

Abstract
Platelets are rapidly responsive sentinel cells that patrol the bloodstream and contribute to the host response to infection. Platelets have been reported to form heterotypic aggregates with leukocytes and may modulate their function. Here, we have investigated platelet-neutrophil complex formation and neutrophil function in response to distinct agonists. The endogenous platelet activator thrombin gave rise to platelet-dependent neutrophil activation, resulting in enhanced phagocytosis and bacterial killing. Streptococcus pyogenes is an important causative agent of severe infectious disease, which can manifest as sepsis and septic shock. M1 protein from S. pyogenes also mediated platelet-neutrophil complex formation; however, these neutrophils were dysfunctional and exhibited diminished chemotactic ability and bacterial killing. This reveals an important agonist-dependent neutrophil dysfunction during platelet-neutrophil complex formation and highlights the role of platelets during the immune response to streptococcal infection.
INTRODUCTION
The predominant role of platelets is to maintain hemostasis; however, these cells can also contribute to host defense and inflammation (1). Platelet activation and aggregation have been reported to occur in response to a number of bacterial pathogens (2). Platelets have been reported to bind to leukocytes, and activated platelets release proinflammatory substances that stimulate leukocyte function (3). Platelets form complexes more avidly with monocytes than neutrophils (4), and platelet-neutrophil complexes (PNCs) have been less investigated. There exist conflicting reports on the relative contribution of neutrophil activation to PNC formation. Peters et al. have demonstrated that neutrophil activation does not influence PNC formation (5), while other studies have reported that neutrophil activation can decrease complex formation (6). PNC formation has mainly been investigated in disease and is proposed to contribute to the pathogenesis of allergy (7), murine lung injury (8), and sepsis (9, 10); however, the importance of these complexes for neutrophil function is not clear. Significant cross talk occurs between coagulation and inflammation, and modulation of these systems contributes to the pathophysiology of sepsis (11). Streptococcus pyogenes is a leading human pathogen, and invasive infections by this organism, such as sepsis and septic shock, are an important cause of morbidity and mortality (12). The pathogenesis of invasive streptococcal infection involves dysregulation of multiple host defense systems, including leukocyte function and coagulation (13). M protein is a cell wall-associated bacterial protein that contributes to multiple aspects of streptococcal pathogenesis (14). M protein can also be released from the bacterial surface in a functionally active, soluble form. M1 protein released from S. pyogenes AP1 forms a complex with plasma fibrinogen, which directly stimulates neutrophil granule release (15, 16) and the formation of neutrophil extracellular traps (NETs) (17). M1 protein in complex with fibrinogen also mediates platelet activation (18). Neutrophil activation and platelet activation in direct response to M1 protein are also dependent on Fc-mediated activation of the cells by specific IgG toward the M1 protein (16, 18). In the present study, we investigate neutrophil function in platelet-neutrophil complexes formed in response to thrombin and M1 protein. We demonstrate that platelet-dependent PNC formation occurs in response to both agonists; however, these PNCs are functionally distinct. Thrombin gave rise to platelet-dependent neutrophil activation, while M1 protein generated dysfunctional neutrophils entrapped in a platelet and fibrinogen network.
MATERIALS AND METHODS
Blood collection.
Blood samples from seven healthy volunteers, five males and two females aged 25 to 39 years of age, were used throughout the study. The regional Ethical Review Board in Lund, Sweden, approved the recruitment of healthy blood donors to the study (reference number 657/2008). The donors had not taken antiplatelet medication in the preceding 10 days. Platelet-rich plasma (PRP) was prepared by centrifugation at 150 × g for 15 min. Washed platelets were prepared by gel filtration of PRP. Neutrophils were isolated using Lymphoprep (Axis Shield) according to the manufacturer's instructions.
Materials.
Thrombin was purchased from Chrono-Log and induces both platelet activation and fibrin polymerization; therefore, throughout this study, all experiments with thrombin were carried out in the presence of the peptide inhibitor of fibrin polymerization GPRP, from Bachem. ADP and formyl-methionyl-leucyl phenylalanine (fMLF) were purchased from Sigma. M1 protein was purified from the growth medium of an isogenic mutant strain that lacks the membrane-spanning region and secretes a soluble form of the protein (19). Anti-human CD62P (clone 9E1) for blocking experiments was purchased from R&D systems, and the control antibody (mouse IgG1 kappa isotype control clone P3) was from eBiosciences. The platelet inhibitor prostacyclin (PGI2) was from Bayer. CD61, CD42PerCP, CD62P PE, PAC1 FITC, CD11bPE-Cy5, and CD61PE were purchased from BD Biosciences, and CD11b.Activation epitope-FITC was from eBiosciences. Secondary antibodies were from Jackson ImmunoResearch. The enzyme-linked immunosorbent assay (ELISA) kits for neutrophil myeloperoxidase (MPO) and human neutrophil gelatinase-associated lipocalin (N-Gal) were from Hycult Biotech. The ELISA kit for neutrophil matrix metalloproteinase 9 (MMP-9) was from Nordic Biosite. Heparin-binding protein (HBP) release was determined using an in-house ELISA as previously described (20). Fibrinopeptide A generation was measured using an ELISA kit from Hyphen Biomed.
Flow cytometry.
Citrated whole blood was diluted in HEPES buffer (10 mM HEPES, 150 mM NaCl, 5 mM KCl, 1 mM MgSO4, pH 7.4) and incubated with agonists for 5 min: ADP (10 μM), thrombin (0.5 U/ml + GPRP), fMLF (1 μM), ADP + fMLF, or M1 protein (5 μg/ml). For platelet blockade experiments, diluted whole blood was incubated with anti-human CD62P (5 μg/ml), isotype control antibody (5 μg/ml), or prostacyclin (240 pg/ml) for 20 min, prior to stimulation with agonists. After incubation, antibodies were added to study the platelet population (CD42-PerCP, CD62P-PE, and PAC1-FITC). In parallel, samples were incubated with another set of antibodies to study the neutrophil population (CD61-PE, CD11b.Activation epitope-FITC, or CD11b-PerCP). Incubations were terminated after 15 min by addition of Uti-Lyse erythrocyte lysis (DakoCytomation). Samples were analyzed on a BD FacsCalibur flow cytometer. PNCs were determined as platelet-positive events (CD61) within the neutrophil population. The activation status of the neutrophil was determined using an antibody against the active conformation of the integrin CD11b (CD11b.Activation epitope).
ELISA.
Diluted whole blood was incubated with platelet and neutrophil agonists as described for flow cytometry. Cells were pelleted at 2,500 × g for 15 min, and the supernatant was subjected to ELISA for determination of neutrophil granule release products: MMP-9, N-Gal, MPO, and HBP. The fibrinopeptide A concentrations in the supernatant of thrombin and M1-activated samples were also measured by ELISA. Anti-M1 IgG antibodies were detected in plasma samples from healthy donors by ELISA as described previously (16).
Immunofluorescence microscopy.
PNC formation was induced by addition of platelet and neutrophil agonists as described for flow cytometry. The red cells were lysed, and the samples were fixed with 1% paraformaldehyde for 45 min at room temperature (RT) and washed with quenching buffer (Dulbecco's phosphate-buffered saline [D-PBS], 5% bovine serum albumin [BSA], 50 mM glycine). Primary antibodies (1:500 of mouse anti-human CD61; clone VI-PL2) or goat anti-human fibrinogen (polyclonal) were incubated with the samples for 60 min at RT. After washing with block buffer (D-PBS, 5% BSA), fluorochrome-conjugated secondary antibodies (1:1,000) were incubated with the samples for 60 min at RT. The samples were washed twice, adhered onto poly-l-lysine-coated coverslips, and mounted using DAPI (4′,6-diamidino-2-phenylindole) ProLong Gold medium. Acquisition of images was performed using a fluorescence microscope (Nikon Eclipse TE300 equipped with a Hamamatsu C4742-95 cooled charge-coupled-device [CCD] camera, using a Plan Apochromat 100× objective with numerical aperture [NA] of 1.4). To increase depth resolution, z-series were captured and out-of-focus light was removed by deconvolution (NIS-Elements 3; Nikon).
Neutrophil phagocytosis.
Neutrophil phagocytosis was investigated using flow cytometry as previously described (21). S. pyogenes AP1 was grown to mid-exponential phase (4 to 5 h) in Todd-Hewitt medium, washed twice in PBS, heat killed, and labeled with Alexa Fluor 647. Washed bacteria were suspended in RPMI medium containing 2% human serum for opsonization. Purified neutrophils (5 × 106/ml) were suspended in RPMI or PRP. Thrombin (0.5 U/ml + GPRP) was added to one sample of neutrophils in PRP to induce PNC formation. As a negative control, the platelet inhibitor prostacyclin was added to a sample of neutrophils in PRP in order to prevent platelet activation and PNC formation. After 20 min at RT, those platelets not associated with neutrophils were removed by washing at 200 × g for 5 min at 4°C, and the pellet was suspended in RPMI. Isolated neutrophils in the absence of platelets were either left untreated or primed with fMLF (1 μM). Neutrophils that had been suspended in platelets were not primed with fMLF. Labeled bacteria were added at a multiplicity of infection (MOI) of 15. Phagocytosis was initiated by incubation at 37°C with rotation and terminated after 60 min by placing the tubes on ice. DyLight 488-conjugated goat anti-human IgG was then added (1:500) for 30 min on ice to label extracellular bacteria. Samples were analyzed on a BD Accuri flow cytometer.
In a parallel experiment, bacterial killing was assessed using a modified Lancefield assay. Thrombin (0.5 U/ml + GPRP) or M1 protein (5 μg/ml) was added to 1 ml of citrated blood to induce PNC formation. After 15 min, washed S. pyogenes AP1 bacteria (2 × 104) were added. The bacterial load was determined by plating samples immediately upon addition of bacteria and after 1 h of incubation with rotation at 37°C. The multiplication factor was determined by dividing the bacterial count after 60 min by the bacterial count at the start of the incubation.
Scanning electron microscopy.
PNC formation was induced in undiluted whole blood by addition of platelet and neutrophil agonists. The red cells were lysed, and the cells were pelleted at 1,000 × g for 15 min, suspended in fixation solution (4% formaldehyde and 2.5% glutaraldehyde in sodium cacodylate), and absorbed onto poly-l-lysine-coated glass coverslips for 1 h. Samples were processed as previously described (22) and examined in a Philips/FEI XL30 FEG scanning electron microscope at an acceleration voltage of 5 kV and a working distance of 10 mm.
Neutrophil chemotaxis.
Purified neutrophils (2 × 106/ml) were suspended in PRP, and PNC formation was induced by addition of platelet and neutrophil agonists as described for neutrophil phagocytosis. Neutrophils were applied to a microchemotaxis chamber (NeuroProbe) for 30 min at 37°C, and directed migration toward fMLF (10 nM) was assessed according to the leading front principle with three cells in one high-power field (23, 24). Three fields were read per well, and the mean migration was calculated.
Neutrophil extracellular trap formation.
Purified neutrophils (2 × 106/ml) were suspended in PRP from M1 protein responders. PNC formation was induced by addition of buffer or 5 μg/ml M1 protein for 45 min. As a positive control, 100 mU/ml of glucose oxidase was added to purified neutrophils for 2.5 h. After incubation, all samples were applied to glass slides by cytospin, and the samples were mounted using mounting medium (DAPI ProLong Gold). Acquisition of images was performed using a fluorescence microscope (Nikon Eclipse TE300 equipped with a Hamamatsu C4742-95 cooled CCD camera, using a Plan Apochromat objective).
Statistics.
For flow cytometry data, variance among different treatments of blood samples was assessed with a one-way analysis of variance (ANOVA). Differences between various treatments were then assessed with a t test. For ELISA data, the difference between treatments was assessed with a t test. Differences among various treatments for immunofluorescence and phagocytosis assays were assessed with the Mann-Whitney test. All P values are uncorrected for multiple comparisons. All statistical analyses were performed in Graphpad Prism version 6.
RESULTS
Platelet activation results in platelet-dependent PNC formation and neutrophil activation.
Platelet and neutrophil activation in whole blood was determined using flow cytometry. ADP and thrombin generated platelet activation as expected (see Fig. S1 in the supplemental material), and both agonists gave rise to PNC formation (Fig. 1A). Thrombin generated significantly more PNCs and significantly greater neutrophil integrin activation than did ADP alone (Fig. 1A and B). The neutrophil agonist fMLF specifically activated neutrophils (Fig. 1B) but did not cause platelet activation (see Fig. S1 in the supplemental material) or PNC formation (Fig. 1A).
FIG 1.

PNC formation and subsequent neutrophil activation are dependent on platelet activation and CD62P translocation. Citrated whole blood was incubated with buffer, ADP (10 μM), thrombin (0.5 U/ml + GPRP peptide), fMLF (1 μM), or ADP + fMLF. Flow cytometry was used to determine PNC formation (A) and neutrophil CD11b integrin activation (CD11b-Activation) (B). The mean fluorescent intensity (MFI) was determined for each antibody, and this value for agonists was divided by the MFI for buffer alone to generate a fold increase for each agonist. The MFI for PNC formation in buffer alone was 4 ± 0.5, and for CD11b-Activation the MFI in buffer alone was 22 ± 5. The means ± standard errors of the means (SEM) from at least five experiments are shown. For inhibition experiments, citrated whole blood was left untreated (black bars) or preincubated with anti-CD62P (light gray bars) or prostacyclin (dark gray bars) before the addition of agonists and determination of PNC formation (C) and neutrophil CD11b integrin activation (D). The MFI for each antibody in the untreated sample was set as the maximum (100%), and all other values were expressed as a percentage of this value. The means ± SEM from four experiments are shown. (E) Neutrophils in whole blood were stimulated with the supernatant of resting washed platelets, thrombin-activated washed platelets, or fMLF (1 μM), and neutrophil CD11b integrin activation was determined. The MFI for fMLF was set as the maximum (100%), and all other values were expressed as a percentage of this value. The means ± SEM from four experiments are shown. (F) Isolated neutrophils in RPMI were stimulated with buffer, thrombin (0.5 U/ml + GPRP peptide), or fMLF (1 μM), and neutrophil CD11b integrin activation was determined. The means ± SEM from six experiments are shown. A one-tailed paired t test was used to analyze differences in all panels: *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001; ns, nonsignificant.
PNC formation in response to both ADP and thrombin was abolished in the presence of prostacyclin or blockade of platelet CD62P (Fig. 1C), demonstrating that PNC formation was strictly dependent on platelet activation and CD62P translocation. An isotype control antibody had no effect on PNC formation in response to ADP or thrombin (data not shown). In the presence of prostacyclin, neutrophil integrin activation in response to the platelet agonist thrombin was inhibited (Fig. 1D), while prostacyclin had no significant effect on neutrophil activation in response to the neutrophil agonist fMLF (Fig. 1D). Importantly, thrombin also failed to directly activate purified neutrophils in the absence of platelets (Fig. 1F), providing further evidence for platelet-mediated neutrophil integrin activation. Thrombin-mediated neutrophil integrin activation was significantly decreased, but not abolished, when the physical contact between the cells was blocked by anti-CD62P (Fig. 1D). In order to test the hypothesis that platelet release products also contribute to neutrophil activation, the supernatant of thrombin-activated isolated platelets was added to neutrophils in whole blood and neutrophil integrin activation was determined. These experiments were carried out in the presence of prostacyclin in order to prevent de novo activation of platelets. The supernatant of resting platelets failed to mediate neutrophil integrin activation, while the supernatant of thrombin-activated platelets generated significant neutrophil integrin activation (Fig. 1E), illustrating that platelet-dependent neutrophil integrin activation involves both direct contact with platelets and release of soluble mediators from platelets.
Platelet-dependent activation of neutrophils results in limited granule release.
Neutrophil activation results in mobilization of subsets of granules in a tightly regulated fashion. Flow cytometry was used to determine the translocation of CD11b (Fig. 2A) from the tertiary granules. ELISA was used to determine release of MMP-9 (Fig. 2B) from the tertiary granules, of N-Gal from the secondary granules (Fig. 2C), and of MPO (Fig. 2D) and HBP (Fig. 2E) from the primary granules. ADP or thrombin treatment of whole blood resulted in CD11b and MMP9 release but failed to mobilize N-Gal, MPO, or HBP from the neutrophils (Fig. 2). The neutrophil agonist fMLF alone or in combination with cytochalasin B was used as a positive control and mediated release of granule proteins (Fig. 2).
FIG 2.

Platelet activation results in limited neutrophil granule release. Citrated whole blood was stimulated with agonists: buffer, ADP (10 μM), thrombin (0.5 U/ml + GPRP peptide), fMLF (1 μM), fMLF + ADP, or fMLF + cytochalasin B (cytb); then, the supernatants were harvested, and neutrophil granule release products were determined for CD11b (A), MMP-9 (B), N-Gal (C), MPO (D), and HBP (E). Flow cytometry was used to determine CD11b release, and data are expressed as mean fluorescent intensity (A). MMP-9, N-Gal, and MPO levels were determined by ELISA, and data are expressed in nanograms per milliliter. HBP levels were determined by ELISA, and data are expressed as the percentage of the maximum release obtained by cell lysis with Triton X-100. The means ± SEM from at least 4 experiments are shown. A two-tailed paired t test was used to analyze differences between agonist- and buffer-treated samples: **, P ≤ 0.01; *, P ≤ 0.05; ns, nonsignificant.
M1 protein from S. pyogenes generates platelet-dependent PNCs.
We have previously reported that M1 protein mediates platelet activation (18) and neutrophil activation (15, 16). PNC formation was then investigated in response to M1 protein using flow cytometry. While ADP + fMLF induced PNC formation in all donors, an interindividual variation was observed in response to M1 protein (Fig. 3A). Some individuals responded vigorously with substantial PNC formation, whereas others gave no PNC formation above background (Fig. 3A). Based on these results, donors were categorized as responders (fold increase > 1) or nonresponders (fold increase = 1) to M1 protein (Fig. 3A). Furthermore, M1 protein induced neutrophil degranulation and upregulation of CD11b on the neutrophils of only those individuals that responded with PNC formation (Fig. 3B), whereas ADP + fMLF induced an upregulation of CD11b in all donors (Fig. 3B). The ability to respond with PNC formation correlated with the titer of IgG against M1 protein present in the donor's plasma (Fig. 3C). PNC formation in response to M1 protein was abolished in the presence of the platelet inhibitor prostacyclin or blockade of platelet CD62P (Fig. 3D). An isotype control antibody had no effect on PNC formation in response to M1 protein (data not shown). The activation status of the neutrophil, as judged by upregulation of CD11b, was not significantly decreased despite inhibition of PNC formation (Fig. 3D). This demonstrates that PNC formation is dependent on platelets, despite the presence of significant direct neutrophil activation by M1 protein.
FIG 3.

M1 protein from S. pyogenes generates platelet-dependent PNCs. Citrated whole blood was incubated with buffer, M1 protein (5 μg/ml), or fMLF + ADP. Flow cytometry was used to determine PNC formation (A) and neutrophil CD11b presentation on the neutrophil surface (B). The median fluorescent intensity (MFI) was determined for each antibody, and this value for M1 protein or fMLF + ADP stimulation was divided by the MFI for buffer alone to generate a fold increase. A fold increase of 1 reflects no change above buffer and an inability to respond to a stimulus (nonresponder). The amount of PNCs formed in response to M1 protein in different donors (n = 10) was measured and correlated to the titer of specific IgG against M1 protein from the same individual (C). Whole blood from four responder donors was left untreated (black bars) or preincubated with antibodies against CD62P (light gray bars) or prostacyclin (dark gray bars) prior to stimulation with M1 protein (5 μg/ml) (D). The MFI for each antibody in the untreated sample was set as the maximum (100%), and all other values were expressed as a percentage of this value. Data are expressed as means ± SEM from four independent donor experiments performed in duplicate. A two-tailed paired t test was used to analyze differences: ***, P ≤ 0.001; ns, nonsignificant. unact, unactivated (buffer).
Immunofluorescence microscopy was used to visualize the direct interaction between platelets and neutrophils. Single platelets and platelet aggregates associated with each neutrophil (Fig. 4). For each stimulus and each of three independent experiments, the number of platelets per neutrophil was determined for 60 neutrophils. Only platelets that were in direct contact with the neutrophil were counted, and while platelet aggregates were often observed (Fig. 4A), these were determined only as one point of contact and therefore one platelet per neutrophil. Consequently, a median of 2 platelets per neutrophil was determined for thrombin, ADP + fMLF, and M1 protein (Fig. 4B).
FIG 4.
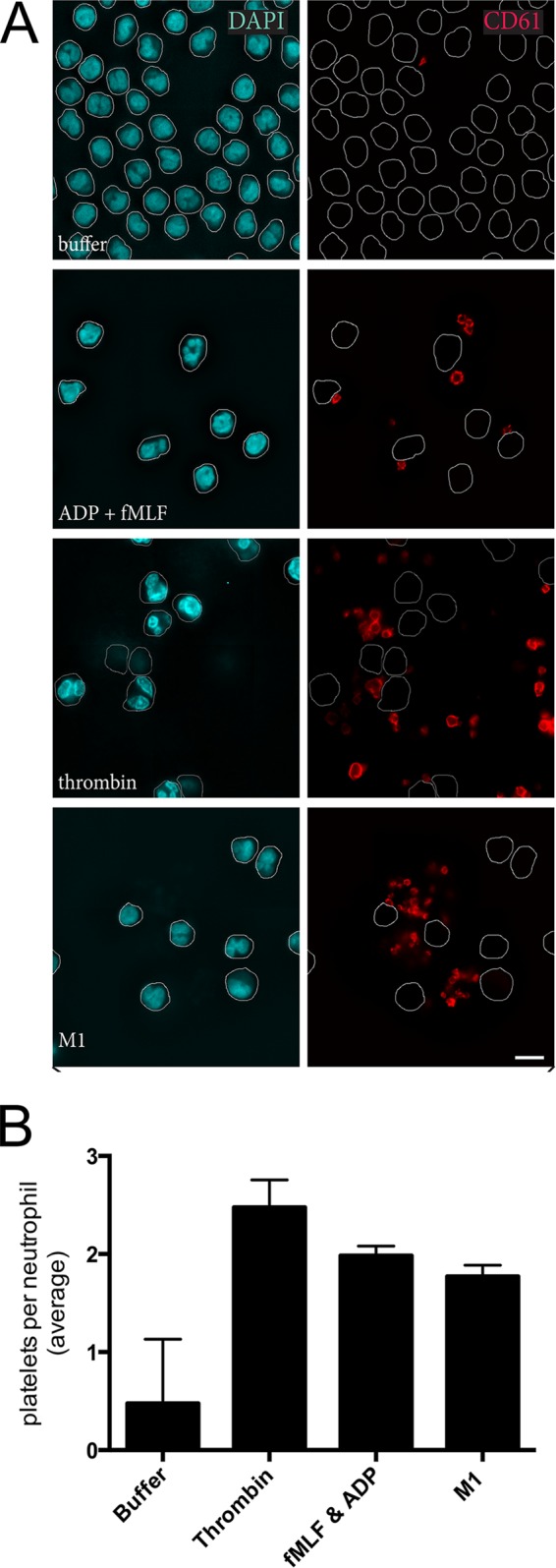
Platelets and platelet aggregates associate with neutrophils in PNCs. Immunofluorescence microscopy was used to visualize CD61-stained platelets and DAPI-stained neutrophils from whole blood stimulated with fMLF + ADP, thrombin + GPRP peptide, or M1 protein (A). For each stimulus and each of three independent experiments, the number of platelets per neutrophil was determined for 60 neutrophils. Data are expressed as medians ± interquartile ranges (B).
PNC formation in response to thrombin results in enhanced phagocytosis.
Neutrophils were purified, suspended in RPMI or PRP, and incubated with labeled bacteria preopsonized with IgG. Phagocytosis was assessed using flow cytometry to distinguish extracellular bacteria from intracellular bacteria (see Fig. S2 in the supplemental material). fMLF-primed neutrophils in RPMI performed efficient phagocytosis (Fig. 5B). Nonprimed neutrophils in PRP in the absence of PNC formation (PRP + inhibitor) did not perform phagocytosis (Fig. 5B), while PNC formation in response to thrombin significantly enhanced the phagocytic ability of nonprimed neutrophils (Fig. 5B). The phagocytosis assay could not be performed with M1 protein-treated samples due to the formation of large cellular aggregates that could not be subjected to flow cytometry. Scanning electron microscopy was used to investigate the morphology of these aggregates. PNCs were previously visualized using immunofluorescence microscopy (Fig. 4) of diluted blood; however, samples for electron microscopy were prepared in undiluted blood in order to have cell numbers equivalent to those used in the phagocytosis assay. No PNCs were observed in buffer-treated samples (Fig. 5B subpanel A). PNCs occurred in the presence of ADP, ADP + fMLF, and thrombin (Fig. 5B subpanels B, C, and D). M1 protein generated PNCs that were composed of almost all available neutrophils and platelets enmeshed in large aggregates (Fig. 5B subpanel E). The large aggregates generated in the M1 protein-mediated PNCs are not due to fibrin formation since fibrinopeptide A was not generated in samples treated with M1 protein (see Fig. S3 in the supplemental material).
FIG 5.

Neutrophil phagocytosis and bacterial killing are stimulated by PNC formation in response to thrombin. Neutrophils were suspended in RPMI alone (RPMI), in RPMI containing 1 μM fMLF (fMLF), in platelet-rich plasma in the presence of prostacyclin (PRP + inhibitor), or in platelet-rich plasma in the presence of 0.5 U/ml thrombin + GPRP peptide (PRP + thrombin). The neutrophils were incubated with opsonized labeled bacteria before determination of phagocytosis by flow cytometry. Data are expressed as the percentage of neutrophils that have intracellular bacteria (A). The medians ± the interquartile ranges from six experiments are shown. The Mann-Whitney test was used to analyze differences; **, P ≤ 0.01; *, P ≤ 0.05. (B) Citrated whole blood was incubated with buffer (subpanel A), ADP (10 μM) (subpanel B), thrombin (0.5 U/ml + GPRP peptide) (subpanel C), fMLF + ADP (subpanel D), or M1 protein (5 μg/ml) (subpanel E), and the morphology of the complexes formed was investigated using scanning electron microscopy. Citrated whole blood was incubated with buffer (unactivated), thrombin + GPRP peptide (thrombin), or M1 protein (M1) prior to addition of washed S. pyogenes bacteria. Bacterial growth was determined by viable count determination, and a multiplication factor was generated by dividing the bacterial count after 60 min by the bacterial count at the start of the incubation (C). The mean of three experiments per donor is represented as a symbol for each donor (filled circle, donor 1; filled square, donor 2; filled diamond, donor 3).
A Lancefield assay was performed with whole blood in order to assess the bactericidal effects of neutrophils in PNCs. S. pyogenes exhibits a donor-dependent ability to grow in human blood (25); therefore, at least a doubling of the bacteria in blood during 1 h was set as the cutoff for bacterial growth in our experiments. Using this criterion, S. pyogenes AP1 was determined to grow in the blood of three of the five donors that had previously been demonstrated to generate PNCs in response to M1 protein. For these three donors, bacterial growth was decreased upon formation of thrombin-induced PNCs compared with the negative control, while bacterial growth was increased upon formation of M1 protein-induced PNCs (Fig. 5C). This suggests that thrombin-induced PNCs enhance the bactericidal effects of neutrophils, while M1 protein-induced PNCs fail to do so.
PNC formation results in agonist-dependent inhibition of chemotaxis.
Purified neutrophils were suspended in PRP, and their chemotactic ability toward fMLF was assessed. As a negative control, neutrophils were suspended in PRP in the presence of prostacyclin, which inhibits PNC formation (PRP + inhibitor), and the distance migrated under these conditions was set to 100% for each experiment. The migration of neutrophils treated with other agonists was expressed as a percentage of this value. PNC formation in response to thrombin or ADP + fMLF did not influence neutrophil chemotaxis (Fig. 6A). PNC formation in response to M1 protein resulted in fewer neutrophils entering into the chemotaxis membrane, and the neutrophils at the leading front migrated a significantly shorter distance (Fig. 6B). This was a platelet-dependent effect since M1 protein did not inhibit chemotaxis of neutrophils in platelet-poor plasma (PPP + M1) (Fig. 6B). NET formation did not occur when neutrophils were suspended in PRP treated with M1 protein, while the positive control, glucose oxidase, generated significant NET formation (Fig. 6C). In order to further investigate the role of NET formation, the preformed PNCs were treated with DNase at concentrations that disintegrate NETs (200 mU at 37°C for 30 min) prior to chemotaxis. The mean migration of neutrophils in M1 protein-stimulated PNCs in the presence of DNase remained significantly decreased (Fig. 6B).
FIG 6.

Neutrophil chemotaxis is diminished by PNC formation in response to M1 protein from S. pyogenes. Neutrophils were allowed to migrate toward fMLF (10 nM), and the mean migration (in micrometers) was determined. The effect of platelets on chemotaxis was assessed by suspension of isolated neutrophils in platelet-rich plasma in the presence of prostacyclin (PRP + inhibitor), thrombin + GPRP peptide (PRP + thrombin), or fMLF+ADP (PRP + fMLF&ADP). The mean migration (in micrometers) of neutrophils in PRP + inhibitor was set to 100% for each experiment, and the migration of all other agonists was expressed as a percentage of this value (A). In parallel experiments, neutrophils were suspended in platelet-rich plasma in the presence of prostacyclin (PRP + inhibitor), M1 protein (PRP + M1), or M1 protein + DNase (PRP + M1+ DNase). As an additional control for M1 protein, neutrophils were suspended in platelet-poor plasma in the presence of M1 protein (PPP + M1) (B). Data are expressed as means ± SEM from at least 3 experiments. A two-tailed paired t test was used to analyze the differences; **, P ≤ 0.01. (C) NET formation was assessed by immunofluorescence microscopy of DAPI-stained neutrophils in RPMI (control) or platelet-rich plasma in the presence of M1 protein (PRP +M1) after 1 h to mimic the conditions of the chemotaxis assay. As an additional control for NET formation, neutrophils in RPMI were left unstimulated or stimulated with 100 mU/ml of glucose oxidase for 2.5 h prior to DAPI staining.
Fibrinogen is associated with PNCs formed in response to M1 protein.
Platelet and neutrophil activation by M1 protein has previously been demonstrated to be dependent on the formation of a complex between M1 protein and plasma fibrinogen. Immunofluorescence microscopy was used to determine the association of plasma fibrinogen with thrombin- or M1 protein-stimulated PNCs. Fibrinogen was associated with platelet aggregates in response to both stimuli; however, only in the case of M1 protein were large amounts of fibrinogen incorporated into the PNCs (Fig. 7). The tight interactions between neutrophils, platelets, and fibrinogen were evident only in the M1-stimulated samples (Fig. 7, lower panels).
FIG 7.

PNCs formed in response to M1 protein from S. pyogenes contain aggregates of fibrinogen. Immunofluorescence microscopy was used to visualize CD61-stained platelets, DAPI-stained neutrophils, and fibrinogen in whole blood stimulated with M1 protein or thrombin + GPRP peptide. The images were scaled and acquired with the same settings. Representative deconvoluted images from two experiments are shown. The upper panels are collapsed three-dimensional (3D) stacks to visualize all stained neutrophils, platelets, and fibrinogens in the field of view. The lower panels are 3D representations to exemplify the tight interactions between all three components.
DISCUSSION
The underlying pathophysiology of invasive infectious disease, such as sepsis and septic shock, is complex, and multiple host response systems become dysregulated. PNCs have been demonstrated to occur during the pathogenesis of diverse cardiovascular diseases, including sepsis (26); however, neutrophil function within these complexes has not been elucidated. We report that agonist-dependent modulation of neutrophil function occurs during PNC formation. Thrombin-mediated PNCs resulted in platelet-dependent neutrophil activation and enhanced neutrophil phagocytosis and killing. PNC formation in response to M1 protein generated neutrophils that were enmeshed in large heterotypic aggregates with platelets and fibrinogen and were compromised in their functional ability.
The potent platelet agonist thrombin generated neutrophil activation through physical contact but also through release of soluble factor(s). Although neutrophils have protease-activated receptors (PARs) (27), they do not respond directly to thrombin (Fig. 1F). Our findings imply that in the absence of a direct neutrophil agonist, activated platelets stimulate phagocytosis and bacterial killing by neutrophils. This reveals an important proinflammatory role for thrombin-activated platelets. Aggregates of activated platelets adhere to the endothelium and facilitate neutrophil adherence to and transmigration across the endothelium (28, 29), and neutrophils have recently been demonstrated to scan the intravascular space for activated platelets prior to transmigration (30). Activation of CD11b is an important step in neutrophil adhesion and transmigration of the endothelium (31); however, thrombin-mediated PNC formation in the circulation, as investigated in our study, did not stimulate neutrophil chemotaxis. This suggests that distinct neutrophil responses are generated by PNC formation in the circulation compared with PNC formation on the activated endothelium.
PNC formation occurs during sepsis; however, there are conflicting data with regard to the relative contribution of platelets and neutrophils to this PNC formation. Peters et al. have reported that neutrophils promote PNC formation (32), while Gawaz et al. have reported platelets to be the most important players in platelet-neutrophil adhesion and in the development of organ dysfunction in sepsis (9). In our study, it is clear that PNC formation is strictly dependent on platelet activation and translocation of CD62P, even in the presence of potent direct neutrophil activation by either fMLF or M1 protein.
M protein is an important virulence factor for S. pyogenes and contributes to multiple aspects of the dysregulated immune response during invasive infection (14). Our study reveals an important aspect of the proinflammatory repertoire of the soluble form of M1 protein. PNCs formed in response to M1 protein contain neutrophils that exhibit diminished chemotaxis and bacterial killing compared with neutrophils in thrombin-induced PNCs. The majority of experimental models of sepsis have been performed with purified lipopolysaccharide (LPS) from Gram-negative bacteria; however, these results cannot be applied to Gram-positive pathogens. PNCs have been reported to occur in response to LPS, resulting in the formation of platelet-dependent NETs that may ensnare bacteria but also cause collateral damage at the local site of inflammation (33). We demonstrate that NET formation does not contribute to neutrophil dysfunction in M1 protein-induced PNCs. The interaction between soluble M1 protein and plasma fibrinogen leads to the incorporation of large amounts of fibrinogen in M1 protein-induced PNCs, in contrast to thrombin (Fig. 7). This fibrinogen entrapment likely explains the neutrophil dysfunction associated with M1 protein and is in agreement with other reports on the pathological effects of complex formation between M1 protein and fibrinogen (34, 35). The ability to acquire plasma fibrinogen is relatively conserved for streptococcal M proteins; therefore, the mechanisms described here may be conserved for other serotypes. There is an interindividual variation in the ability to respond with PNC formation, and this is due to specific IgG against the M1 protein present in only certain individuals. This is in line with our previous findings that direct neutrophil activation and direct platelet activation in response to M1 protein are IgG dependent (16, 18). Individuals with IgG against certain regions of the M1 protein produced during a primary infection may therefore be more at risk of the severe manifestations of sepsis in response to reinfection with S. pyogenes.
Vascular dysfunction and organ damage are a hallmark of streptococcal sepsis (36), and neutrophils play a central role in the pathogenesis of vascular dysfunction (37). We propose that PNCs formed in response to M1 protein may adhere to the inflamed endothelium and occlude the microvasculature. Interestingly, the administration of M1 protein to mice has previously been demonstrated to give rise to vascular dysfunction (15, 34, 35). We have recently reported that PNCs are formed in the circulation in response to S. pyogenes and that platelets contribute to the pathogenesis of infection in a mouse model of streptococcal sepsis (38). Furthermore, PNCs formed in response to M1 protein occur at a distance from the bacteria; therefore, the bacteria can avoid entrapment and exposure to antimicrobial effector mechanisms within the complexes. Collectively, the data demonstrate that the functional consequences of PNC formation for the neutrophil are agonist dependent and that PNC formation may be an important event in the pathogenesis of sepsis.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported in part by the Swedish Research Council (projects 21112 and 21587), The Royal Physiographic Society in Lund, and the foundations of Åke-Wiberg, Crafoord, and Österlund.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00508-15.
REFERENCES
- 1.Semple JW, Freedman J. 2010. Platelets and innate immunity. Cell Mol Life Sci 67:499–511. doi: 10.1007/s00018-009-0205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox D, Kerrigan SW, Watson SP. 2011. Platelets and the innate immune system: mechanisms of bacterial-induced platelet activation. J Thromb Haemost 9:1097–1107. doi: 10.1111/j.1538-7836.2011.04264.x. [DOI] [PubMed] [Google Scholar]
- 3.Ghasemzadeh M, Hosseini E. 2013. Platelet-leukocyte crosstalk: linking proinflammatory responses to procoagulant state. Thromb Res 131:191–197. doi: 10.1016/j.thromres.2012.11.028. [DOI] [PubMed] [Google Scholar]
- 4.Michelson AD, Barnard MR, Krueger LA, Valeri CR, Furman MI. 2001. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation 104:1533–1537. doi: 10.1161/hc3801.095588. [DOI] [PubMed] [Google Scholar]
- 5.Peters MJ, Heyderman RS, Hatch DJ, Klein NJ. 1997. Investigation of platelet-neutrophil interactions in whole blood by flow cytometry. J Immunol Methods 209:125–135. doi: 10.1016/S0022-1759(97)00139-7. [DOI] [PubMed] [Google Scholar]
- 6.Rinder HM, Tracey JL, Rinder CS, Leitenberg D, Smith BR. 1994. Neutrophil but not monocyte activation inhibits P-selectin-mediated platelet adhesion. Thromb Haemost 72:750–756. [PubMed] [Google Scholar]
- 7.Pitchford SC, Yano H, Lever R, Riffo-Vasquez Y, Ciferri S, Rose MJ, Giannini S, Momi S, Spina D, O'connor B, Gresele P, Page CP. 2003. Platelets are essential for leukocyte recruitment in allergic inflammation. J Allergy Clin Immunol 112:109–118. doi: 10.1067/mai.2003.1514. [DOI] [PubMed] [Google Scholar]
- 8.Zarbock A, Singbartl K, Ley K. 2006. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest 116:3211–3219. doi: 10.1172/JCI29499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gawaz M, Dickfeld T, Bogner C, Fateh-Moghadam S, Neumann FJ. 1997. Platelet function in septic multiple organ dysfunction syndrome. Intensive Care Med 23:379–385. doi: 10.1007/s001340050344. [DOI] [PubMed] [Google Scholar]
- 10.Russwurm S, Vickers J, Meier-Hellmann A, Spangenberg P, Bredle D, Reinhart K, Lösche W. 2002. Platelet and leukocyte activation correlate with the severity of septic organ dysfunction. Shock 17:263–268. doi: 10.1097/00024382-200204000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Opal SM, Esmon CT. 2003. Bench-to-bedside review: functional relationships between coagulation and the innate immune response and their respective roles in the pathogenesis of sepsis. Crit Care 7:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carapetis J, Steer A, Mulholland E, Weber M. 2005. The global burden of group A streptococcal diseases. Lancet Infect Dis 5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 13.Walker MJ, Barnett TC, Mcarthur JD, Cole JN, Gillen CM, Henningham A, Sriprakash KS, Sanderson-Smith ML, Nizet V. 2014. Disease manifestations and pathogenic mechanisms of group A streptococcus. Clin Microbiol Rev 27:264–301. doi: 10.1128/CMR.00101-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oehmcke S, Shannon O, Mörgelin M, Herwald H. 2010. Streptococcal M proteins and their role as virulence determinants. Clin Chim Acta 411:1172–1180. doi: 10.1016/j.cca.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 15.Herwald H, Cramer H, Mörgelin M, Russell W, Sollenberg U, Norrby-Teglund A, Flodgaard H, Lindbom L, Björck L. 2004. M protein, a classical bacterial virulence determinant, forms complexes with fibrinogen that induce vascular leakage. Cell 116:367–379. doi: 10.1016/S0092-8674(04)00057-1. [DOI] [PubMed] [Google Scholar]
- 16.Kahn F, Mörgelin M, Shannon O, Norrby-Teglund A, Herwald H, Olin AI, Björck L. 2008. Antibodies against a surface protein of Streptococcus pyogenes promote a pathological inflammatory response. PLoS Pathog 4(9):e1000149. doi: 10.1371/journal.ppat.1000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oehmcke S, Mörgelin M, Herwald H. 2009. Activation of the human contact system on neutrophil extracellular traps. J Innate Immun 1:225–230. doi: 10.1159/000203700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shannon O, Hertzén E, Norrby-Teglund A, Mörgelin M, Sjöbring U, Björck L. 2007. Severe streptococcal infection is associated with M protein-induced platelet activation and thrombus formation. Mol Microbiol 65:1147–1157. doi: 10.1111/j.1365-2958.2007.05841.x. [DOI] [PubMed] [Google Scholar]
- 19.Collin M, Olsén A. 2000. Generation of a mature streptococcal cysteine proteinase is dependent on cell wall-anchored M1 protein. Mol Microbiol 36:1306–1318. [DOI] [PubMed] [Google Scholar]
- 20.Tapper HH, Karlsson AA, Mörgelin MM, Flodgaard HH, Herwald HH. 2002. Secretion of heparin-binding protein from human neutrophils is determined by its localization in azurophilic granules and secretory vesicles. Blood 99:1785–1793. doi: 10.1182/blood.V99.5.1785. [DOI] [PubMed] [Google Scholar]
- 21.Nordenfelt P. 2014. Quantitative assessment of neutrophil phagocytosis using flow cytometry. Methods Mol Biol 1124:279–289. doi: 10.1007/978-1-62703-845-4_18. [DOI] [PubMed] [Google Scholar]
- 22.Svensson L, Baumgarten M, Mörgelin M, Shannon O. 2014. Platelet activation by Streptococcus pyogenes leads to entrapment in platelet aggregates, from which bacteria subsequently escape. Infect Immun 82:4307–4314. doi: 10.1128/IAI.02020-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nybo M, Sørensen O, Leslie R, Wang P. 1998. Reduced expression of C5a receptors on neutrophils from cord blood. Arch Dis Child Fetal Neonatal Ed 78:F129–F132. doi: 10.1136/fn.78.2.F129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Falk W, Goodwin RH, Leonard EJ. 1980. A 48-well micro chemotaxis assembly for rapid and accurate measurement of leukocyte migration. J Immunol Methods 33:239–247. [DOI] [PubMed] [Google Scholar]
- 25.Lancefield RC. 1962. Current knowledge of type-specific M antigens of group A streptococci. J Immunol 89:307–313. [PubMed] [Google Scholar]
- 26.Totani L, Evangelista V. 2010. Platelet-leukocyte interactions in cardiovascular disease and beyond. Arterioscler Thromb Vasc Biol 30:2357–2361. doi: 10.1161/ATVBAHA.110.207480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Howells GL, Macey MG, Chinni C, Hou L, Fox MT, Harriott P, Stone SR. 1997. Proteinase-activated receptor-2: expression by human neutrophils. J Cell Sci 110(Part 7):881–887. [DOI] [PubMed] [Google Scholar]
- 28.Diacovo TG, Roth SJ, Buccola JM, Bainton DF, Springer TA. 1996. Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the beta 2-integrin CD11b/CD18. Blood 88:146–157. [PubMed] [Google Scholar]
- 29.Zwaginga JJ, Torres HI, Lammers J, Sixma JJ, Koenderman L, Kuijper PH. 1999. Minimal platelet deposition and activation in models of injured vessel wall ensure optimal neutrophil adhesion under flow conditions. Arterioscler Thromb Vasc Biol 19:1549–1554. doi: 10.1161/01.ATV.19.6.1549. [DOI] [PubMed] [Google Scholar]
- 30.Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, Nácher M, Pitaval C, Radovanovic I, Fukui Y, McEver RP, Filippi M-D, Lizasoain I, Ruiz-Cabello J, Zarbock A, Moro MA, Hidalgo A. 2014. Neutrophils scan for activated platelets to initiate inflammation. Science 346:1234–1238. doi: 10.1126/science.1256478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zen K, Guo Y-L, Li L-M, Bian Z, Zhang C-Y, Liu Y. 2011. Cleavage of the CD11b extracellular domain by the leukocyte serprocidins is critical for neutrophil detachment during chemotaxis. Blood 117:4885–4894. doi: 10.1182/blood-2010-05-287722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peters MJ, Heyderman RS, Faust S, Dixon GLJ, Inwald DP, Klein NJ. 2003. Severe meningococcal disease is characterized by early neutrophil but not platelet activation and increased formation and consumption of platelet-neutrophil complexes. J Leukoc Biol 73:722–730. doi: 10.1189/jlb.1002509. [DOI] [PubMed] [Google Scholar]
- 33.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, Devinney R, Doig CJ, Green FHY, Kubes P. 2007. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 13:463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 34.McNamara C, Zinkernagel AS, Macheboeuf P, Cunningham MW, Nizet V, Ghosh P. 2008. Coiled-coil irregularities and instabilities in group A Streptococcus M1 are required for virulence. Science 319:1405–1408. doi: 10.1126/science.1154470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macheboeuf P, Buffalo C, Fu C-Y, Zinkernagel AS, Cole JN, Johnson JE, Nizet V, Ghosh P. 2011. Streptococcal M1 protein constructs a pathological host fibrinogen network. Nature 472:64–68. doi: 10.1038/nature09967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stevens DL. 2003. Group A streptococcal sepsis. Curr Infect Dis Rep 5:379–386. doi: 10.1007/s11908-003-0017-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edens HA, Parkos CA. 2003. Neutrophil transendothelial migration and alteration in vascular permeability: focus on neutrophil-derived azurocidin. Curr Opin Hematol 10:25–30. doi: 10.1097/00062752-200301000-00005. [DOI] [PubMed] [Google Scholar]
- 38.Kahn F, Hurley S, Shannon O. 2013. Platelets promote bacterial dissemination in a mouse model of streptococcal sepsis. Microbes Infect 15:669–677. doi: 10.1016/j.micinf.2013.05.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.