed] [Google Scholar]
- 77.Kowaltowski A.J., Castilho R.F., Vercesi A.E. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001;495(1–2):12–15. doi: 10.1016/s0014-5793(01)02316-x. [DOI] [PubMed] [Google Scholar]
- 78.Ronchi J.A., Vercesi A.E., Castilho R.F. Reactive oxygen species and permeability transition pore in rat liver and kidney mitoplasts. J. Bioenergy Biomembr. 2011;43(6):709–715. doi: 10.1007/s10863-011-9384-1. [DOI] [PubMed] [Google Scholar]
- 79.Halestrap A.P., Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Biophys. Acta. 2009;1787(11):1402–1415. doi: 10.1016/j.bbabio.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 80.Lee R.C., Feinbaum R.L., Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 81.Wightman B., Ha I., Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75(5):855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 82.Kozomara A., Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42(Database issue):D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim W. Histone acetyltransferase GCN5 interferes with the miRNA pathway in Arabidopsis. Cell Res. 2009;19(7):899–909. doi: 10.1038/cr.2009.59. [DOI] [PubMed] [Google Scholar]
- 84.Ying S.Y., Lin S.L. Intron-derived microRNAs—fine tuning of gene functions. Gene. 2004;342(1):25–28. doi: 10.1016/j.gene.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 85.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 86.Lagos-Quintana M. Identification of novel genes coding for small expressed RNAs. Science. 2001;294(5543):853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 87.Bartel D.P. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cheng X., Ku C.H., Siow R.C. Regulation of the Nrf2 antioxidant pathway by microRNAs: new players in micromanaging redox homeostasis. Free Radic. Biol. Med. 2013;64:4–11. doi: 10.1016/j.freeradbiomed.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 89.Stachurska A. Cross-talk between microRNAs, nuclear factor E2-related factor 2, and heme oxygenase-1 in ochratoxin A-induced toxic effects in renal proximal tubular epithelial cells. Mol. Nutr. Food Res. 2013;57(3):504–515. doi: 10.1002/mnfr.201200456. [DOI] [PubMed] [Google Scholar]
- 90.Narasimhan M. Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal SH-SY5Y cells. PLoS One. 2012;7(12):e51111. doi: 10.1371/journal.pone.0051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Papp D. The NRF2-related interactome and regulome contain multifunctional proteins and fine-tuned autoregulatory loops. FEBS Lett. 2012;586(13):1795–1802. doi: 10.1016/j.febslet.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eades G. miR-200 a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J. Biol. Chem. 2011;286(47):40725–40733. doi: 10.1074/jbc.M111.275495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hou W. MicroRNA-196 represses Bach1 protein and hepatitis C virus gene expression in human hepatoma cells expressing hepatitis C viral proteins. Hepatology. 2010;51(5):1494–1504. doi: 10.1002/hep.23401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sangokoya C., Telen M.J., Chi J.T. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood. 2010;116(20):4338–4348. doi: 10.1182/blood-2009-04-214817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang M. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res. Treat. 2011;129(3):983–991. doi: 10.1007/s10549-011-1604-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang H. Activation of a novel c-Myc-miR27-prohibitin 1 circuitry in cholestatic liver injury inhibits glutathione synthesis in mice. Antioxid. Redox Signal. 2015;22(3):259–274. doi: 10.1089/ars.2014.6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Do M.T. Metformin induces microRNA-34 a to downregulate the Sirt1/Pgc-1alpha/Nrf2 pathway, leading to increased susceptibility of wild-type p53 cancer cells to oxidative stress and therapeutic agents. Free Radic. Biol. Med. 2014;74:21–34. doi: 10.1016/j.freeradbiomed.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 98.Huang X. The role of miR-34 a in the hepatoprotective effect of hydrogen sulfide on ischemia/reperfusion injury in young and old rats. PLoS One. 2014;9(11):e113305. doi: 10.1371/journal.pone.0113305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li N. Increased expression of miR-34 a and miR-93 in rat liver during aging, and their impact on the expression of Mgst1 and Sirt1. Mech. Ageing Dev. 2011;132(3):75–85. doi: 10.1016/j.mad.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 100.Yang J.J. MicroRNA-200 a controls Nrf2 activation by target Keap1 in hepatic stellate cell proliferation and fibrosis. Cell Signal. 2014;26(11):2381–2389. doi: 10.1016/j.cellsig.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 101.Pulkkinen K.H., Yla-Herttuala S., Levonen A.L. Heme oxygenase 1 is induced by miR-155 via reduced BACH1 translation in endothelial cells. Free Radic. Biol. Med. 2011;51(11):2124–2131. doi: 10.1016/j.freeradbiomed.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 102.Boesch-Saadatmandi C. Effect of quercetin and its metabolites isorhamnetin and quercetin-3-glucuronide on inflammatory gene expression: role of miR-155. J. Nutr. Biochem. 2011;22(3):293–299. doi: 10.1016/j.jnutbio.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 103.Li S. microRNA-155 silencing inhibits proliferation and migration and induces apoptosis by upregulating BACH1 in renal cancer cells. Mol. Med. Rep. 2012;5(4):949–954. doi: 10.3892/mmr.2012.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Espinosa-Diez C. Targeting of gamma-glutamyl-cysteine ligase by miR-433 reduces glutathione biosynthesis and promotes TGF-beta-dependent fibrogenesis. Antioxid. Redox Signal. 2014 doi: 10.1089/ars.2014.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ungvari Z. Aging-induced dysregulation of dicer1-dependent microRNA expression impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J. Gerontol. A Biol. Sci. Med. Sci. 2013;68(8):877–891. doi: 10.1093/gerona/gls242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wiesen J.L., Tomasi T.B. Dicer is regulated by cellular stresses and interferons. Mol. Immunol. 2009;46(6):1222–1228. doi: 10.1016/j.molimm.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.O’Connell R.M. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010;10(2):111–122. doi: 10.1038/nri2708. [DOI] [PubMed] [Google Scholar]
- 108.Leung A.K., Sharp P.A. MicroRNA functions in stress responses. Mol. Cell. 2010;40(2):205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sato F. MicroRNAs and epigenetics. FEBS J. 2011;278(10):1598–1609. doi: 10.1111/j.1742-4658.2011.08089.x. [DOI] [PubMed] [Google Scholar]
- 110.Cyr A.R., Domann F.E. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid. Redox Signal. 2011;15(2):551–589. doi: 10.1089/ars.2010.3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fu Y. Regulation of NADPH oxidase activity is associated with miRNA-25-mediated NOX4 expression in experimental diabetic nephropathy. Am. J. Nephrol. 2010;32(6):581–589. doi: 10.1159/000322105. [DOI] [PubMed] [Google Scholar]
- 112.Chan S.Y. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009;10(4):273–284. doi: 10.1016/j.cmet.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tak H. miR-27 regulates mitochondrial networks by directly targeting the mitochondrial fission factor. Exp. Mol. Med. 2014;46:e123. doi: 10.1038/emm.2014.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bai X.Y. miR-335 and miR-34 a Promote renal senescence by suppressing mitochondrial antioxidative enzymes. J. Am. Soc. Nephrol. 2011;22(7):1252–1261. doi: 10.1681/ASN.2010040367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li R. MicroRNA-145 protects cardiomyocytes against hydrogen peroxide (H(2)O(2))-induced apoptosis through targeting the mitochondria apoptotic pathway. PLoS One. 2012;7(9):e44907. doi: 10.1371/journal.pone.0044907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang H. MicroRNA-181 c targets Bcl-2 and regulates mitochondrial morphology in myocardial cells. J. Cell Mol. Med. 2015 doi: 10.1111/jcmm.12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dang K., Myers K.A. The role of hypoxia-induced miR-210 in cancer progression. Int. J. Mol. Sci. 2015;16(3):6353–6372. doi: 10.3390/ijms16036353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yamamoto H. MicroRNA-494 regulates mitochondrial biogenesis in skeletal muscle through mitochondrial transcription factor A and Forkhead box j3. Am. J. Physiol. Endocrinol. Metab. 2012;303(12):E1419–E1427. doi: 10.1152/ajpendo.00097.2012. [DOI] [PubMed] [Google Scholar]
- 119.Bartoszewska S. Regulation of the unfolded protein response by microRNAs. Cell. Mol. Biol. Lett. 2013;18(4):555–578. doi: 10.2478/s11658-013-0106-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Byrd A.E., Brewer J.W. Micro(RNA)managing endoplasmic reticulum stress. IUBMB Life. 2013;65(5):373–381. doi: 10.1002/iub.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Greco S., Gaetano C., Martelli F. HypoxamiR regulation and function in ischemic cardiovascular diseases. Antioxid. Redox Signal. 2014;21(8):1202–1219. doi: 10.1089/ars.2013.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yao M. Dicer mediating the expression of miR-143 and miR-155 regulates hexokinase II associated cellular response to hypoxia. Am. J. Physiol.—Lung Cell. Mol. Physiol. 2014;307(11):L829–L837. doi: 10.1152/ajplung.00081.2014. [DOI] [PubMed] [Google Scholar]
- 123.Qin Q., Furong W., Baosheng L. Multiple functions of hypoxia-regulated miR-210 in cancer. J. Exp. Clin. Cancer Res. 2014;33:50. doi: 10.1186/1756-9966-33-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rink C., Khanna S. MicroRNA in ischemic stroke etiology and pathology. Physiol. Genomics. 2011;43(10):521–528. doi: 10.1152/physiolgenomics.00158.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kauppinen A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013;25(10):1939–1948. doi: 10.1016/j.cellsig.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 126.Song M.A. Differential expression of microRNAs in ischemic heart disease. Drug Discov. Today. 2015;20(2):223–235. doi: 10.1016/j.drudis.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kim H.J. Carbon monoxide protects against hepatic ischemia/reperfusion injury by modulating the miR-34 a/SIRT1 pathway. Biochim. Biophys. Acta. 2015;1852(7):1550–1559. doi: 10.1016/j.bbadis.2015.04.017. [DOI] [PubMed] [Google Scholar]
- 128.Forman H.J., Traber M., Ursini F. Antioxidants: GRABbing new headlines. Free Radic. Biol. Med. 2014;66:1–2. doi: 10.1016/j.freeradbiomed.2013.10.808. [DOI] [PubMed] [Google Scholar]
- 129.Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–183. doi: 10.1016/j.redox.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jones D.P., Sies H. The redox code. Antioxid. Redox Signal. 2015 doi: 10.1089/ars.2015.6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.van der Vlies D. Oxidation of ER resident proteins upon oxidative stress: effects of altering cellular redox/antioxidant status and implications for protein maturation. Antioxid. Redox Signal. 2003;5(4):381–387. doi: 10.1089/152308603768295113. [DOI] [PubMed] [Google Scholar]
- 132.Fewell S.W. The action of molecular chaperones in the early secretory pathway. Annu. Rev. Genet. 2001;35:149–191. doi: 10.1146/annurev.genet.35.102401.090313. [DOI] [PubMed] [Google Scholar]
- 133.Gorlach A., Klappa P., Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 2006;8(9–10):1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- 134.Ramming T., Appenzeller-Herzog C. The physiological functions of mammalian endoplasmic oxidoreductin 1: on disulfides and more. Antioxid. Redox Signal. 2012;16(10):1109–1118. doi: 10.1089/ars.2011.4475. [DOI] [PubMed] [Google Scholar]
- 135.Hatahet F., Ruddock L.W. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 2009;11(11):2807–2850. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 136.Dorner A.J., Wasley L.C., Kaufman R.J. Increased synthesis of secreted proteins induces expression of glucose-regulated proteins in butyrate-treated Chinese hamster ovary cells. J. Biol. Chem. 1989;264(34):20602–20607. [PubMed] [Google Scholar]
- 137.Kozutsumi Y. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332(6163):462–464. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- 138.Harding H.P., Zhang Y., Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 139.Travers K.J. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101(3):249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 140.Kim P.S., Arvan P. Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: disorders of protein trafficking and the role of ER molecular chaperones. Endocrine Rev. 1998;19(2):173–202. doi: 10.1210/edrv.19.2.0327. [DOI] [PubMed] [Google Scholar]
- 141.Aridor M., Hannan L.A. Traffic jams II: an update of diseases of intracellular transport. Traffic (Copenhagen, Denmark) 2002;3(11):781–790. doi: 10.1034/j.1600-0854.2002.31103.x. [DOI] [PubMed] [Google Scholar]
- 142.Cullinan S.B., Diehl J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004;279(19):20108–20117. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- 143.Tu B.P., Weissman J.S. Oxidative protein folding in eukaryotes: mechanisms and consequences. J. Cell Biol. 2004;164(3):341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Harding H.P. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 2003;11(3):619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 145.Cabibbo A. ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. J. Biol. Chem. 2000;275(7):4827–4833. doi: 10.1074/jbc.275.7.4827. [DOI] [PubMed] [Google Scholar]
- 146.Pagani M. Endoplasmic reticulum oxidoreductin 1-lbeta (ERO1-Lbeta), a human gene induced in the course of the unfolded protein response. J. Biol. Chem. 2000;275(31):23685–23692. doi: 10.1074/jbc.M003061200. [DOI] [PubMed] [Google Scholar]
- 147.Pagani M. The C-terminal domain of yeast Ero1p mediates membrane localization and is essential for function. FEBS Lett. 2001;508(1):117–120. doi: 10.1016/s0014-5793(01)03034-4. [DOI] [PubMed] [Google Scholar]
- 148.Mezghrani A. Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J. 2001;20(22):6288–6296. doi: 10.1093/emboj/20.22.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Molteni S.N. Glutathione limits Ero1-dependent oxidation in the endoplasmic reticulum. J. Biol. Chem. 2004;279(31):32667–32673. doi: 10.1074/jbc.M404992200. [DOI] [PubMed] [Google Scholar]
- 150.Gilady S.Y. Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM) Cell Stress Chaperones. 2010;15(5):619–629. doi: 10.1007/s12192-010-0174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tu B.P., Weissman J.S. The FAD- and O(2)-dependent reaction cycle of Ero1-mediated oxidative protein folding in the endoplasmic reticulum. Mol. Cell. 2002;10(5):983–994. doi: 10.1016/s1097-2765(02)00696-2. [DOI] [PubMed] [Google Scholar]
- 152.Gross E. Structure of Ero1p, source of disulfide bonds for oxidative protein folding in the cell. Cell. 2004;117(5):601–610. doi: 10.1016/s0092-8674(04)00418-0. [DOI] [PubMed] [Google Scholar]
- 153.Tavender T.J., Springate J.J., Bulleid N.J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 2010;29(24):4185–4197. doi: 10.1038/emboj.2010.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zito E. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Mol. Cell. 2010;40(5):787–797. doi: 10.1016/j.molcel.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ozawa K. Expression of the oxygen-regulated protein ORP150 accelerates wound healing by modulating intracellular VEGF transport. J. Clin. Investig. 2001;108(1):41–50. doi: 10.1172/JCI11772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tobisawa S. Mutant SOD1 linked to familial amyotrophic lateral sclerosis, but not wild-type SOD1, induces ER stress in COS7 cells and transgenic mice. Biochem. Biophys. Res. Commun. 2003;303(2):496–503. doi: 10.1016/s0006-291x(03)00353-x. [DOI] [PubMed] [Google Scholar]
- 157.Tu P.H. Oxidative stress, mutant SOD1, and neurofilament pathology in transgenic mouse models of human motor neuron disease. Lab. Investig. J. Tech. Methods Pathol. 1997;76(4):441–456. [PubMed] [Google Scholar]
- 158.Bruening W. Up-regulation of protein chaperones preserves viability of cells expressing toxic Cu/Zn-superoxide dismutase mutants associated with amyotrophic lateral sclerosis. J. Neurochem. 1999;72(2):693–699. doi: 10.1046/j.1471-4159.1999.0720693.x. [DOI] [PubMed] [Google Scholar]
- 159.Wong P.C. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14(6):1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 160.Shiu R.P., Pouyssegur J., Pastan I. Glucose depletion accounts for the induction of two transformation-sensitive membrane proteinsin Rous sarcoma virus-transformed chick embryo fibroblasts. Proc. Natl. Acad. Sci. USA. 1977;74(9):3840–3844. doi: 10.1073/pnas.74.9.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Heacock C.S., Sutherland R.M. Enhanced synthesis of stress proteins caused by hypoxia and relation to altered cell growth and metabolism. Br. J. Cancer. 1990;62(2):217–225. doi: 10.1038/bjc.1990.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ozawa K. 150-kDa oxygen-regulated protein (ORP150) suppresses hypoxia-induced apoptotic cell death. J. Biol. Chem. 1999;274(10):6397–6404. doi: 10.1074/jbc.274.10.6397. [DOI] [PubMed] [Google Scholar]
- 163.Blais J.D. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol. Cell. Biol. 2004;24(17):7469–7482. doi: 10.1128/MCB.24.17.7469-7482.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Estes S.D., Stoler D.L., Anderson G.R. Normal fibroblasts induce the C/EBP beta and ATF-4 bZIP transcription factors in response to anoxia. Exp. Cell Res. 1995;220(1):47–54. doi: 10.1006/excr.1995.1290. [DOI] [PubMed] [Google Scholar]
- 165.Liu Q. A Fenton reaction at the endoplasmic reticulum is involved in the redox control of hypoxia-inducible gene expression. Proc. Natl. Acad. Sci. USA. 2004;101(12):4302–4307. doi: 10.1073/pnas.0400265101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Koivunen P. An endoplasmic reticulum transmembrane prolyl 4-hydroxylase is induced by hypoxia and acts on hypoxia-inducible factor alpha. J. Biol. Chem. 2007;282(42):30544–30552. doi: 10.1074/jbc.M704988200. [DOI] [PubMed] [Google Scholar]
- 167.Gess B. The cellular oxygen tension regulates expression of the endoplasmic oxidoreductase ERO1-Lalpha. Eur. J. Biochem./FEBS. 2003;270(10):2228–2235. doi: 10.1046/j.1432-1033.2003.03590.x. [DOI] [PubMed] [Google Scholar]
- 168.Xue X. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J. Biol. Chem. 2005;280(40):33917–33925. doi: 10.1074/jbc.M505818200. [DOI] [PubMed] [Google Scholar]
- 169.Yoneda T. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J. Biol. Chem. 2001;276(17):13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- 170.Nakagawa T. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403(6765):98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 171.Rao R.V. Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J. Biol. Chem. 2001;276(36):33869–33874. doi: 10.1074/jbc.M102225200. [DOI] [PubMed] [Google Scholar]
- 172.Matsuzawa A. Physiological roles of ASK1-mediated signal transduction in oxidative stress- and endoplasmic reticulum stress-induced apoptosis: advanced findings from ASK1 knockout mice. Antioxid. Redox Signal. 2002;4(3):415–425. doi: 10.1089/15230860260196218. [DOI] [PubMed] [Google Scholar]
- 173.Nishitoh H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16(11):1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Urano F. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 175.Tobiume K. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2(3):222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Oono K. JAB1 participates in unfolded protein responses by association and dissociation with IRE1. Neurochem. Int. 2004;45(5):765–772. doi: 10.1016/j.neuint.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 177.Burwell L.S. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem. J. 2006;394(3):627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Dahm C.C., Moore K., Murphy M.P. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: implications for the interaction of nitric oxide with mitochondria. J. Biol. Chem. 2006;281(15):10056–10065. doi: 10.1074/jbc.M512203200. [DOI] [PubMed] [Google Scholar]
- 179.Galkin A., Moncada S. S-nitrosation of mitochondrial complex I depends on its structural conformation. J. Biol. Chem. 2007;282(52):37448–37453. doi: 10.1074/jbc.M707543200. [DOI] [PubMed] [Google Scholar]
- 180.Shiva S. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J. Exp. Med. 2007;204(9):2089–2102. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nadtochiy S.M., Burwell L.S., Brookes P.S. Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia–reperfusion injury. J. Mol. Cell. Cardiol. 2007;42(4):812–825. doi: 10.1016/j.yjmcc.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Murphy M.P. Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 2012;16(6):476–495. doi: 10.1089/ars.2011.4289. [DOI] [PubMed] [Google Scholar]
- 183.Prime T.A. A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia–reperfusion injury. Proc. Natl. Acad. Sci. USA. 2009;106(26):10764–10769. doi: 10.1073/pnas.0903250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chouchani E.T. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013;19(6):753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Borutaite V., Brown G.C. S-nitrosothiol inhibition of mitochondrial complex I causes a reversible increase in mitochondrial hydrogen peroxide production. Biochim. Biophys. Acta. 2006;1757(5–6):562–566. doi: 10.1016/j.bbabio.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 186.Skyschally A. Across-species transfer of protection by remote ischemic preconditioning with species-specific myocardial signal transduction by RISK and SAFE pathways. Circ. Res. 2015 doi: 10.1161/CIRCRESAHA.117.306878. [DOI] [PubMed] [Google Scholar]
- 187.Imai S. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403(6771):795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 188.Shinmura K. Post-translational modification of mitochondrial proteins by caloric restriction: possible involvement in caloric restriction-induced cardioprotection. Trends Cardiovasc. Med. 2013;23(1):18–25. doi: 10.1016/j.tcm.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 189.Anedda A. The transcription factor Nrf2 promotes survival by enhancing the expression of uncoupling protein 3 under conditions of oxidative stress. Free Radic. Biol. Med. 2013;61:395–407. doi: 10.1016/j.freeradbiomed.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 190.Leonard M.O. Reoxygenation-specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia–reperfusion injury. FASEB J. 2006;20(14):2624–2626. doi: 10.1096/fj.06-5097fje. [DOI] [PubMed] [Google Scholar]
- 191.Wu J.X. Curcumin pretreatment and post-treatment both improve the antioxidative ability of neurons with oxygen-glucose deprivation. Neural Regen. Res. 2015;10(3):481–489. doi: 10.4103/1673-5374.153700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wang N.P. Curcumin promotes cardiac repair and ameliorates cardiac dysfunction following myocardial infarction. Br. J. Pharmacol. 2012;167(7):1550–1562. doi: 10.1111/j.1476-5381.2012.02109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Trujillo J. Renoprotective effect of the antioxidant curcumin: recent findings. Redox Biol. 2013;1:448–456. doi: 10.1016/j.redox.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Li W. Novel curcumin analogue 14 p protects against myocardial ischemia reperfusion injury through Nrf2-activating anti-oxidative activity. Toxicol. Appl. Pharmacol. 2015;282(2):175–183. doi: 10.1016/j.taap.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 195.Zhao H.D. Sulforaphane protects liver injury induced by intestinal ischemia reperfusion through Nrf2-ARE pathway. World J. Gastroenterol. 2010;16(24):3002–3010. doi: 10.3748/wjg.v16.i24.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Soane L. Sulforaphane protects immature hippocampal neurons against death caused by exposure to hemin or to oxygen and glucose deprivation. J. Neurosci. Res. 2010;88(6):1355–1363. doi: 10.1002/jnr.22307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Danilov C.A. Sulforaphane protects astrocytes against oxidative stress and delayed death caused by oxygen and glucose deprivation. Glia. 2009;57(6):645–656. doi: 10.1002/glia.20793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Piao C.S. Sulforaphane protects ischemic injury of hearts through antioxidant pathway and mitochondrial K(ATP) channels. Pharmacol. Res. 2010;61(4):342–348. doi: 10.1016/j.phrs.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 199.Yoon H.Y. Sulforaphane protects kidneys against ischemia–reperfusion injury through induction of the Nrf2-dependent phase 2 enzyme. Biochem. Pharmacol. 2008;75(11):2214–2223. doi: 10.1016/j.bcp.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 200.Li Z. Sulforaphane protects hearts from early injury after experimental transplantation. Ann. Transplant. 2013;18:558–566. doi: 10.12659/AOT.889342. [DOI] [PubMed] [Google Scholar]
- 201.Anderson E.J., Katunga L.A., Willis M.S. Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clin. Exp. Pharmacol. Physiol. 2012;39(2):179–193. doi: 10.1111/j.1440-1681.2011.05641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ma H. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur. Heart J. 2011;32(8):1025–1038. doi: 10.1093/eurheartj/ehq253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zhang Y. 4-hydroxy-2-nonenal protects against cardiac ischemia–reperfusion injury via the Nrf2-dependent pathway. J. Mol. Cell. Cardiol. 2010;49(4):576–586. doi: 10.1016/j.yjmcc.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 204.Lopez-Bernardo E. 4-Hydroxynonenal induces Nrf2-mediated UCP3 upregulation in mouse cardiomyocytes. Free Radic. Biol. Med. 2015 doi: 10.1016/j.freeradbiomed.2015.03.032. [DOI] [PubMed] [Google Scholar]
- 205.Cordeiro J.V., Jacinto A. The role of transcription-independent damage signals in the initiation of epithelial wound healing. Nat. Rev. Mol. Cell Biol. 2013;14(4):249–262. doi: 10.1038/nrm3541. [DOI] [PubMed] [Google Scholar]
- 206.den Hertog J., Groen A., van der Wijk T. Redox regulation of protein-tyrosine phosphatases. Arch. Biochem. Biophys. 2005;434(1):11–15. doi: 10.1016/j.abb.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 207.Salmeen A. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423(6941):769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 208.van Montfort R.L. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423(6941):773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 209.Claiborne A. Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38(47):15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 210.Gopalakrishna R., Jaken S. Protein kinase C signaling and oxidative stress. Free Radic. Biol. Med. 2000;28(9):1349–1361. doi: 10.1016/s0891-5849(00)00221-5. [DOI] [PubMed] [Google Scholar]
- 211.Marshall C.J. MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr. Opin. Genet. Dev. 1994;4(1):82–89. doi: 10.1016/0959-437x(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 212.Catarzi S. Redox regulation of ERK1/2 activation induced by sphingosine 1-phosphate in fibroblasts: involvement of NADPH oxidase and platelet-derived growth factor receptor. Biochim. Biophys. Acta. 2011;1810(4):446–456. doi: 10.1016/j.bbagen.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 213.Meng D. Insulin-like growth factor-I (IGF-I) induces epidermal growth factor receptor transactivation and cell proliferation through reactive oxygen species. Free Radic. Biol. Med. 2007;42(11):1651–1660. doi: 10.1016/j.freeradbiomed.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 214.Nagai H. Pathophysiological roles of ASK1-MAP kinase signaling pathways. J. Biochem. Mol. Biol. 2007;40(1):1–6. doi: 10.5483/bmbrep.2007.40.1.001. [DOI] [PubMed] [Google Scholar]
- 215.Zhou J.Y., Liu Y., Wu G.S. The role of mitogen-activated protein kinase phosphatase-1 in oxidative damage-induced cell death. Cancer Res. 2006;66(9):4888–4894. doi: 10.1158/0008-5472.CAN-05-4229. [DOI] [PubMed] [Google Scholar]
- 216.Yu R. Activation of mitogen-activated protein kinase pathways induces antioxidant response element-mediated gene expression via a Nrf2-dependent mechanism. J. Biol. Chem. 2000;275(51):39907–39913. doi: 10.1074/jbc.M004037200. [DOI] [PubMed] [Google Scholar]
- 217.Kim S.U. Peroxiredoxin I is a ROS/p38 MAPK-dependent inducible antioxidant that regulates NF-kappaB-mediated iNOS induction and microglial activation. J. Neuroimmunol. 2013;259(1–2):26–36. doi: 10.1016/j.jneuroim.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 218.Dhar A., Young M.R., Colburn N.H. The role of AP-1, NF-kappaB and ROS/NOS in skin carcinogenesis: the JB6 model is predictive. Mol. Cell. Biochem. 2002;234–235(1–2):185–193. [PubMed] [Google Scholar]
- 219.Chen C. Activation of antioxidant-response element (ARE), mitogen-activated protein kinases (MAPKs) and caspases by major green tea polyphenol components during cell survival and death. Arch. Pharm. Res. 2000;23(6):605–612. doi: 10.1007/BF02975249. [DOI] [PubMed] [Google Scholar]
- 220.Brown G.C., Borutaite V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim. Biophys. Acta. 2004;1658(1–2):44–49. doi: 10.1016/j.bbabio.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 221.Hill B.G., Darley-Usmar V.M. S-nitrosation and thiol switching in the mitochondrion: a new paradigm for cardioprotection in ischaemic preconditioning. Biochem. J. 2008;412(2):e11–e13. doi: 10.1042/BJ20080716. [DOI] [PubMed] [Google Scholar]
- 222.Taylor E.R. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J. Biol. Chem. 2003;278(22):19603–19610. doi: 10.1074/jbc.M209359200. [DOI] [PubMed] [Google Scholar]
- 223.Chen C.L. Site-specific S-glutathiolation of mitochondrial NADH ubiquinone reductase. Biochemistry. 2007;46(19):5754–5765. doi: 10.1021/bi602580c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kang P.T. Protein thiyl radical mediates S-glutathionylation of complex I. Free Radic. Biol. Med. 2012;53(4):962–973. doi: 10.1016/j.freeradbiomed.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Hurd T.R. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of CYS residues in decreasing oxidative damage. J. Biol. Chem. 2008;283(36):24801–24815. doi: 10.1074/jbc.M803432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Brookes P.S. Mitochondria: regulators of signal transduction by reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2002;33(6):755–764. doi: 10.1016/s0891-5849(02)00901-2. [DOI] [PubMed] [Google Scholar]
- 227.Murray J. Oxidative damage to mitochondrial complex I due to peroxynitrite: identification of reactive tyrosines by mass spectrometry. J. Biol. Chem. 2003;278(39):37223–37230. doi: 10.1074/jbc.M305694200. [DOI] [PubMed] [Google Scholar]
- 228.Abello N. Protein tyrosine nitration: selectivity, physicochemical and biological consequences, denitration, and proteomics methods for the identification of tyrosine-nitrated proteins. J. Proteome Res. 2009;8(7):3222–3238. doi: 10.1021/pr900039c. [DOI] [PubMed] [Google Scholar]
- 229.Chen C.L. Protein tyrosine nitration of the flavin subunit is associated with oxidative modification of mitochondrial complex II in the post-ischemic myocardium. J. Biol. Chem. 2008;283(41):27991–28003. doi: 10.1074/jbc.M802691200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Yarian C.S., Rebrin I., Sohal R.S. Aconitase and ATP synthase are targets of malondialdehyde modification and undergo an age-related decrease in activity in mouse heart mitochondria. Biochem. Biophys. Res. Commun. 2005;330(1):151–156. doi: 10.1016/j.bbrc.2005.02.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Benderdour M. Cardiac mitochondrial NADP+-isocitrate dehydrogenase is inactivated through 4-hydroxynonenal adduct formation: an event that precedes hypertrophy development. J Biol Chem. 2003;278(46):45154–45159. doi: 10.1074/jbc.M306285200. [DOI] [PubMed] [Google Scholar]
- 232.Lee J.H., Yang E.S., Park J.W. Inactivation of NADP+-dependent isocitrate dehydrogenase by peroxynitrite. Implications for cytotoxicity and alcohol-induced liver injury. J. Biol. Chem. 2003;278(51):51360–51371. doi: 10.1074/jbc.M302332200. [DOI] [PubMed] [Google Scholar]
- 233.Lucas D.T., Szweda L.I. Declines in mitochondrial respiration during cardiac reperfusion: age-dependent inactivation of alpha-ketoglutarate dehydrogenase. Proc. Natl. Acad. Sci. USA. 1999;96(12):6689–6693. doi: 10.1073/pnas.96.12.6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.McLain A.L. Glutathionylation of alpha-ketoglutarate dehydrogenase: the chemical nature and relative susceptibility of the cofactor lipoic acid to modification. Free Radic. Biol. Med. 2013;61:161–169. doi: 10.1016/j.freeradbiomed.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bulteau A.L. Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc. Natl. Acad. Sci. USA. 2005;102(17):5987–5991. doi: 10.1073/pnas.0501519102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Stachowiak O. Mitochondrial creatine kinase is a prime target of peroxynitrite-induced modification and inactivation. J. Biol. Chem. 1998;273(27):16694–16699. doi: 10.1074/jbc.273.27.16694. [DOI] [PubMed] [Google Scholar]
- 237.Kim J. Oxidative modification of cytochrome c by singlet oxygen. Free Radic. Biol. Med. 2008;44(9):1700–1711. doi: 10.1016/j.freeradbiomed.2007.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Zarse K. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial l-proline catabolism to induce a transient ROS signal. Cell Metab. 2012;15(4):451–465. doi: 10.1016/j.cmet.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Sena L.A., Chandel N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell. 2012;48(2):158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Yun J., Finkel T. Mitohormesis. Cell Metab. 2014;19(5):757–766. doi: 10.1016/j.cmet.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Finkel T. Signal transduction by mitochondrial oxidants. J. Biol. Chem. 2012;287(7):4434–4440. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Chandel N.S. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA. 1998;95(20):11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Scherz-Shouval R. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26(7):1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Adlam V.J. Targeting an antioxidant to mitochondria decreases cardiac ischemia–reperfusion injury. FASEB J. 2005;19(9):1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 245.Dare A.J. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 2015;5:163–168. doi: 10.1016/j.redox.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Zhao W.Y. Mitochondria-targeted antioxidant peptide SS31 prevents hypoxia/reoxygenation-induced apoptosis by down-regulating p66Shc in renal tubular epithelial cells. Cell. Physiol. Biochem. 2013;32(3):591–600. doi: 10.1159/000354463. [DOI] [PubMed] [Google Scholar]
- 247.Dai W. Bendavia, a mitochondria-targeting peptide, improves postinfarction cardiac function, prevents adverse left ventricular remodeling, and restores mitochondria-related gene expression in rats. J. Cardiovasc. Pharmacol. 2014;64(6):543–553. doi: 10.1097/FJC.0000000000000155. [DOI] [PubMed] [Google Scholar]
- 248.Tabara L.C. Mitochondria-targeted therapies for acute kidney injury. Expert Rev. Mol. Med. 2014;16:e13. doi: 10.1017/erm.2014.14. [DOI] [PubMed] [Google Scholar]
- 249.Ong S.B. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121(18):2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 250.Qi X. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013;126(3):789–802. doi: 10.1242/jcs.114439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kroemer G., Marino G., Levine B. Autophagy and the integrated stress response. Mol. Cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Andres A.M. Mitophagy is required for acute cardioprotection by simvastatin. Antioxid. Redox Signal. 2014;21(14):1960–1973. doi: 10.1089/ars.2013.5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Sies H. Role of metabolic H2O2 generation: redox signaling and oxidative stress. J. Biol. Chem. 2014;289(13):8735–8741. doi: 10.1074/jbc.R113.544635. [DOI] [PMC free article] [PubMed] [Google Scholar]