

Abstract
Several bacterial species recruit the complement regulators C4b-binding protein, factor H, and vitronectin, resulting in resistance against the bactericidal activity of human serum. It was recently demonstrated that bacteria also bind plasminogen, which is converted to plasmin that degrades C3b and C5. In this study, we found that a series of clinical isolates (n = 58) of the respiratory pathogen Moraxella catarrhalis, which is commonly isolated from preschool children and adults with chronic obstructive pulmonary disease (COPD), significantly binds human plasminogen. Ubiquitous surface protein A2 (UspA2) and hybrid UspA2 (UspA2H) were identified as the plasminogen-binding factors in the outer membrane proteome of Moraxella. Furthermore, expression of a series of truncated recombinant UspA2 and UspA2H proteins followed by a detailed analysis of protein-protein interactions suggested that the N-terminal head domains bound to the kringle domains of plasminogen. The binding affinity constant (KD) values of full-length UspA230–539 (amino acids 30 to 539 of UspA2) and full-length UspA2H50–720 for immobilized plasminogen were 4.8 × 10−8 M and 3.13 × 10−8 M, respectively, as measured by biolayer interferometry. Plasminogen bound to intact M. catarrhalis or to recombinant UspA2/UspA2H was readily accessible for a urokinase plasminogen activator that converted the zymogen into active plasmin, as verified by the specific substrate S-2251 and a degradation assay with fibrinogen. Importantly, plasmin bound at the bacterial surface also degraded C3b and C5, which consequently may contribute to reduced bacterial killing. Our findings suggest that binding of plasminogen to M. catarrhalis may lead to increased virulence and, hence, more efficient colonization of the host.
INTRODUCTION
Moraxella catarrhalis is a Gram-negative human respiratory pathogen that is associated with acute otitis media in children and exacerbations in patients with chronic obstructive pulmonary disease (COPD) (1–3). It is most commonly found in a polymicrobial community with other pathogens such as Streptococcus pneumoniae and nontypeable Haemophilus influenzae. An efficient vaccine against M. catarrhalis has not yet been developed. However, several outer membrane proteins (OMPs) of M. catarrhalis have been shown to be immunogenic and are thus suggested as potential components in a future subunit vaccine (4).
The complement system is the first line of immune defense and consists of >50 different proteins and effector components that clear invading pathogens through the formation of the membrane attack complex (MAC) or by assisting macrophages in opsonophagocytosis. Complement activation consists of a cascade of reactions that are triggered by the formation of an antigen-antibody complex (classical pathway), recognition of bacterial surface carbohydrates (lectin-mediated pathway), or spontaneous activation (alternative pathway). The activation of one of these pathways leads to the cleavage of C3 into C3a and C3b, where C3b is deposited on the surface of the pathogen. Deposition of C3b activates the C5 convertase to cleave C5 into C5a and C5b. C5a and C3a induce a proinflammatory host cell response, whereas C5b initiates the formation of the MAC (5, 6). M. catarrhalis clinical isolates are highly serum resistant, and this is accomplished mainly by hijacking complement regulators in order to inhibit the formation of the MAC (4). M. catarrhalis recruits vitronectin from serum and ultimately inhibits the assembly of the C5b-C7 complex and polymerization of C9 (7–10). In addition, C4b-binding protein (C4BP) is utilized by M. catarrhalis for inhibition of the classical pathway of complement activation (11). M. catarrhalis also binds factor H, one of the regulators of the alternative pathway (12), and directly interacts with complement (13, 14), strategies that help increase bacterial survival.
M. catarrhalis ubiquitous surface proteins (Usps) belong to a family of trimeric autotransporters that are found in several Gram-negative species. The proteins in this group are composed of membrane anchor, stalk, neck, and head domains and thus appear as “lollipops” on the bacterial outer membrane (4, 15, 16). Ubiquitous surface proteins are generally categorized into three distinct subgroups: UspA1 is present in all clinical isolates, whereas UspA2 and hybrid UspA2 (UspA2H) are found in 75% and 25% of strains, respectively. Both UspA1 and UspA2 function as adhesins and interact with extracellular matrix (ECM) proteins such as laminin and fibronectin (17, 18). In addition, UspA1 and UspA2 neutralize C3 and C3d and attract C4BP and vitronectin to protect M. catarrhalis from the bactericidal activity of serum (8, 11).
Plasminogen is a 92-kDa single-chain glycoprotein that circulates in blood as a zymogen. The conversion of plasminogen into plasmin (active protease) is catalyzed by tissue-type urokinase plasminogen activator (tPA) or urokinase plasminogen activator (uPA) (19). The basic function of plasmin is to degrade fibrin (fibrinolysis), which is involved in homeostatic processes such as blood coagulation, cell migration, wound repair, and remodeling of the ECM. Excess activity of plasmin is regulated by alpha-2 antiplasmin and alpha-2 macroglobulin (20). Plasminogen was recently also reported to be a regulator of the complement system, efficiently degrading complement factors, including C3b and C5 (21). In addition, plasminogen activates host procollagenases and matrix metalloproteinases that subsequently degrade various components of the ECM. The protease activity mediated by plasminogen is occasionally utilized by bacterial pathogens in order to promote virulence (20, 22).
In this study, we show that a series of M. catarrhalis clinical isolates had the capacity to attract plasminogen from human serum and use it for inactivation of complement components. Plasminogen binding at the bacterial surface occurred via UspA2/UspA2H and was easily activated by urokinase plasminogen activator. The interaction of UspA2/UspA2H with plasminogen consisted of ionic interactions and the lysine-binding kringle domains of plasminogen. Active plasmin degraded the complement components C3b and C5, which ultimately enhanced the survival of serum-susceptible M. catarrhalis. Taken together, our results thus shed light upon how M. catarrhalis acquires serum resistance through recruitment of plasminogen.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
M. catarrhalis RH4 and Bc5 and other Moraxella clinical isolates (8) were cultured on chocolate agar plates or in brain heart infusion (BHI) broth, followed by incubation at 37°C in a humid atmosphere with 5% CO2. Details of the M. catarrhalis clinical isolates selected for this study were described previously by Su et al. (8). M. catarrhalis uspA1, uspA2, and mid single and multiple mutants were obtained from our laboratory (11). M. catarrhalis mutants were grown in 1.5 μg ml−1 chloramphenicol, 7 μg ml−1 zeocin, or 20 μg ml−1 kanamycin (10). Double and multiple mutants were grown in combinations of antibiotics. Escherichia coli DH5α and E. coli BL21(DE3) were cultured in Luria-Bertani (LB) broth or on LB agar plates at 37°C. E. coli cells containing pET26b expression vectors with various truncated uspA2 or uspA2H genes (8, 9) were grown in LB medium supplemented with 50 μg ml−1 kanamycin.
Plasminogen direct binding assay.
Plasminogen binding at the surface of bacteria was analyzed by a direct ligand binding assay. For this purpose, plasminogen was labeled with 125I by using chloramine-T as described previously (23). Approximately 107 M. catarrhalis cells were blocked with phosphate-buffered saline (PBS)–2.5% bovine serum albumin (BSA) and added to microtiter plates. 125I-labeled plasminogen was added at increasing concentrations to bacteria and incubated for 1 h at 37°C. In inhibition experiments, cold ligands were added to samples prior to the addition of 125I-labeled plasminogen. In the next step, bacteria were washed 3 times with PBS to remove unbound 125I-labeled plasminogen. Plates were harvested in a 96-well plate harvester (Tomtec, Hamden, CT) and counted with a liquid scintillation counter (Trilux, Microbeta 1450; PerkinElmer, Waltham, MA).
Protein purification and enzyme-linked immunosorbent assay (ELISA).
Full-length UspA2 and UspA2H proteins and fragments were expressed and purified as described previously (8, 17). An iron-binding periplasmic protein (YfeA) from Yersinia pestis was used as a negative control. Similar to the UspA2 and UspA2H fragments, this protein was also expressed in the pET26b(+) expression vector and purified by using Ni-nitrilotriacetic acid (NTA) affinity chromatography. PolySorp microtiter plates (Nunc-Immuno, Roskilde, Denmark) were coated with purified UspA2 and UspA2H (both at 50 nM) in the presence of 100 mM Tris-HCl (pH 9.0) for 15 h at 4°C. Plates were washed 3 times with PBS and blocked with PBS–2.5% BSA for 1 h at room temperature (RT). Plasminogen or kringle domains (24) were added at increasing concentrations to plates and incubated for 1 h at room temperature. In blocking assays, NaCl, ε-amino caproic acid (εACA), laminin, fibronectin, and vitronectin were added to the wells prior to the addition of plasminogen. Unbound plasminogen was removed by washes with PBS containing 0.05% Tween 20 (PBS-T). Thereafter, sheep anti-human plasminogen polyclonal antibody (PAb) (dilution, 1:1,000; AbD Serotec, Oxford, United Kingdom) in PBS containing 2.5% BSA (PBS-BSA) was added. After 1 h of incubation at RT, plates were washed 4 times with PBS-T and incubated with secondary horseradish peroxidase (HRP)-conjugated donkey anti-sheep PAb (dilution, 1:1,000; AbD Serotec) in PBS-BSA. Finally, plates were washed 4 times with PBS-T and developed. Plates were read in a 96-well microtiter plate reader (Sunrise Tecan, Männedorf, Germany) at 450 nm.
Measurement of protein-protein interactions by biolayer interferometry.
The interaction between UspA2, UspA2H, and plasminogen was analyzed by using a biolayer interferometry technique (Octet Red96; ForteBio, CA). Plasminogen was immobilized in an amine-reactive (AR2G) sensor. Sensors were activated by submerging them into a mixture of 0.2 M 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 0.05 M N-hydroxysulfosuccinimide (NHS) solutions followed by loading of the sensors with plasminogen. The sensor thickness was achieved between 2-4 nm. Unoccupied groups on the sensor layer were quenched with 2 M ethanolamine. An empty sensor neutralized by ethanolamine and a plasminogen-bound sensor in PBS were considered controls. UspA2 and UspA2H were dialyzed in PBS, and 0.1 to 2.0 μM was used for analysis of binding kinetics. Similar to UspA2 and UspA2H, we also included YfeA as a negative control. Data were collected by using Octet data acquisition software, and analysis of the data was performed by using the Data Analysis 8.1 module.
Measurement of plasmin activity.
Plasminogen bound to the bacterial surface or to recombinant UspA2/UspA2H was activated by using urokinase plasminogen activator (uPA). The activity of plasmin was measured by using the S-2251 chromogenic substrate (Chromogenix, Bedford, MA). Bacteria (107) or 100 nM UspA2/UspA2H that was used to coat microtiter plates was allowed to bind 5 μg plasminogen in the presence of 2.5% BSA in PBS. The unbound fraction was washed, and samples were incubated in a plasmin activity assay buffer containing 32 mM Tris-HCl (pH 7.5) and 175 mM NaCl. Finally, 5 U uPA was added to this solution, and the mixture was incubated at 37°C. The absorbance of the solutions was measured in a spectrophotometer at 405 nm every 30 min for up to 6 h. Control mixtures without uPA and plasminogen were prepared in parallel. We also measured plasmin activity by using a fibrinogen degradation assay and Western blotting. Bacteria (107) or UspA2/UspA2H bound to plasminogen was added to 2 μg fibrinogen and 5 U uPA. Controls were also prepared without plasminogen and uPA. The degradation of fibrinogen (fibrin) was monitored by stopping the reaction at different time points (10 to 30 min) by adding SDS loading dye to the mixture. Samples were boiled at 95°C for 10 min, and supernatants were separated by 12% SDS-PAGE. Thereafter, the gels were blotted onto a polyvinylidene difluoride (PVDF) membrane. Blots were blocked with 5% milk for 1 h at RT and incubated with primary sheep anti-human fibrinogen PAb (Sigma) diluted 1:1,000 in 5% milk. After 1 h of incubation at RT, membranes were washed with PBS-T. Subsequently, the membranes were incubated with HRP-conjugated donkey anti-sheep/goat PAb (AbD Serotec) diluted 1:1,000 in 5% milk. Finally membranes were washed in PBS-T and developed by using an ECL Western blotting kit (Pierce, IL).
C3b and C5 degradation by plasmin.
Plasmin modulates the activity of the complement system by degrading complement factors C3b and C5 (21, 25). We analyzed the degradation of C3b and C5 by plasminogen bound at the surface of M. catarrhalis or bound to UspA2/UspA2H by Western blotting. Plasminogen was added together with 5 U uPA or 2 μg C3b or C5 to microtiter plates containing bacteria (107) or coated with UspA2/UspA2H, followed by incubation at 37°C. Reactions were terminated by the addition of SDS loading dye to the mixture at different time intervals (1 to 20 h). Reaction mixtures were boiled for 10 min at 95°C. Supernatants were separated on 10% or 12% SDS-PAGE gels and blotted onto a PVDF membrane. One set of gels was stained with Coomassie R-250 stain in order to monitor the loading control. Blots were developed by using sheep anti-human C3b (CompTech, Tyler, TX) or sheep anti-human C5 (CompTech) primary monoclonal antibodies (MAbs), both at a 1:1,000 dilution. Finally, the membranes were incubated with diluted (1:1,000) HRP-conjugated donkey anti-sheep/goat PAb, and blots were developed by using an ECL Western blotting kit (Pierce, IL).
Serum resistance assay.
We also tested whether normal human serum (NHS) that had been pretreated with UspA2/UspA2H would have decreased bactericidal activity. Dynabeads (Novex, Life Technologies, Norway) were coated with UspA2/UspA2H and washed thoroughly with PBS. Protein-coated Dynabeads (10 μl) were incubated with 10 μg plasminogen for 1 h at 37°C, and unbound plasminogen was thoroughly washed with PBS. Beads were resuspended in dextrose-gelatin-Veronal buffer (DGVB) (pH 7.3) containing 140 mM glucose, 0.1% (wt/vol) gelatin, 1 mM MgCl2, and 0.15 mM CaCl2. Subsequently, 10% NHS and 20 U uPA were added to this solution. The reaction mixture was incubated at 37°C for 2 h. This NHS preparation was used for the bactericidal activity assay. The negative-control protein YfeA (Yersinia iron-binding protein) was also used to coat Dynabeads and processed similarly to the UspA2/UspA2H-coated beads. In parallel, control beads without any coating (beads only) that were treated with 2.5% BSA and washed in PBS were also included.
RESULTS
UspA2 and UspA2H are the major plasminogen-binding proteins in Moraxella.
We observed that plasminogen binds to the surface of M. catarrhalis and therefore wanted to identify the plasminogen-binding protein(s) of this particular pathogen. Whole-cell proteins of M. catarrhalis wild-type (WT) and ΔuspA1 and ΔuspA2 mutant strains were analyzed by native PAGE and far-Western blotting with plasminogen as bait. The result clearly indicated that UspA2 was a high-molecular-weight protein that bound plasminogen (Fig. 1A). The loading controls are shown in Fig. S1A in the supplemental material. M. catarrhalis Bc5 and isogenic mutants were analyzed for plasminogen binding by using a pulldown assay. Bacteria were incubated with purified plasminogen or normal human serum. The unbound proteins were washed, and total proteins were separated by SDS-PAGE (see Fig. S1B and S1C in the supplemental material) and blotted. The presence of plasminogen was analyzed by using an anti-plasminogen PAb. Interestingly, deletion of UspA2 from the bacterial surface resulted in reduced binding of purified or serum-derived plasminogen (Fig. 1B). M. catarrhalis WT and isogenic mutant strains were also tested for 125I-labeled plasminogen by a direct binding assay. Deletion of uspA2 from M. catarrhalis Bc5 caused a 35.6% decrease in 125I-labeled plasminogen binding, as can be seen in Fig. 1C. In contrast to the ΔuspA2 mutants, no decreased plasminogen binding was observed with the ΔuspA1 and Δmid mutants. Moraxella IgD-binding protein (MID) (26) mutants were also included as a control. The Δmid mutants of M. catarrhalis RH4 and Bc5 did not show any reduced plasminogen binding.
FIG 1.

M. catarrhalis UspA2 and UspA2H are plasminogen-binding proteins. (A) Whole-cell proteins of M. catarrhalis Bc5 and mutants were separated by native PAGE, and plasminogen binding was analyzed by far-Western blotting. High-molecular-mass proteins (UspA2 and UspA2H) revealed binding to plasminogen that was absent in the M. catarrhalis Bc5 ΔuspA2 and ΔuspA1 ΔuspA2 mutants. The control for loading of proteins is shown in Fig. S1A in the supplemental material. (B) Binding of pure plasminogen (Plg) and plasminogen from serum (dilution, 1:50) to M. catarrhalis Bc5 and isogenic mutants. Binding was detected by Western blotting using an anti-plasminogen PAb. Similarly to the M. catarrhalis uspA2 mutant, the double (ΔuspA1 ΔuspA2) and triple (ΔuspA1 ΔuspA2 Δmid) mutants also showed decreased plasminogen binding. (C) Comparative binding of 125I-labeled plasminogen to M. catarrhalis Bc5 and the corresponding isogenic mutants. (D) Whole-cell proteins of M. catarrhalis and mutants were separated by native PAGE. The high-molecular-mass protein UspA2H was detected as a plasminogen-binding protein by far-Western blotting. (E) M. catarrhalis RH4 and the corresponding Usp mutants were incubated with serum or purified plasminogen. (F) Comparison of 125I-plasminogen binding to M. catarrhalis RH4 and isogenic mutants. Experiments for data in panels C and F were repeated three times in triplicates. Experiments with blots shown in panels A, B, D, and E were repeated twice, and one representative blot is shown. Statistical analyses for panels C and F were performed by using one-way analysis of variance, where the wild type was compared with each mutant. **, P ≤ 0.01; ***, P ≤ 0.001.
One-fourth of M. catarrhalis clinical isolates express UspA2H at their surface. Therefore, we selected M. catarrhalis RH4, which carries UspA2H, and its isogenic mutants to detect plasminogen-binding proteins by far-Western blotting. In parallel to UspA2, UspA2H was identified as a major plasminogen-binding protein in M. catarrhalis RH4 (Fig. 1D to F). Taken together, our direct binding assays and Western blot analyses strongly suggested that M. catarrhalis recruits plasminogen at its surface by using the trimeric autotransporter UspA2 or UspA2H.
Moraxella catarrhalis clinical isolates bind plasminogen at their surface.
To prove that plasminogen binding is a common characteristic of all M. catarrhalis isolates, we analyzed a series of Moraxella isolates by using the direct binding assay. M. catarrhalis isolates (n = 58) were incubated with 125I-labeled plasminogen, and bound radioactivity was measured. All M. catarrhalis isolates tested bound plasminogen, but the binding capacity varied among isolates. The majority (≈50%) were high-binding isolates; that is, ≥10% of added plasminogen bound to bacteria. Of note, 6 clinical isolates displayed an extraordinarily high plasminogen-binding capacity (Fig. 2A). Since both UspA2 and UspA2H were involved in interactions with plasminogen, M. catarrhalis Bc5 and RH4 were selected for further detailed analysis as representatives for UspA2 and UspA2H, respectively. Dose-dependent binding of 125I-labeled plasminogen was seen with both M. catarrhalis RH4 and Bc5 (Fig. 2B and C). The binding of 125I-labeled plasminogen to bacteria was not completely saturable. The reason for this could be due to other low-affinity plasminogen receptors at the surface of M. catarrhalis. Multiple bacterium-host protein interactions are often observed (9, 27).
FIG 2.

M. catarrhalis clinical isolates interact with plasminogen. (A) Direct binding assay showing binding of 125I-labeled plasminogen to Moraxella clinical isolates (n = 58). (B) Dose-dependent binding of 125I-labeled plasminogen to M. catarrhalis Bc5, which expresses UspA2. (C) Binding of 125I-labeled plasminogen to UspA2H-expressing M. catarrhalis strain RH4 is also dose dependent. Mean values of data from triplicates of three independent experiments are presented, and error bars indicate standard deviations. According to the plasminogen binding detected, isolates that bound ≥10% of the added radioactivity (≥6,000 cpm) were considered high-binding strains, those that bound between 4,000 and 6,000 cpm were considered medium-binding strains, and those that bound ≤4,000 cpm were considered low-plasminogen-binding Moraxella strains.
The N-terminal head domains of UspA2 and UspA2H bind plasminogen.
UspA2 and UspA2H are trimeric autotransporters composed of head, neck, stalk, and hydrophobic membrane-anchoring domains (14). We mapped the plasminogen-binding region of UspA2 and UspA2H by using truncated proteins that were recombinantly expressed in E. coli. As shown in Fig. 3A, full-length UspA230–539 (amino acids 30 to 539 of UspA2) of M. catarrhalis Bc5 bound plasminogen in a dose-dependent manner, as determined by an ELISA. To pinpoint the plasminogen-binding region of UspA2, a series of truncated recombinant proteins were used (8, 9, 14). Our results revealed that all UspA2 fragments, except for UspA2165–318 and UspA2302–458, significantly bound plasminogen in comparison with the negative-control protein YfeA. However, UspA230–539, UspA230–177, and UspA2101–318 had the highest levels of binding compared to the other truncated fragments (Fig. 3B). Similar to UspA230–539, full-length UspA2H50–720 also bound plasminogen in a concentration-dependent manner (Fig. 3C). The recombinant full-length UspA2H50–720 and truncated UspA2H50–296 proteins had the highest levels of interaction with plasminogen in comparison to the other UspA2H fragments (Fig. 3D). These results indicated that the plasminogen-binding region was located at the N-terminal head domains of UspA2 (amino acids 100 to 177) and UspA2H (amino acids 50 to 296). The binding affinity constant (KD) values of UspA230–539 and UspA2H50–720 for immobilized plasminogen were 4.8 × 10−8 M and 3.13 × 10−8 M, respectively, as measured by biolayer interferometry (Octet Red) (Fig. 3E and F).
FIG 3.

The head regions of UspA2 and UspA2H interact with plasminogen. (A) Dose-dependent binding of plasminogen to UspA230–539 as revealed by an ELISA. (B) Schematic representation of constructs used for recombinant expression of UspA2 in E. coli. The right panel shows ELISA results demonstrating the binding of the plasminogen to UspA2 fragments. (C) Dose-dependent binding of plasminogen to UspA2H50–720 as revealed by an ELISA. (D) Schematic representation of constructs used for recombinant expression of UspA2H in E. coli and binding of these proteins to plasminogen. (E and F) Affinities of binding between plasminogen and UspA2 (E) and UspA2H (F) were measured by biolayer interferometry (Octet Red96). In panels B and D, results represent means of data from triplicates of three independent experiments, and error bars indicate standard deviations. Data shown in panels A and C were analyzed by two-way analysis of variance, and data shown in panels B and D were analyzed by one-way analysis of variance. Plasminogen binding was significantly (P ≤ 0.001) higher than that for the controls (panels A and C). *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
We previously reported that M. catarrhalis UspA2 interacts with several ECM proteins, including vitronectin, fibronectin, and laminin (9, 17, 18). The UspA2 N-terminal head domain binds to laminin and vitronectin, whereas the neck region of UspA2 binds to fibronectin. To analyze whether plasminogen binds to the same region as the ECM proteins, we performed a competitive inhibition experiment using an ELISA. Microtiter plates were coated with full-length UspA230–539, and the specific binding of plasminogen was measured in the presence of fibronectin, vitronectin, laminin, or UspA230–539, which were all added simultaneously. Laminin (2.5 nM) inhibited the binding of plasminogen to UspA2 by 59.3% (Fig. 4A), whereas the addition of vitronectin (2.5 nM) inhibited plasminogen binding by 34.5%. In contrast, fibronectin (2.5 nM) inhibited the binding of plasminogen to UspA2 by only 19.9%, and there was no further significant reduction with up to 100 nM fibronectin. In parallel with the data obtained with UspA2, similar results were found with UspA2H (Fig. 4B). YfeA did not show any decrease of plasminogen binding compared to UspA2 or UspA2H in those competitive inhibition experiments. Our results thus clearly indicated that plasminogen shares the vitronectin- and laminin-binding regions on UspA2 and UspA2H, as schematically outlined in Fig. 3B and D (top). In contrast, fibronectin has distinct binding regions, that is, the neck and stalk of UspA2, which partially overlap and hence did not significantly compete with plasminogen.
FIG 4.
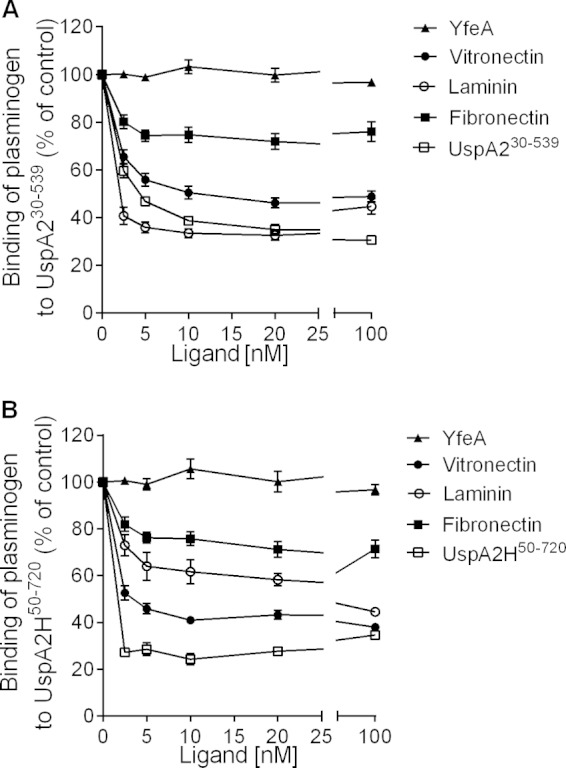
Plasminogen binds to the same regions of UspA2 and UspA2H as vitronectin and laminin. (A) ELISA showing inhibition of plasminogen binding to UspA230–539 with increasing concentrations of vitronectin, laminin, and UspA230–539. In contrast, fibronectin inhibited the interaction partially. (B) ELISA showing inhibition of plasminogen binding to UspA2H50–720 in the presence of increasing concentrations of vitronectin, fibronectin, laminin, and UspA2H50–720. Statistical analyses were performed by two-way analysis of variance. Data shown are the means of data from triplicates of three independent experiments, and error bars indicate standard deviations. All ligand concentrations between 2.5 and 20 nM showed statistically significant decreases in plasminogen binding to UspA230–539 and UspA2H50–720 compared with samples without ligands (0 nM). Fibronectin (2.5 nM) showed significant inhibition (P ≤ 0.001) in comparison to the control. However, further addition of fibronectin did not reveal any significant decrease compared to 2.5 nM fibronectin.
The interaction of Moraxella catarrhalis with plasminogen depends on lysine-binding residues and ionic strength.
Plasminogen consists of five kringle domains (K1 to K5) that have affinity for lysine or lysine analogues. We have recently shown that lysine-binding residues of plasminogen are important for the interaction with protein E of H. influenzae (27). In addition, we noticed that ionic forces are involved in the interaction, since binding of plasminogen to protein E was dependent on NaCl. To analyze whether UspA2 and UspA2H bind to plasminogen in a way similar to that of protein E, M. catarrhalis was incubated with 125I-labeled plasminogen in the presence of the lysine analogue ε-amino caproic acid (εACA). As shown in Fig. 5A, the addition of εACA at 0.05 μM resulted in a 12.9% decrease in plasminogen binding to M. catarrhalis RH4, whereas εACA at the same concentration significantly decreased binding (59.9%) to M. catarrhalis Bc5. At 1 mM εACA, 54.9% and 79.5% reductions in plasminogen binding to M. catarrhalis RH4 and Bc5, respectively, were observed (Fig. 5A). Finally, the addition of NaCl also resulted in inhibited 125I-plasminogen binding to both M. catarrhalis strains (Fig. 5B).
FIG 5.

Plasminogen binds to M. catarrhalis via kringle domains and involves ionic interactions. (A) 125I-plasminogen binding to M. catarrhalis strains is inhibited by the addition of increasing concentrations of εACA. (B) Increasing concentrations of NaCl inhibit the interaction of 125I-labeled plasminogen with M. catarrhalis. Binding at 150 mM NaCl was set as 100%, which represents physiological conditions. (C) εACA inhibits the plasminogen-UspA2/UspA2H interaction as determined by an ELISA. (D) Binding of plasminogen to UspA2/UspA2H is inhibited by NaCl, as demonstrated by an ELISA. Mean values from three replicates of three independent experiments are shown, and error bars indicate standard deviations. (E) Schematic outline of truncated plasminogen fragments used in this study. (F) Binding of different plasminogen kringle domains to UspA2 and UspA2H as demonstrated by an ELISA. Shown are means of data from three independent experiments in triplicate, and error bars denote standard deviations. Statistical analyses were performed by two-way analysis of variance. Plasminogen and K1-K5 samples were compared with other kringle domain samples. Plasminogen binding was significantly (P ≤ 0.001) inhibited with 0.05 to 10 mM εACA (A and C) and 0.25 to 2.15 M NaCl (B and D). ***, P ≤ 0.001.
To further study protein-protein interactions, a microtiter plate was coated with recombinant UspA2H50–720 and UspA2H50–296 (both at 50 nM). Plasminogen (20 nM) was added in the presence of εACA at increasing concentrations. εACA (0.1 mM) reduced the binding of plasminogen to UspA2 and UspA2H (Fig. 5C). In parallel, with whole bacteria, the addition of NaCl also inhibited plasminogen binding to both recombinant UspA2 and UspA2H (Fig. 5D).
Finally, we used a set of recombinant plasminogen constructs encompassing different combinations of kringle domains (Fig. 5E). The plasminogen K1 to K5 domains interacted with UspA2 and UspA2H in a way similar to that of the commercial full-length plasminogen (Fig. 5F). When K1 and K2 were deleted, almost completely reduced binding to UspA2 and UspA2H was observed. In conclusion, our results indicate that ionic interactions are important for plasminogen binding to M. catarrhalis UspA2 and UspA2H and also that the lysine-binding residues of plasminogen K1 and K2 are crucial for this interaction.
Plasminogen bound at the surface of bacteria is converted to active plasmin.
The conversion of plasminogen to plasmin is regulated by tissue-type plasminogen activator (tPA) or urokinase plasminogen activator (uPA). To analyze whether plasminogen can be converted into plasmin at the surface of M. catarrhalis, plasmin activity was measured by using the chromogenic substrate S-2251, which is cleaved by plasmin. Interestingly, most of the surface-bound plasminogen was converted into plasmin within 3 h in the presence of uPA (Fig. 6A). We also tested the activation of plasminogen bound to recombinant UspA2 and UspA2H (Fig. 6B). Here, plasmin activity was detected in the UspA2- and UspA2H-coated microtiter wells, whereas the negative control (recombinant YfeA), which did not bind plasminogen, lacked any proteolytic (plasmin) activity.
FIG 6.

Plasminogen bound at the bacterial surface can be converted into plasmin. (A) Chromogenic assay showing time-dependent conversion of plasminogen to plasmin at the surface of M. catarrhalis strains. The positive control consisted of 1 μg plasminogen and 5 U uPA. Negative controls without uPA or without plasminogen (uPA only) were included. (B) Time-dependent conversion of plasminogen to plasmin when plasminogen is bound to UspA2 and UspA2H. The positive control consisted of 50 ng plasminogen and 2 U uPA. Experiments were repeated twice with triplicate wells, and results from one typical experiment are shown. (C) Plasmin activity is retained at the bacterial surface, as analyzed by fibrinogen degradation. Bacteria (107) with plasminogen bound at their surface were incubated with 2 μg fibrinogen and uPA. Samples were collected at different time points and analyzed by Western blotting. The fibrinogen α-chain has a molecular mass of ∼63.5 kDa, the β-chain has a molecular mass of ∼56 kDa, and the γ-chain has a molecular mass of ∼47 kDa. Plasmin degrades α- and β-chains predominantly. Therefore, in blots, the degradation of α- and β-chains is shown. (D) Control reactions performed in the absence of bacteria. (E) Plasmin activity is retained when it is bound to recombinant UspA2 or UspA2H. A microtiter plate was coated with recombinant proteins, and plasminogen was added. In the next step, uPA and fibrinogen were added, and degradation was monitored by Western blotting. (F) Control reactions were performed in the absence of UspA2 and UspA2H. Experiments with the blots shown in panels C to F were repeated twice, and one set of blots is shown here. Plg, plasminogen; Fgn, fibrinogen.
We also measured plasmin activity by including human fibrinogen, which is a natural substrate and has a high sensitivity for digestion by plasmin. M. catarrhalis Bc5 and RH4 were incubated with plasminogen, followed by the addition of fibrinogen together with uPA. The reaction was stopped at various time points, and the total protein content was analyzed by SDS-PAGE and Western blotting. One set of the gels was stained with Coomassie R-250 to visualize uniform loading of proteins (see Fig. S2 in the supplemental material). The fibrinogen α-chain has a molecular mass of ∼63.5 kDa, the β-chain has a molecular mass of ∼56 kDa, and the γ-chain has a molecular mass of ∼47 kDa. Plasmin predominantly digests the α- and β-chains in comparison to the γ-chain. Therefore, in blots, degradation of α- and β-chains is shown. Importantly, M. catarrhalis RH4 bound high concentrations of plasminogen that degraded fibrinogen very efficiently (Fig. 6C), and intact fibrinogen disappeared after 20 min of incubation. In contrast, M. catarrhalis Bc5, which bound less plasminogen (Fig. 2), caused a slower degradation of fibrinogen. Control reactions without bacteria were run in parallel and proved that the experimental setup was functional (Fig. 6D). Fibrinogen degradation was also analyzed when plasminogen was bound to purified recombinant UspA2 or UspA2H (Fig. 6E). More efficient degradation of fibrinogen was observed for reactions with plasminogen and UspA2H than for reactions with UspA2. Data for the appropriate controls in the presence or absence of plasminogen are shown in Fig. 6F. Taken together, our results directly revealed that plasminogen bound to the bacterial surface via UspA2/UspA2H is fully accessible to uPA and hence can be converted to functional plasmin.
C3b and C5 are degraded by plasmin bound at the bacterial surface.
C3 is an important component of the alternative complement pathway and is cleaved into C3b and further on into C3a after activation. Plasminogen interacts with C3b and inactivates it by cleavage (21, 25). To analyze the degradation capacity of C3b when plasmin was bound to the surface of M. catarrhalis, plasminogen bound at the bacterial surface was activated with uPA, followed by the addition of pure C3b. Degradation of C3b was measured at different time points (2 to 20 h) by separation on SDS-PAGE gels followed by Western blotting. One set of gels was stained with Coomassie blue, in order to verify that equivalent protein concentrations were loaded (see Fig. S3A in the supplemental material). C3b constitutes α′- and β-chains (101 and 75 kDa, respectively) connected with disulfide bonds. Degradation of C3b chains by plasmin results in low-molecular-mass products of the α′-chain ranging from 30 to 68 kDa (21). In our hands, C3b degradation products appeared within 2 h, and the intensity of degraded C3b fragments increased with further incubation (Fig. 7A). RH4 has a higher plasminogen-binding capacity and hence showed more efficient C3b degradation than did Bc5 (Fig. 7A). The plasminogen bound to the recombinant proteins UspA230–539 and UspA2H50–720 in a microtiter plate also digested C3b after it was converted into active plasmin (see Fig. S4A in the supplemental material).
FIG 7.

Plasminogen bound to the surface of M. catarrhalis cleaves the complement proteins C3b and C5. (A) M. catarrhalis strains Bc5 and RH4 loaded with plasminogen degrade C3b. Bacteria (107) bound to plasminogen were incubated with 5 μg C3b and 5 U uPA at 37°C. At the indicated time points, the cleavage of C3b was analyzed by Western blotting. When plasminogen is converted into active plasmin, it cleaves the α′-chain of C3b into 68-kDa, 40-kDa, 36-kDa, and 30-kDa products, as illustrated with arrows. (B) Degradation of C5 by plasmin bound to the M. catarrhalis surface. Settings similar to those used in the experiment for panel A were used, except that C3b was replaced with C5. C5 is composed of a 115-kDa α-chain and a 75-kDa β-chain. The α-chain is degraded by plasmin to produce 25- to 65-kDa α′-fragments, as illustrated. Degradation was monitored at different time points by Western blotting. Positive controls consisted of 1 μg plasminogen, and negative controls consisted of only uPA or only plasminogen. All experiments were repeated twice, and one representative blot is shown. The loading controls are shown in Fig. S3 in the supplemental material.
We also tested plasmin-mediated C5 degradation related to plasminogen bound at the M. catarrhalis surface. Plasminogen bound to both M. catarrhalis RH4 and Bc5 degraded C5 within 2 h of incubation, and low-molecular-mass products ranging from 65 to 25 kDa appeared. As expected, M. catarrhalis RH4 expressing UspA2H degraded the C5 α-chain more intensely than did M. catarrhalis Bc5 (Fig. 7B). One set of gels was stained with Coomassie blue in order to verify that the same concentrations of protein were loaded (see Fig. S3B in the supplemental material). In addition, the plasmin bound to recombinant UspA2 or UspA2H also degraded C5 (see Fig. S4B in the supplemental material). These results, taken together, indicate that plasminogen bound at the surface of M. catarrhalis is converted into active plasmin and degrades components of the innate immune system.
UspA2/UspA2H-dependent plasminogen binding protects M. catarrhalis from the bactericidal activity of human serum.
Our experiments suggested that C3b and C5 were efficiently degraded by plasmin derived from plasminogen at the M. catarrhalis surface. We therefore tested whether bound plasminogen protected Moraxella from the bactericidal activity of serum. Dynabeads were coated with recombinant UspA2, UspA2H, or YfeA, followed by the addition of plasminogen. Thereafter, we added NHS together with uPA to the beads and incubated the beads for 2 h at 37°C. M. catarrhalis RH4 and Bc5 are serum resistant, whereas UspA2- and UspA2H-deficient mutants are highly susceptible to the bactericidal effect of NHS (9). NHS preincubated with beads coated with the various proteins was tested for bactericidal activity against the M. catarrhalis Bc5 ΔuspA2 and RH4 ΔuspA2H strains and two other serum-sensitive M. catarrhalis strains (KR509 and KR539). Importantly, beads with immobilized UspA2H/plasminogen neutralized the bactericidal effect of NHS and rescued 40.1 to 78.3% of the bacteria, in comparison to NHS or serum treated with the YfeA-coated control beads (Fig. 8). In parallel, UspA2/plasminogen-coated beads protected 15.05 to 78.60% of serum-sensitive M. catarrhalis bacteria. Samples consisting of UspA2/plasminogen-coated or UspA2H/plasminogen-coated beads without uPA showed only minor protection of serum-sensitive M. catarrhalis bacteria (Fig. 8). Beads treated with NHS only also showed results similar to those for the YfeA-coated beads. This result supported our hypothesis that M. catarrhalis utilizes human plasminogen to survive lethal attacks from the complement system.
FIG 8.

Plasminogen bound to UspA2 or UspA2H impairs serum bactericidal activity. Recombinant UspA2, UspA2H, or YfeA was immobilized on Dynabeads and incubated with plasminogen. NHS (10%) supplemented with uPA (20 U) was added to beads, followed by incubation for 2 h at 37°C. The serum was tested for bactericidal activity against serum-sensitive M. catarrhalis Bc5 ΔuspA2, RH4 ΔuspA2H, KR509, and KR539 strains. Beads were also coated with the control protein YfeA, which does not bind plasminogen. Bacterial survival in the absence of serum was set as 100%. In parallel, UspA2, UspA2H, and YfeA immobilized on Dynabeads and incubated with NHS in the absence of uPA were included as controls. Dynabeads were also treated with BSA (2.5% BSA in PBS), washed with PBS, and then added to NHS. The results shown are means of data from three independent experiments with triplicate samples, and bars denote standard errors of the means. Statistical analyses were performed by two-way analysis of variance. Statistical significance between data for NHS and those for other serum samples is shown. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
DISCUSSION
The majority of M. catarrhalis strains are considered to be noninvasive, and only in rare cases does Moraxella cause invasive disease. Importantly, M. catarrhalis does not secrete any proteases or toxins that potentially would damage the epithelial cell layer and the extracellular matrix (ECM) for deep tissue penetration (4). Despite this fact, M. catarrhalis successfully adheres to and resides at the surface of the host epithelium, resulting in the induction of a proinflammatory response. This bacterial species has a unique capacity to inactivate the acute-phase reactant and protease inhibitor α1-antichymotrypsin and thereby potentiates the effect of the protease chymotrypsin (28). This cunning strategy may induce excessive inflammation, resulting in more exposed ECM, which is beneficial for bacterial colonization. Another approach used by microbes to degrade the ECM is to attract the zymogen plasminogen. It has been reported that when plasminogen is bound to pathogens, it contributes to adherence to host tissues (29), degradation of ECM proteins resulting in increased invasion (30), and, finally, degradation of complement (25). Interestingly, our present results also suggest that M. catarrhalis utilizes host plasminogen to escape from the innate host defense. It was previously shown that plasminogen bound at the surface of cells of the respiratory pathogen H. influenzae degrades C3b (27). In parallel, Borrelia burgdorferi recruits plasminogen and inactivates C3b as well as C5 to escape the innate immune system (25). Similar to those findings, we observed that M. catarrhalis with plasmin at the bacterial surface also degrades C3b and C5, which most likely contributes to increased resistance against serum-mediated killing.
M. catarrhalis clinical isolates exhibit a high degree of serum resistance (7–9). This serum resistance is dependent on the acquisition of host serum complement regulators such as vitronectin, factor H, and C4BP and the complement components C3 and C3d (9, 11–14). Clinical isolates of M. catarrhalis have variable serum resistance that cannot be correlated with the recruitment of a single complement inhibitor. Hence, serum resistance is dependent on the recruitment of multiple complement regulators at the same time. For instance, one strain may acquire a substantial amount of vitronectin and less of the other factors, while another strain may acquire much more C4BP than vitronectin. In addition to the interaction of the well-known complement inhibitors of M. catarrhalis, our study suggests that plasminogen can also be used as a tool to inactivate the host complement system.
UspA1 and UspA2/UspA2H are major surface proteins of M. catarrhalis involved in interactions with fibronectin, vitronectin, laminin, C4BP, C3, C3d, and α1-antichymotrypsin (9, 11, 14, 17, 18, 28). In our hands, plasminogen binding occurred particularly with UspA2 and UspA2H. The head domains of UspA2 and UspA2H bind plasminogen that overlaps the vitronectin-, laminin-, and fibronectin-binding regions. Our inhibition experiments also suggested that plasminogen binding to UspA2 and UspA2H is inhibited by laminin and vitronectin, whereas fibronectin has the least inhibitory effect (Fig. 4). Fibronectin binds below the head region of UspA2 (18) and therefore blocks plasminogen binding only partially. Since the head regions of UspA2 and UspA2H bind multiple host proteins, binding to plasminogen might be conditional or dependent on the availability of putative ligands. When multiple ligands are present, as in the case where bacteria are incubated with NHS, UspA2- and UspA2H-mediated plasminogen binding to M. catarrhalis is still clearly observed (Fig. 1).
M. catarrhalis UspA proteins are also receptors for C3 and C3d. C3 binds to the stalk region of the UspA2 molecule, i.e., within the region spanning amino acids 200 to 539. On the other hand, C3d is recognized within the stalk and neck regions of UspA2 (amino acids 101 to 318). The target regions of C3 and C3d on UspA2 are thus separated from the plasminogen-binding site. Binding of C3 and C3d by UspA1 and UspA2 also results in increased serum resistance (13, 14). Since UspA2 attracts C3 or C3d, it may simultaneously result in the degradation of C3b in case the bacterial protein acquires plasminogen in the vicinity.
M. catarrhalis clinical isolates showed variable plasminogen-binding capacities. This variability was not correlated with UspA2 or UspA2H. UspA2 and UspA2H are diverse in their amino acid sequences and also in the sizes of their head regions (8), which may further influence plasminogen interactions. We have previously shown that UspA2 expression levels in clinical isolates are variable (14). Hence, the expression level of M. catarrhalis UspA2/UspA2H may also be a determining factor for plasminogen binding.
Respiratory pathogens, including Streptococcus pneumoniae, Mycobacterium tuberculosis, Streptococcus pyogenes, Staphylococcus aureus, Neisseria meningitidis, H. influenzae, and Mycoplasma pneumoniae, are known for hijacking plasminogen from the host (31). Bacterial plasminogen surface receptors have diverse structures and functions. Some bacteria have multiple plasminogen-binding proteins at their surface, for example, S. pneumoniae, which recruits plasminogen via choline-binding protein E (CBPE), PavB, PfbB, and PfbA. Moreover, S. pyogenes binds plasminogen via M and M-like proteins and type I fimbriae. S. aureus attracts plasminogen to its surface by using immunoglobulin-binding protein (Sbi) and extracellular fibrinogen-binding protein (31). Similarly, H. influenzae protein E and aspartase also bind plasminogen (27, 32). While diversity is the most common feature of bacterial plasminogen receptors, homologues of type I fimbriae, M-like proteins of Streptococcus spp., glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and enolases of multiple pathogens have the capacity to attract plasminogen (31). M. catarrhalis has not been tested previously for interactions with plasminogen. Our study shows that the trimeric autotransporters UspA2 and UspA2H are major plasminogen-binding proteins. However, it cannot be excluded that other proteins with a lower affinity for plasminogen might also be present at the surface of M. catarrhalis.
Plasminogen consists of five different homologous kringle domains (K1 to K5). These kringle domains, except for K3 (31), have the capacity to bind free lysine or lysine-like compounds. The highest lysine-binding affinity has been reported for K1, followed by the K4, K5, and K2 domains. Since lysine-like ligands, e.g., εACA and tranexamic acid, bind to kringle domains and mask them, these compounds have been used to verify bacterial interactions. We found that the binding of plasminogen to UspA2 and UspA2H can easily be inhibited by using εACA (Fig. 5). Moreover, the truncated fragments of plasminogen suggested that kringle domains 1 and 2 are involved in binding of UspA2 and UspA2H (Fig. 5E and F).
In conclusion, we have shown that the multifunctional proteins UspA2 and UspA2H recruit plasminogen to the surface of M. catarrhalis. The surface-bound plasminogen can be converted into highly active plasmin by host factors, and we postulate that this may contribute to bacterial virulence.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Foundations of Alfred Österlund, Anna and Edwin Berger, Greta and Johan Kock, Åke Wiberg, and O. E. and Edla Johansson, from the Lars Hierta Foundation, from the Swedish Medical Research Council (grant number K2015-57X-03163-43-4 [http://www.vr.se/]), from the Cancer Foundation at the University Hospital in Malmö, from the Physiographical Society (Forssman's Foundation), and from the Skåne County Council research and development foundation.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00310-15.
REFERENCES
- 1.Catlin BW. 1990. Branhamella catarrhalis: an organism gaining respect as a pathogen. Clin Microbiol Rev 3:293–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy TF. 1996. Branhamella catarrhalis: epidemiology, surface antigenic structure, and immune response. Microbiol Rev 60:267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karalus R, Campagnari A. 2000. Moraxella catarrhalis: a review of an important human mucosal pathogen. Microbes Infect 2:547–559. doi: 10.1016/S1286-4579(00)00314-2. [DOI] [PubMed] [Google Scholar]
- 4.Su YC, Singh B, Riesbeck K. 2012. Moraxella catarrhalis: from interactions with the host immune system to vaccine development. Future Microbiol 7:1073–1100. doi: 10.2217/fmb.12.80. [DOI] [PubMed] [Google Scholar]
- 5.Lambris JD, Ricklin D, Geisbrecht BV. 2008. Complement evasion by human pathogens. Nat Rev Microbiol 6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zipfel PF. 2009. Complement and immune defense: from innate immunity to human diseases. Immunol Lett 126:1–7. doi: 10.1016/j.imlet.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 7.Attia AS, Ram S, Rice PA, Hansen EJ. 2006. Binding of vitronectin by the Moraxella catarrhalis UspA2 protein interferes with late stages of the complement cascade. Infect Immun 74:1597–1611. doi: 10.1128/IAI.74.3.1597-1611.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su YC, Hallstrom BM, Bernhard S, Singh B, Riesbeck K. 2013. Impact of sequence diversity in the Moraxella catarrhalis UspA2/UspA2H head domain on vitronectin binding and antigenic variation. Microbes Infect 15:375–387. doi: 10.1016/j.micinf.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 9.Singh B, Blom AM, Unal C, Nilson B, Morgelin M, Riesbeck K. 2010. Vitronectin binds to the head region of Moraxella catarrhalis ubiquitous surface protein A2 and confers complement-inhibitory activity. Mol Microbiol 75:1426–1444. doi: 10.1111/j.1365-2958.2010.07066.x. [DOI] [PubMed] [Google Scholar]
- 10.Singh B, Su YC, Riesbeck K. 2010. Vitronectin in bacterial pathogenesis: a host protein used in complement escape and cellular invasion. Mol Microbiol 78:545–560. doi: 10.1111/j.1365-2958.2010.07373.x. [DOI] [PubMed] [Google Scholar]
- 11.Nordstrom T, Blom AM, Forsgren A, Riesbeck K. 2004. The emerging pathogen Moraxella catarrhalis interacts with complement inhibitor C4b binding protein through ubiquitous surface proteins A1 and A2. J Immunol 173:4598–4606. doi: 10.4049/jimmunol.173.7.4598. [DOI] [PubMed] [Google Scholar]
- 12.Bernhard S, Fleury C, Su YC, Zipfel PF, Koske I, Nordstrom T, Riesbeck K. 2014. Outer membrane protein OlpA contributes to Moraxella catarrhalis serum resistance via interaction with factor H and the alternative pathway. J Infect Dis 210:1306–1310. doi: 10.1093/infdis/jiu241. [DOI] [PubMed] [Google Scholar]
- 13.Nordstrom T, Blom AM, Tan TT, Forsgren A, Riesbeck K. 2005. Ionic binding of C3 to the human pathogen Moraxella catarrhalis is a unique mechanism for combating innate immunity. J Immunol 175:3628–3636. doi: 10.4049/jimmunol.175.6.3628. [DOI] [PubMed] [Google Scholar]
- 14.Hallstrom T, Nordstrom T, Tan TT, Manolov T, Lambris JD, Isenman DE, Zipfel PF, Blom AM, Riesbeck K. 2011. Immune evasion of Moraxella catarrhalis involves ubiquitous surface protein A-dependent C3d binding. J Immunol 186:3120–3129. doi: 10.4049/jimmunol.1002621. [DOI] [PubMed] [Google Scholar]
- 15.Lyskowski A, Leo JC, Goldman A. 2011. Structure and biology of trimeric autotransporter adhesins. Adv Exp Med Biol 715:143–158. doi: 10.1007/978-94-007-0940-9_9. [DOI] [PubMed] [Google Scholar]
- 16.Singh B, Su YC, Al-Jubair T, Mukherjee O, Hallstrom T, Morgelin M, Blom AM, Riesbeck K. 2014. A fine-tuned interaction between trimeric autotransporter Haemophilus surface fibrils and vitronectin leads to serum resistance and adherence to respiratory epithelial cells. Infect Immun 82:2378–2389. doi: 10.1128/IAI.01636-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan TT, Forsgren A, Riesbeck K. 2006. The respiratory pathogen Moraxella catarrhalis binds to laminin via ubiquitous surface proteins A1 and A2. J Infect Dis 194:493–497. doi: 10.1086/505581. [DOI] [PubMed] [Google Scholar]
- 18.Tan TT, Nordstrom T, Forsgren A, Riesbeck K. 2005. The respiratory pathogen Moraxella catarrhalis adheres to epithelial cells by interacting with fibronectin through ubiquitous surface proteins A1 and A2. J Infect Dis 192:1029–1038. doi: 10.1086/432759. [DOI] [PubMed] [Google Scholar]
- 19.Lahteenmaki K, Kuusela P, Korhonen TK. 2001. Bacterial plasminogen activators and receptors. FEMS Microbiol Rev 25:531–552. [DOI] [PubMed] [Google Scholar]
- 20.Singh B, Fleury C, Jalalvand F, Riesbeck K. 2012. Human pathogens utilize host extracellular matrix proteins laminin and collagen for adhesion and invasion of the host. FEMS Microbiol Rev 36:1122–1180. doi: 10.1111/j.1574-6976.2012.00340.x. [DOI] [PubMed] [Google Scholar]
- 21.Barthel D, Schindler S, Zipfel PF. 2012. Plasminogen is a complement inhibitor. J Biol Chem 287:18831–18842. doi: 10.1074/jbc.M111.323287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh B, Al-Jubair T, Morgelin M, Thunnissen MM, Riesbeck K. 2013. The unique structure of Haemophilus influenzae protein E reveals multiple binding sites for host factors. Infect Immun 81:801–814. doi: 10.1128/IAI.01111-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh B, Jalalvand F, Morgelin M, Zipfel P, Blom AM, Riesbeck K. 2011. Haemophilus influenzae protein E recognizes the C-terminal domain of vitronectin and modulates the membrane attack complex. Mol Microbiol 81:80–98. doi: 10.1111/j.1365-2958.2011.07678.x. [DOI] [PubMed] [Google Scholar]
- 24.Hallstrom T, Morgelin M, Barthel D, Raguse M, Kunert A, Hoffmann R, Skerka C, Zipfel PF. 2012. Dihydrolipoamide dehydrogenase of Pseudomonas aeruginosa is a surface-exposed immune evasion protein that binds three members of the factor H family and plasminogen. J Immunol 189:4939–4950. doi: 10.4049/jimmunol.1200386. [DOI] [PubMed] [Google Scholar]
- 25.Koenigs A, Hammerschmidt C, Jutras BL, Pogoryelov D, Barthel D, Skerka C, Kugelstadt D, Wallich R, Stevenson B, Zipfel PF, Kraiczy P. 2013. BBA70 of Borrelia burgdorferi is a novel plasminogen-binding protein. J Biol Chem 288:25229–25243. doi: 10.1074/jbc.M112.413872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nordstrom T, Jendholm J, Samuelsson M, Forsgren A, Riesbeck K. 2006. The IgD-binding domain of the Moraxella IgD-binding protein MID (MID962-1200) activates human B cells in the presence of T cell cytokines. J Leukoc Biol 79:319–329. [DOI] [PubMed] [Google Scholar]
- 27.Barthel D, Singh B, Riesbeck K, Zipfel PF. 2012. Haemophilus influenzae uses the surface protein E to acquire human plasminogen and to evade innate immunity. J Immunol 188:379–385. doi: 10.4049/jimmunol.1101927. [DOI] [PubMed] [Google Scholar]
- 28.Manolov T, Tan TT, Forsgren A, Riesbeck K. 2008. Moraxella-dependent alpha 1-antichymotrypsin neutralization: a unique virulence mechanism. Am J Respir Cell Mol Biol 38:609–617. doi: 10.1165/rcmb.2007-0289OC. [DOI] [PubMed] [Google Scholar]
- 29.Bergmann S, Schoenen H, Hammerschmidt S. 2013. The interaction between bacterial enolase and plasminogen promotes adherence of Streptococcus pneumoniae to epithelial and endothelial cells. Int J Med Microbiol 303:452–462. doi: 10.1016/j.ijmm.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Attali C, Frolet C, Durmort C, Offant J, Vernet T, Di Guilmi AM. 2008. Streptococcus pneumoniae choline-binding protein E interaction with plasminogen/plasmin stimulates migration across the extracellular matrix. Infect Immun 76:466–476. doi: 10.1128/IAI.01261-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanderson-Smith ML, De Oliveira DM, Ranson M, McArthur JD. 2012. Bacterial plasminogen receptors: mediators of a multifaceted relationship. J Biomed Biotechnol 2012:272148. doi: 10.1155/2012/272148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sjostrom I, Grondahl H, Falk G, Kronvall G, Ullberg M. 1997. Purification and characterisation of a plasminogen-binding protein from Haemophilus influenzae. Sequence determination reveals identity with aspartase. Biochim Biophys Acta 1324:182–190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.