

Abstract
We previously reported that Neisseria meningitidis internalization into human brain microvasocular endothelial cells (HBMEC) was triggered by the influx of extracellular l-glutamate via the GltT-GltM l-glutamate ABC transporter, but the underlying mechanism remained unclear. We found that the ΔgltT ΔgltM invasion defect in assay medium (AM) was alleviated in AM without 10% fetal bovine serum (FBS) [AM(−S)]. The alleviation disappeared again in AM(−S) supplemented with 500 μM glutamate. Glutamate uptake by the ΔgltT ΔgltM mutant was less efficient than that by the wild-type strain, but only upon HBMEC infection. We also observed that both GltT-GltM-dependent invasion and accumulation of ezrin, a key membrane-cytoskeleton linker, were more pronounced when N. meningitidis formed larger colonies on HBMEC under physiological glutamate conditions. These results suggested that GltT-GltM-dependent meningococcal internalization into HBMEC might be induced by the reduced environmental glutamate concentration upon infection. Furthermore, we found that the amount of glutathione within the ΔgltT ΔgltM mutant was much lower than that within the wild-type N. meningitidis strain only upon HBMEC infection and was correlated with intracellular survival. Considering that the l-glutamate obtained via GltT-GltM is utilized as a nutrient in host cells, l-glutamate uptake via GltT-GltM plays multiple roles in N. meningitidis internalization into HBMEC.
INTRODUCTION
Neisseria meningitidis is a Gram-negative microorganism and an exclusive human pathogen. It usually exists in an asymptomatic nasopharyngeal carriage state at a rate of 0.4% to 25% in human populations (1, 2). However, N. meningitidis can cause devastating invasive diseases, such as septicemia or meningitis, by penetration of the mucosal tissue, invasion of the bloodstream, and colonization of the meninges. Human genome-wide association studies (3, 4) identified single nucleotide polymorphisms in complement factor H (CFH) and CFH-related protein 3 (CFHR3), and CFHR3 promotes immune activation by acting as an antagonist of CFH (5). Environmental conditions, such as smoking (6, 7) and climate (8), are also likely to be important in determining the outcome of infection. However, the exact reasons why the diseases occur in some individuals and not in others remain unclear.
Many attempts to identify bona fide virulence factors in N. meningitidis have been reported. Surface molecules, such as Opa, Opc, pili, lipooligosaccharide, and the polysaccharide capsule, have been identified as adhesive and invasive factors in N. meningitidis (9–12). In addition, genome informatics also revealed several minor adhesion and invasion proteins, such as App (13), NhhA (14), MspA (15), and NadA (16). Other attempts to find meningococcal virulence factors by genome informatics involved comparisons between commensal and pathogenic bacteria (17) and the modes of bacterial virulence evolution (18). However, many of the so-called meningococcal “virulence genes” are also present in commensal neisserial species (19, 20), and attempts to identify meningococcal virulence factors that are restricted only to hyperinvasive strains have so far failed (21). Likewise, a prominent virulence system, such as the type III secretion machinery present in many enteric pathogens, has not been found in N. meningitidis (22). Thus, the pathogenic factors that determine the virulence of N. meningitidis remain to be elucidated.
l-Glutamate is an important molecule for all living organisms, in which it plays various roles. For example, in mammals, l-glutamate is the predominant excitatory neurotransmitter in the nervous system (23). In many prokaryotes, glutamate is an intermediate product for ammonium assimilation (24, 25). In N. meningitidis, glutamate is essential for growth in chemically defined medium (26), as well as in the intracellular milieu (27). N. meningitidis has two glutamate transporters: the high-sodium (Na+)-dependent glutamate transporter GltS (28, 29) and the l-glutamate ABC-type transporter GltT-GltM, which functions under low-Na+ conditions (28–30). GltT-GltM is reportedly required for survival in HeLa cells (27, 28), as well as for resistance to neutrophil oxidative burst (29).
In contrast to neutrophils and macrophages, endothelial cells have classically been perceived as playing a quiescent role in the host immune response by acting as barriers to the influx of microorganisms and by releasing chemoattractants that recruit professional phagocytes to sites of infection. In the past decade, however, accumulated data have begun to portray endothelial cells as more active participants in host defense. After exposure to infectious agents, endothelial cells can generate increased concentrations of reactive oxygen species (ROS) (31–34). There is indirect evidence that in neisseriae, cervical epithelial cells may have an oxidative defense capacity, since several mutants of Neisseria gonorrhoeae that are susceptible to ROS killing in vitro also have decreased survival within the cells (35). In N. meningitidis, two superoxide dismutases (encoded by sodB and sodC), glutathione peroxidase (encoded by gpxA), and catalase (encoded by kat) function as antioxidants against ROS (36), but the mechanism of meningococcal survival against intracellular ROS has remained unclear.
We previously reported that meningococcal internalization into human endothelial and epithelial cells is triggered by the influx of extracellular l-glutamate via the GltT-GltM l-glutamate ABC transporter (30), but the mechanism was unknown. In this study, we found several indirect lines of evidence suggesting that GltT-GltM-dependent meningococcal internalization into human brain microvasocular endothelial cells (HBMEC) is induced by the reduction of the environmental glutamate concentration upon infection. Moreover, we also found that glutamate uptake via GltT-GltM enhanced glutathione production, which resulted in increased meningococcal survival in HBMEC. Considering the fact that the glutamate imported via GltT-GltM was utilized as a nutrient for meningococcal survival in host cells (27), the uptake of glutamate via GltT-GltM in N. meningitidis seems to perform many biological functions in meningococcal internalization into host cells.
MATERIALS AND METHODS
Bacterial growth conditions.
N. meningitidis strains (stored at −80°C) were routinely grown on GC agar plates at 37°C in a 5% CO2 atmosphere (37). Brain heart infusion (Becton-Dickinson) agar containing 3% defibrinated horse blood (Nihon Biotest, Japan), was used for selecting kanamycin-resistant N. meningitidis strains (38). Escherichia coli was grown on Luria (L) plates or in L broth liquid cultures at 37°C. When required, antibiotics were added at the following concentrations: kanamycin at 150 μg/ml, erythromycin at 4 μg/ml, and spectinomycin at 75 μg/ml for N. meningitidis; kanamycin at 50 μg/ml, ampicillin at 50 μg/ml, and spectinomycin at 75 μg/ml for E. coli. All of the meningococcal strains used in this study were derivatives of N. meningitidis strain HT1125. The HT1125 strain is an unencapsulated (ΔsiaB-ΔsiaD::kan) mutant of an NIID280 N. meningitidis strain, which is untypeable (Opc− Pili+ Opa+) and belongs to the ST-2032 complex (38). The N. meningitidis strains used in this study are listed in Table 1.
TABLE 1.
N. meningitidis mutant strains used in this study
| Strain | Genotype | Parent strain | Reference |
|---|---|---|---|
| HT1125 | ΔsiaB-ΔsiaD::kan | NIID280 | 38 |
| HT1414 | ΔsiaB-ΔsiaD::kan ΔgltT-ΔgltM::ermC | HT1125 | 30 |
| HT1156 | ΔsiaB-ΔsiaD::kan pilE::ermC | HT1125 | 38 |
| HT1688 | ΔsiaB-ΔsiaD::kan ΔpilV::ermC | HT1125 | 42 |
| HT1876 | ΔsiaB-ΔsiaD::kan ΔgshB::spc | HT1125 | This study |
| HT1520 | ΔsiaB-ΔsiaD::kan gltS::spc | HT1125 | 30 |
Construction of meningococcal mutants.
To construct the N. meningitidis mutant with a gshB deletion, a 2-kb DNA fragment from N. meningitidis HT1125 chromosomal DNA containing the gshB gene (1 kb) and its upstream (0.5 kb) and downstream (0.5 kb) regions was amplified with the primers gshB-1 (CCCAAAGGCTTCAAACGCCTGATT) and gshB-2 (ACACCAGCAGTACGTTCAGCCACA) with PrimeStar Max DNA polymerase (TaKaRa Bio, Japan) and cloned into the SmaI site of the pTWV228 vector (4 kb; TaKaRa Bio) to construct pHT1055 (6 kb). The 5-kb DNA region of pHT1055, which lacked the region including the gshB gene, was amplified again with the primers gshB-3 (ATTTCCTTTCCGGTGTGCCGAATG) and gshB-4 (ACCGATGCCGTCTGAAAGGCTTTT) with PrimeStar Max DNA polymerase and was ligated to a spectinomycin resistance gene (spc) to construct pHT1058. The linearized pHT1058 (500 ng) was transformed into HT1125, and spectinomycin-resistant (Spr) clones were selected, resulting in the gshB deletion mutant of HT1125, named HT1876 (Table 1). Transformation of the N. meningitidis strains was performed as described previously (37).
Tissue culture.
HBMEC were cultivated as described previously (39). For a given experiment, HBMEC were seeded in a culture flask or dish or on a coverglass and used within 2 days after reaching confluence.
Determination of host cell-associated and internalized bacteria.
HBMEC were cultivated on gelatin-coated 96-well tissue culture plates (Iwaki, Japan) at 37°C in a 5% CO2 atmosphere for 2 days to a concentration of 1 × 104 cells/well. At 2 h prior to bacterial infection, the culture medium was replaced with assay medium (AM), which was MCDB131 (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 90 μg ml−1 heparin, and 3 mM glutamine. AM without 10% FBS [AM(−S)] or AM without FBS and either glutamate or sodium chloride [AM(−S, −G) or AM(−S, −Na)] was used instead of AM where indicated. AM(−S, −G) and AM(−S, −Na) were prepared from 2-fold-concentrated MCDB131 without glutamate and sodium chloride, which was specially manufactured by the Cell Science & Technology Institute (Japan). In a standard experiment, the bacterial suspension was prepared in AM [AM(−S), AM(−S, −G), or AM(−S, −Na) where indicated] at an optical density at 600 nm (OD600) of 0.05, which corresponded to approximately 5 × 106 CFU/100 μl. The multiplicity of infection (MOI) was 500, a condition previously used in other studies (40, 41), to examine efficient N. meningitidis internalization (39). If a lower MOI was required, the bacterial suspension was prepared by further dilution. A 100-μl portion of the bacterial suspension was inoculated onto the host cell monolayers in duplicate for each assay and incubated at 37°C in a 5% CO2 atmosphere for 4 h. To determine bacterial adherence, the monolayers were washed with prewarmed AM(−S) four times to remove nonadherent bacteria. Adherent bacteria were released by the addition of phosphate-buffered saline (PBS) containing 2% saponin, and CFU were determined on GC agar plates with appropriate dilutions to count the cell-adherent bacteria. To determine the internalized bacteria, AM(−S) containing 150 μg ml−1 gentamicin was added, and the cultures were incubated further for 1 h to kill all of the extracellular bacteria. We confirmed that up to 5 × 107 extracellular meningococci were almost completely (>99.999%) killed under these experimental conditions (data not shown). The amounts of internalized bacteria that were not killed by the gentamicin treatment were determined by the addition of PBS containing 2% saponin and plating on GC agar after appropriate dilutions to count the bacteria as CFU. The results are expressed as means and standard deviations (SD), and bacterial numbers were compared statistically using a two-tailed Student t test.
Western blotting.
N. meningitidis, grown on agar plates, was suspended at an OD600 of 0.05 in 10 ml AM and incubated at 37°C in a 5% CO2 atmosphere for 4 h. The bacteria in 4-ml and 1-ml portions of the suspension were harvested by centrifugation at 4°C for analyses of PilV and PilE, respectively. The bacteria were resuspended in 50 μl 1× SDS buffer and boiled for 10 min. SDS-PAGE and Western blotting were performed as described previously (42).
Aggregation assay in AM.
The aggregation assay was performed as described by Brown et al. (43). In brief, N. meningitidis bacteria grown on agar plates were suspended at an OD600 of 0.05 in 100 μl AM and incubated at 37°C in a 5% CO2 atmosphere for 4 h in 96-well coverglass bottom plates (Iwaki, Japan). Aggregate formation on the bottoms of the wells was visualized by phase-contrast microscopy, using an Olympus CKX31 microscope with a 40× objective. Digital images were recorded using an Olympus C-5060 digital camera, with an NY2000S3 superadaptor mounted on the microscope.
Determination of glutamate concentrations in media.
The medium that was used for the infection assay for 4 h was transferred into a 1.5-ml tube and centrifuged at 10,000 × g at 4°C for 2 min. The glutamate concentration in the supernatant was measured with an l-glutamate assay kit II (Yamasa Shoyu, Japan).
Observation of meningococcal colonies on HBMEC.
HBMEC monolayers, grown on a coverglass at 37°C in a 5% CO2 atmosphere for 2 days, were infected with N. meningitidis in AM, AM(−S), or AM(−S, +Glu) at MOIs of 5, 50, and 500 for 4 h. The infected HBMEC monolayers were washed four times with prewarmed AM(−S), fixed with 4% paraformaldehyde in PBS for 15 min, and blocked with 2% bovine serum albumin (BSA) in PBS for 15 min. The resultant monolayers were incubated with 100-fold-diluted anti-meningococcus rabbit serum (unpublished data) for 45 min, followed by the Alexa Fluor 488-conjugated F(ab′) fragment of anti-rabbit IgG (Invitrogen), diluted 200-fold, for 45 min. The glass coverslips were mounted on the glass slides with ProLong Gold Antifade Reagent (Invitrogen). The infected HBMEC and the attached meningococci were observed with a BX51 microscope (Olympus) equipped with a charge-coupled-device (CCD) camera (Orca-ER; Hamamatsu) with a 100× oil immersion objective.
Determination of l-glutamate uptake into bacteria that infected HBMEC.
N. meningitidis strains, grown on GC agar at 37°C in a 5% CO2 atmosphere overnight, were suspended in 1 ml PBS and harvested by centrifugation at 10,000 × g for 2 min. The bacterial pellets were resuspended in 1 ml PBS and centrifuged again. The resultant pellets were suspended in AM(−S, −G) to adjust the bacteria to an OD600 of 0.076. HBMEC layers, seeded in a 24-well plate at a concentration of 7 × 104 cells/well, were washed twice with 1 ml PBS, and then the bacterial suspension (462.5 μl) was added to each well of the 24-well plate. At the same time, a 93.5-μl aliquot of the bacterial suspension was also added to one well of the 96-well plate to measure glutamate uptake by the bacteria only. The assays were initiated by the addition of 37.5 μl and 7.5 μl of 0.4 mM l-[14C]glutamic acid (specific activity, 9.25 MBq/mmol; Perkin-Elmer Life Science) to the 24-well and 96-well plates, respectively, for a final glutamate concentration of 300 μM, and the cells were incubated at 37°C in a 5% CO2 atmosphere for 4 h. The infected HBMEC layers were washed 4 times with 1-ml aliquots of prewarmed AM(−S) and lysed with 500 μl of 2% saponin in PBS. The bacteria that infected the HBMEC in the solution were harvested on a 13-mm-diameter 0.45-μm HAWP membrane (Millipore) by vacuum filtration. The bacterial suspension without HBMEC in the 96-well plate was also collected on the membrane. The filter was washed with a 10-fold volume of PBS and then transferred to a 1.5-ml tube. The bacteria on the filter were suspended in 400 μl H2O, and a 200-μl aliquot of the bacterial suspension was used to measure the radioactivity. The radioactivity was determined by scintillation counting in an 8-ml vial containing 5 ml Filtron-X (National Diagnotics). Background levels of l-[14C]glutamic acid binding to the membrane, HBMEC, or bacteria were determined in control samples and were subtracted from all values. The protein concentration was determined with a bicinchoninic acid (BCA) protein assay kit (Thermo), with bovine serum albumin as the standard. The l-glutamate transport values are expressed as picomoles of substrate transported per microgram of bacterial protein (means and SD).
Observations of meningococci and ezrin accumulation by immunofluorescence staining.
HBMEC monolayers, grown on a coverglass at 37°C in a 5% CO2 atmosphere for 2 days, were infected with N. meningitidis in AM, AM(−S), or AM(−S, +Glu) at an MOI of 500 for 4 h. The infected HBMEC monolayers were washed four times with prewarmed AM(−S), fixed with 4% paraformaldehyde in PBS for 15 min, permeabilized with 0.2% Triton X-100 in PBS, and blocked with 2% BSA in PBS for 15 min. The resultant monolayers were incubated with the anti-ezrin monoclonal antibody 3C12 (Santa Cruz) diluted 100-fold for 45 min and then with the Alexa Fluor 594-conjugated F(ab′) fragment of anti-mouse IgG (Invitrogen) diluted 200-fold for 45 min under moist and dark conditions. To visualize meningococci, the monolayers were further incubated with 100-fold-diluted anti-meningococcus rabbit serum for 45 min and with the Alexa Fluor 488-conjugated F(ab′) fragment of anti-rabbit IgG (Invitrogen) diluted 200-fold for 45 min. The glass coverslips were mounted on the glass slides with ProLong Gold Antifade Reagent. The infected HBMEC and the attached meningococci were observed with a BX51 microscope with a 40× oil immersion objective.
Isolation of RNA, protein, and glutathione from bacteria that infected HBMEC.
HBMEC were seeded on 150-mm culture dishes (Nihon Genetics, Japan) and used within 2 days after reaching confluence (∼7 × 108 cells). HT1125 or HT1414 bacteria, grown on GC agar plates, were suspended in 20 ml AM to adjust the bacteria to an OD600 of 0.05. The bacterial suspensions were added to the HBMEC layers on 150-mm culture dishes and incubated at 37°C in a 5% CO2 atmosphere for 4 h. The infected HBMEC layers were washed twice with 20 ml prewarmed PBS and then twice with 10 ml prewarmed PBS and lysed with 10 ml of 2% saponin in PBS or with 10 ml 2% saponin in PBS containing Complete Ultra (Roche) for protein isolation by incubation at 37°C for 10 min. Bacteria in the suspension were harvested by centrifugation at 3,000 × g at 4°C for 5 min. The resultant pellet was used to isolate bacterial RNA (see Table S1 in the supplemental material), proteins (see Table S2 in the supplemental material), or glutathione (see below).
Monitoring meningococcal survival in HBMEC.
HBMEC (4 × 104 cells/well) were grown in 24-well tissue culture plates (Iwaki) at 37°C in a 5% CO2 atmosphere for 2 days. For this experiment only, the HBMEC monolayers were infected with N. meningitidis strains at an MOI of 5,000 for 2 h in AM(−S), since the maximum number of internalized bacteria was obtained when a high MOI of N. meningitidis was used to infect HBMEC (39). The monolayers were washed with 500 μl prewarmed AM(−S) four times and then incubated with 500 μl AM(−S) containing 150 μg ml−1 gentamicin for 1 h to kill the extracellular bacteria. The infected HBMEC layer was washed with 500 μl AM(−S) four times to remove the residual gentamicin and lysed with 200 μl of 2% saponin in PBS. This time point was defined as the zero hour. The numbers of intracellular bacteria at time zero and 1, 2, 3, and 4 h of incubation after the gentamicin treatment were determined as CFU by plating on GC agar plates after appropriate dilution. The survival percentages (expressed as means and SD) were calculated by the following formula: 102 × (CFU at the indicated h)/(CFU at 0 h).
Determination of the glutathione concentration in bacteria that infected HBMEC.
The bacteria that infected HBMEC were harvested, suspended in 1 ml H2O, and centrifuged at 10,000 × g at 4°C for 2 min. This procedure was repeated 3 times. The resultant pellets were suspended in 100 μl of 0.5% sulfosalicylic acid, and the bacteria were lysed by three freeze/thaw cycles, briefly sonicated, and centrifuged again at 14,000 × g at 4°C for 10 min. The glutathione concentration in the supernatant was measured with a total glutathione quantification kit (Dojindo, Japan). The protein concentration was determined with a BCA protein assay kit (Thermo). The amount of glutathione in bacteria was expressed as nanomoles of glutathione per milligram of bacterial protein (means and SD).
Statistical analyses.
Results are expressed as means and SD. The numbers of adhered and internalized bacteria, ratios of internalized to adhered bacteria, glutamate concentrations, glutathione measurements, l-glutamate uptake, and meningococcal survival in HBMEC were compared using the two-tailed Student t test, and P values of <0.05 were considered to be significant.
RESULTS
The ΔgltT ΔgltM mutation did not considerably affect meningococcal transcription or translation upon HBMEC infection.
In an attempt to evaluate the underlying mechanisms of meningococcal internalization dependent on GltT-GltM, we isolated RNA from N. meningitidis strains that infected HBMEC, and the transcriptional changes in N. meningitidis upon HBMEC infection were investigated by transcriptome analysis (RNA-seq) (see Table S1 in the supplemental material). Comparison of expression between bacteria only and bacteria that infected HBMEC, using the wild-type (WT) N. meningitidis strain HT1125, revealed the altered expression of many genes, especially those related to rRNA and proteins, upon infection (see Table S1A in the supplemental material). Comparison between HT1414 and HT1125, however, revealed lower gene expression numbers, and fewer gene reads (less than 50) were altered between the WT and the ΔgltT ΔgltM mutant (see Table S1B in the supplemental material). Moreover, the identified genes did not seem to belong to a particular biological category, since most of the genes were annotated as “hypothetical gene” (see Table S1B in the supplemental material). We also analyzed the differences in the protein levels between HT1125 and HT1414 upon HBMEC infection by liquid chromatography-tandem mass spectrometry (LC–MS-MS) with Tandem Mass Tag labeling (Thermo Fisher Scientific) (see Table S2 in the supplemental material). Compared to the transcriptome analysis results (see Table S1 in the supplemental material), fewer proteins (61 proteins) were detected, and they showed lower degrees of expression differences (less than 2-fold) between HT1125 and HT1414. Therefore, the results of the transcriptome and proteome analyses suggested that GltT-GltM-dependent meningococcal internalization into HBMEC was not likely to be regulated at either the transcriptional or translational level.
GltT-GltM-dependent internalization into HBMEC was less prominent in AM(−S).
We previously showed that the ΔgltT ΔgltM mutant, HT1414, was approximately 100-fold less efficiently internalized into HBMEC than the wild-type strain, HT1125, at an MOI of 500 in AM (Fig. 1 C) (30). We also examined the expression of PilE and PilV, components of the meningococcal pili that affect meningococcal infection (42, 44), as well as the aggregate formation linked to meningococcal virulence (43), and found that there were no differences between HT1125 and HT1414 (see Fig. S1 in the supplemental material). These results are consistent with those obtained by the proteome analysis. Therefore, we confirmed that the internalization defect of HT1414 was not due to loss of protein or the function of the meningococcal pili in AM.
FIG 1.

Effects of glutamate supplementation in AM on N. meningitidis infection of HBMEC. Shown are adherence (A), internalization (B), and the internalization/adhesion ratio (percent internalized) (C) of N. meningitidis wild-type (HT1125) and ΔgltT ΔgltM (HT1414) strains in HBMEC in AM, AM(−S), AM(−S, −G) supplemented with glutamate (Glu), and AM(−S, −Na) supplemented with 50 mM NaCl at an MOI of 500 (meningococcal colony formation is shown in Fig. 3 and 4). The numbers of bacteria were measured as CFU. Internalized bacteria were determined as bacteria recovered after gentamicin treatment. Each value is the mean and standard deviation of the mean (CFU per 104 HBMEC) from the results of at least four experiments. Under each bar is shown the presence (+) or absence (−) of intact gltT gltM genes in the strain studied. The open and solid bars indicate the numbers of HT1125 and HT1414 bacteria, respectively. Statistical analyses were performed with a two-tailed Student t test; N.S., nonsignificant; *, P < 0.05; **, P < 0.01; #, P < 0.02.
During the course of our studies to elucidate the mechanism of GltT-GltM-dependent invasion, we found that the ΗΤ1414 invasion defect was alleviated in AM(−S), which lacked the 10% FBS (Fig. 1C). This result suggested that some component(s) of FBS might have affected GltT-GltM-dependent meningococcal internalization into HBMEC. Since GltT-GltM is an ABC-type glutamate transporter that can function under low-Na+ conditions (27, 30), we focused on two molecules: sodium chloride and l-glutamate. To examine the possibility that these factors affected GltT-GltM-dependent internalization into HBMEC, we performed infection assays in AM(−S, −Na) supplemented with sodium chloride and in AM(−S, −G) supplemented with glutamate (Fig. 1). The supplementation with 50 mM sodium chloride, which was an approximately 4-fold-higher concentration than that in AM, did not reduce HT1414 internalization as in AM (Fig. 1C). The same finding was observed in AM(−S, −G) supplemented with 100 μM glutamate. However, the efficient invasion of HT1414 in AM(−S) disappeared upon supplementation with 500 μM glutamate in AM(−S, −G) (Fig. 1C). This result suggested that GltT-GltM-dependent internalization into HBMEC could be affected by the environmental glutamate concentration.
Glutamate levels in AM and AM(−S).
We examined whether there was a difference in the glutamate concentrations in AM and AM(−S) (Fig. 2). AM with HBMEC only (without bacteria) contained approximately 100 μM glutamate, while AM(−S) contained less than 50 μM glutamate (Fig. 2). Since approximately 30 μM glutamate is present in the original base medium and the glutamate derived from 10% FBS was estimated to be approximately 50 μM, these results suggested that incubation with HBMEC did not increase the glutamate content. However, the glutamate concentration in AM with both HBMEC and the wild-type N. meningitidis strain was over 150 μM, and that in AM(−S) was approximately 100 μM. These results suggested that incubation with the bacteria might increase the net glutamate content by up to 50 μM, regardless of the medium. The glutamate concentrations in AM and AM(−S) with both HBMEC and the ΔgltT ΔgltM mutant were similar to those with the wild-type strain (Fig. 2). Since the glutamate concentration in whole blood is 150 to 300 μM (45), the glutamate concentration in AM in vitro was likely to be similar to that in vivo. Together, these results suggested that GltT-GltM-dependent meningococcal invasion was observed with the physiological glutamate concentration (150 to 300 μM) and that the dependency could be alleviated under low-glutamate conditions.
FIG 2.
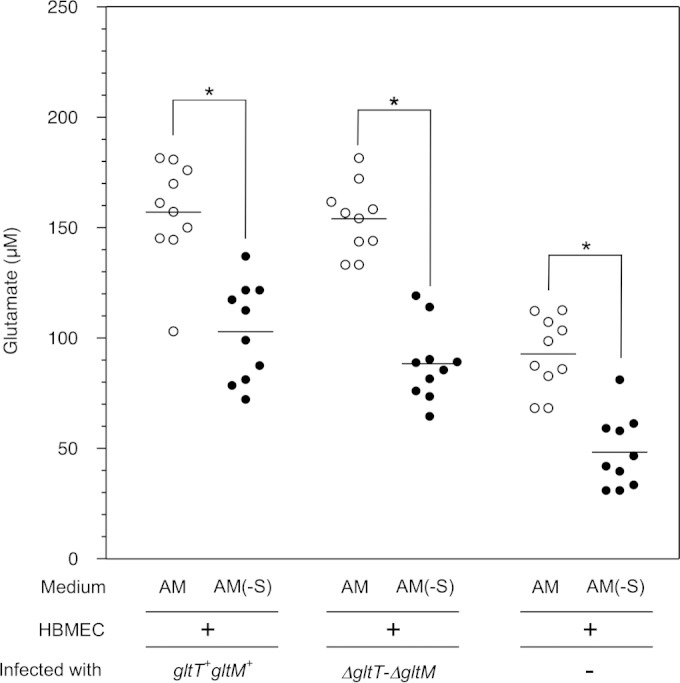
Measurement of glutamate concentrations in AM. The glutamate concentrations were determined in the supernatants of the AM and AM(−S) used for Fig. 1. Each circle represents the glutamate concentration in one sample. The open and solid circles indicate the glutamate concentrations in AM and AM(−S), respectively. Statistical analyses were performed with a two-tailed Student t test; *, P < 0.0001. Horizontal lines indicate the means.
GltT-GltM-dependent meningococcal invasion was observed when N. meningitidis formed large colonies on HBMEC at a physiological glutamate concentration.
To study the relationship between GltT-GltM-dependent meningococcal invasion and colony formation on HBMEC, we further examined the GltT-GltM-dependent meningococcal invasion at lower MOIs and under different glutamate conditions (Fig. 3), since infections at higher MOIs within limited incubation times facilitated the formation of larger colonies on the host cells (39). In AM (physiological glutamate conditions), the ΔgltT ΔgltM mutant, HT1414, was approximately 50-fold less efficiently internalized than the wild-type strain, HT1125, into HBMEC at an MOI of 500 (Fig. 1C and 3C). However, at an MOI of 50, HT1414 was only 5-fold less efficiently internalized than HT1125, and the difference was not observed at an MOI of 5 (Fig. 3C). These results suggested that GltT-GltM-mediated meningococcal invasion was dependent on the MOI. To observe the meningococcal infection of HBMEC, the infected HBMEC were also analyzed by an immunofluorescence assay with microscopy (Fig. 4). At an MOI of 500 in AM, HT1125 and HT1414 formed large colonies on HBMEC (Fig. 4F and L). However, at MOIs of 50 and 5, smaller colonies were observed (Fig. 4D, E, J, and K). These results suggested that, at physiological glutamate concentrations, GltT-GltM-dependent invasion occurred more efficiently when N. meningitidis formed larger colonies on HBMEC. In contrast, in AM(−S) (low-glutamate conditions), ΗΤ1414 was only 5-fold less efficiently internalized than HT1125 at an MOI of 500, and no difference was observed even at an MOI of 50 (Fig. 3C), indicating increased invasion efficiency of HT1414, by approximately 10-fold, in AM(−S) compared with that in AM. Colony formation on HBMEC in AM(−S) was similar to that in AM (data not shown). These results suggested that the defect in GltT-GltM-dependent meningococcal invasion could be suppressed by reducing the environmental glutamate concentration. In AM(−S, +Glu), HT1414 was approximately 50-fold and 5-fold less efficiently internalized than HT1125 at MOIs of 500 and 50, respectively (Fig. 3C), and the colony formation was almost the same as that in AM (data not shown). Considering the results shown in Fig. 3 and 4 together, GltT-GltM-dependent N. meningitidis internalization into HBMEC might be elicited by the reduction of the environmental glutamate concentration when N. meningitidis formed large colonies on HBMEC.
FIG 3.

The ΔgltT ΔgltM invasion defect was observed only under physiological glutamate conditions and only at higher MOIs. Shown are adherence (A), internalization (B), and the internalization/adhesion ratio (percent internalized) (C) of N. meningitidis wild-type (HT1125) and ΔgltT ΔgltM (HT1414) strains in AM, AM(−S), and AM(−S, −G) with 500 μM glutamate [AM(−S, +Glu)] at MOIs of 5, 50, and 500. Internalized bacteria were determined as bacteria recovered after gentamicin treatment. Each value is the mean and standard deviation of the mean (CFU per 104 cells) from the results of at least four experiments. The open and solid bars indicate the numbers of HT1125 and HT1414 bacteria, respectively. Statistical analyses were performed with a two-tailed Student t test; *, P < 0.01; **, P < 0.05.
FIG 4.

N. meningitidis formed larger colonies on HBMEC at higher MOIs. Shown are phase-contrast and immunofluorescent staining with anti-N. meningitidis (α-Nm) rabbit serum and anti-rabbit IgG-Alexa 488, respectively. HBMEC infections were done with wild-type (HT1125) and ΔgltT ΔgltM (HT1414) strains in AM at MOIs of 5, 50, and 500. Magnification, ×1,000.
Enhancement of meningococcal glutamate uptake via GltT-GltM upon HBMEC infection.
We next examined the uptake of environmental glutamate via meningococcal GltT-GltM upon HBMEC infection. The l-glutamate uptake activity levels did not differ significantly between the wild type (HT1125) and the ΔgltT ΔgltM mutant (HT1414) when the bacteria were incubated in AM(−S, −G) supplemented with 300 μM 14C-labeled glutamate (Fig. 5, left). However, l-glutamate uptake by ΗΤ1414 was less efficient than that by HT1125 upon HBMEC infection (Fig. 5, right). These results suggested that meningococcal GltT-GltM was more important for l-glutamate uptake when N. meningitidis infected the host cells than when N. meningitidis grew in the medium.
FIG 5.
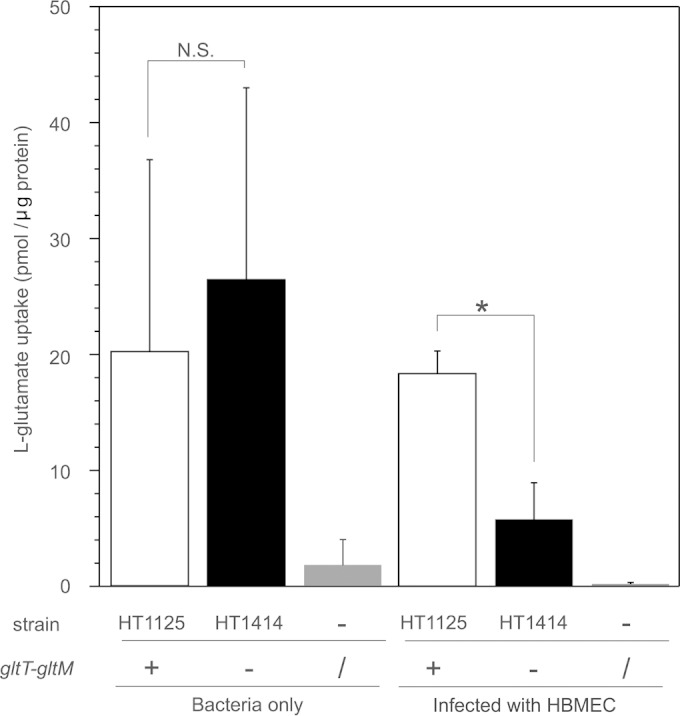
Uptake of glutamate by wild-type and ΔgltT ΔgltM strains with or without HBMEC infection. The assay was performed as described in Materials and Methods in AM(−S, −G) supplemented with 300 μM l-[14C]glutamate. Experiments without bacteria were also analyzed as negative controls. The open, solid, and gray bars indicate the l-glutamate uptake of wild-type (HT1125) and ΔgltT ΔgltM (HT1414) N. meningitidis strains and the negative control, respectively. Each value is the mean and standard deviation of the mean from the results of at least three experiments. Statistical analyses were performed with a two-tailed Student t test; N.S., nonsignificant; *, P < 0.01.
Ezrin accumulation was correlated with the invasion efficiency of the ΔgltT ΔgltM mutant under physiological and low-glutamate conditions.
Meningococcal internalization into endothelial cells is accompanied by the accumulation of ezrin, one of the membrane-associated proteins responsible for linking the plasma membrane to the underlying actin cytoskeleton (46, 47). We examined the changes in the host cell cytoskeleton upon meningococcal infection by using indirect immunofluorescence to monitor the localization of ezrin (Fig. 6). Ezrin accumulated minimally in HBMEC that were not infected with N. meningitidis in any medium, and especially in AM(−S) (Fig. 6B, b), indicating that the reduction of environmental glutamate itself did not induce ezrin accumulation in HBMEC. In AM (physiological glutamate conditions), ezrin did not appreciably accumulate beneath the ΔgltT ΔgltM mutant HT1414 (Fig. 6A, h), while ezrin clearly accumulated beneath the wild-type N. meningitidis strain, HT1125 (Fig. 6A, e). This phenotype was the same as that in the previous report (30) and was consistent with the HT1414 invasion defect in AM (Fig. 1C and 3C). However, in AM(−S) (low-glutamate conditions), ezrin accumulation beneath ΗΤ1414 was more frequently observed than in AM (Fig. 6B, h) and was correlated with the increased invasion efficiency of HT1414 in AM(−S) (Fig. 1C and 3C). Furthermore, in AM(−S, +Glu), ezrin accumulation beneath ΗΤ1414 was less efficient (Fig. 6C, h), and this was also correlated with the low invasion efficiency of HT1414 in AM(−S, +Glu) (Fig. 1C and 3C). Combined with the fact that the functions of the pili in HT1414 and HT1125 were similar (see Fig. S1 in the supplemental material), all of these results suggested that the reduction of the environmental glutamate concentration alleviated the defects in both ezrin accumulation and GltT-GltM-dependent meningococcal invasion.
FIG 6.

Phase-contrast and immunofluorescence microscopy showing ezrin accumulation beneath N. meningitidis-infected HBMEC. Noninfected controls are also shown. HBMEC monolayers were infected with N. meningitidis wild-type (HT1125) and ΔgltT ΔgltM (HT1414) strains in AM (A), AM(−S) (B), and AM(−S, +Glu) (C). N. meningitidis and HBMEC were observed by phase-contrast microscopy (left column). N. meningitidis strains and ezrin were immunostained with two sets of primary and secondary antibodies: anti-N. meningitidis rabbit serum and Alexa Fluor 488-conjugated anti-rabbit IgG (middle column) and anti-ezrin monoclonal antibody (MAb) and Alexa Fluor 594-conjugated anti-mouse IgG (right column). Magnification, ×400.
Infection activity and glutathione synthesis.
N. meningitidis reportedly resists neutrophil oxidative burst by synthesizing glutathione, a key molecule in the control of the redox state in all living cells (48, 49), from l-glutamate obtained via GltT-GltM (29). Since endothelial cells produce oxygen radicals, such as ROS, upon microbial infection (31, 33, 34), we speculated that a relationship might exist between glutathione synthesis and HBMEC invasion ability under our experimental conditions. To examine this possibility, we constructed the gshB (glutathione synthetase) deletion mutant HT1876 and examined its ability to infect HBMEC (Fig. 7). As shown in Fig. 1 and our previous study (30), the internalization of the ΔgltT ΔgltM mutant, HT1414, into HBMEC was approximately 100-fold less efficient than that of the wild-type strain, HT1125 (Fig. 7C). However, ΗΤ1876 was internalized into HBMEC approximately 10-fold less efficiently than HT1125, although the adhesion numbers were similar (Fig. 7C). These results suggested that glutathione production contributed to the GltT-GltM-dependent invasion of HBMEC. Notably, HT1414 was internalized into HBMEC approximately 10-fold less efficiently than HT1876 (Fig. 7C), implying that glutathione-independent invasion was also involved in GltT-GltM-dependent internalization into HBMEC. All of these results suggested that GltT-GltM-dependent meningococcal invasion comprised both glutathione-dependent and glutathione-independent phenotypes.
FIG 7.

Glutathione production contributes to meningococcal survival in HBMEC. Shown are adherence (A and D), internalization (B and E), and the internalization/adhesion ratio (percent internalized) (C and F) of wild-type (HT1125), ΔgltT ΔgltM (HT1414), and ΔgshB (HT1876) N. meningitidis strains in HBMEC examined in AM (A to C) or AM(−S) (D to F). The numbers of bacteria were measured as CFU. Internalized bacteria were determined as bacteria recovered after gentamicin treatment. Each value is the mean and standard deviation of the mean (CFU per 104 cells) from the results of at least four experiments. The open, solid, and gray bars indicate the numbers of HT1125, HT1414, and HT1876 bacteria, respectively. Statistical analyses were performed with a two-tailed Student t test; N.S., nonsignificant; *, P < 0.001.
Suppression of the glutathione-independent invasion defect by AM(−S).
Glutathione-independent invasion was considered to comprise both invasion caused by the environmental glutamate reduction around the meningococcal colony on HBMEC and glutamate acquisition by HBMEC (27, 28). To further examine these possibilities, meningococcal invasion into HBMEC was analyzed in AM(−S) under conditions where only invasion by glutamate reduction would be alleviated (Fig. 1C, 3C, and 7D, E, and F). The ΔgshB mutant, HT1876, was approximately 5-fold less efficiently internalized into HBMEC than the wild-type strain, HT1125 (Fig. 7F), suggesting that a similar glutathione-dependent phenotype was observed in AM(−S). However, a comparison of the ΔgltT ΔgltM mutant, HT1414, and HT1876, in which the glutathione-dependent phenotype was eliminated, revealed that the internalization of HT1414 into HBMEC seemed to be less efficient than that of HT1876, but the difference was not statistically significant (Fig. 7F). Notably, the difference in AM(−S) became smaller than that in AM (Fig. 7C and F), indicating that the defect in glutathione-independent invasion in AM could be suppressed in AM(−S), but not completely.
Meningococcal survival in HBMEC.
Glutathione-dependent invasion is likely to be linked to intracellular survival against ROS in HBMEC. To examine this possibility, we analyzed meningococcal survival in HBMEC (Fig. 8). The survival of the wild-type N. meningitidis strain, HT1125, was approximately 100% at 1 h and gradually declined to approximately 30% at 4 h (Fig. 8). In contrast, the ΔgshB mutant, HT1876, survived less efficiently than HT1125 at every time point, and the survival rate declined to approximately 0.5% at 4 h (Fig. 8). The low survival rate was also observed with the ΔgltT ΔgltM mutant, HT1414 (Fig. 8). These results suggested that glutathione-dependent invasion was correlated with intracellular survival by glutathione synthesis.
FIG 8.
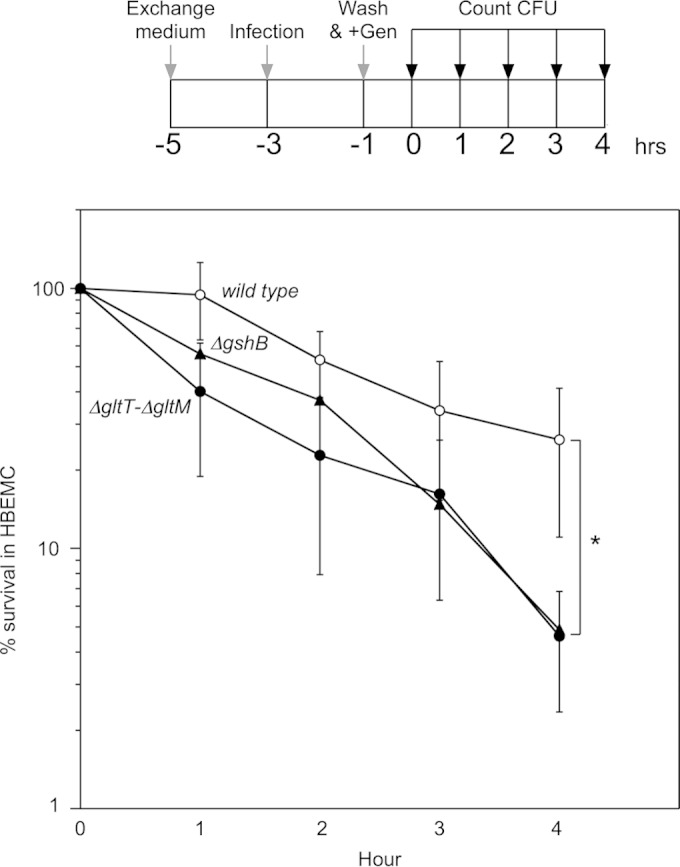
Percent survival of intracellular bacteria after killing of extracellular bacteria with gentamicin. (Top) Protocol to monitor the number of intracellular bacteria after a 2-hour infection. Gen, gentamicin. The detailed procedures are described in Materials and Methods. (Bottom) Percent survival of intracellular bacteria in HBMEC. The percent survival was calculated as follows: (CFU at indicated time/CFU at removal of gentamicin) × 100. The symbols indicate the survival percentages of the HT1125, HT1414, and HT1876 N. meningitidis strains. The error bars represent the standard deviations of the means from the results of at least five experiments. Statistical analyses were performed with a two-tailed Student t test; *, P < 0.02.
GltT-GltM plays a significant role in glutathione synthesis in N. meningitidis upon HBMEC infection.
We further examined the change in the intracellular glutathione content upon HBMEC infection. The intracellular glutathione concentration in the ΔgshB mutant was approximately 40 μmol/mg protein, representing a background glutathione level under our experimental conditions (Fig. 9). Under the same conditions, the intracellular glutathione concentrations in N. meningitidis strains (wild type, ΔgltT ΔgltM, and gltS) were approximately 250 to 300 μmol/mg protein during incubation in AM for 4 h (Fig. 9, left). These results suggested that the amount of intracellular glutathione was not greatly affected by either the ΔgltT ΔgltM or gltS mutation when the bacteria were grown alone. However, when the N. meningitidis strains infected HBMEC, the intracellular glutathione concentration in the ΔgltT ΔgltM mutant was much lower than those in the wild-type and gltS N. meningitidis strains (Fig. 9, right). These results suggested that l-glutamate uptake via GltT-GltM is crucial for glutathione synthesis when N. meningitidis infects host cells, while glutathione synthesis is not affected by GltT-GltM under noninfective conditions.
FIG 9.
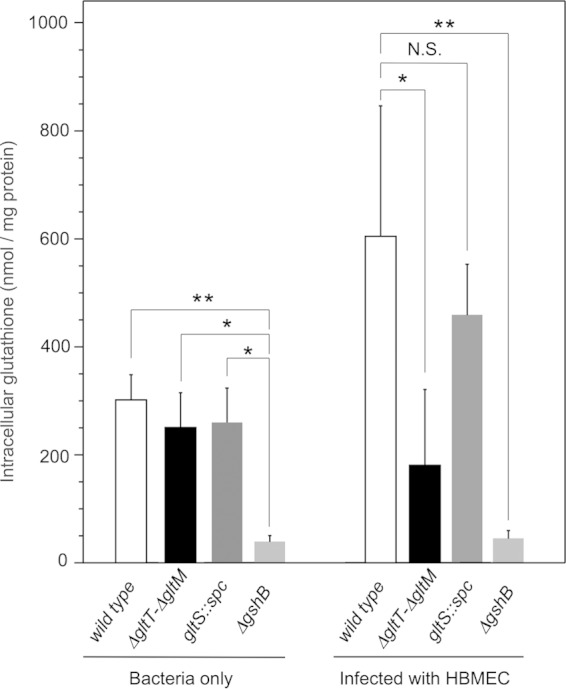
Intracellular glutathione levels in N. meningitidis with or without HBMEC infection. The intracellular glutathione concentration in N. meningitidis was measured as described in Materials and Methods. The glutathione concentration in bacteria is expressed as nanomoles of glutathione per milligram of bacterial protein. The error bars represent the standard deviations of the means from the results of at least four experiments. Statistical analyses were performed with a two-tailed Student t test; N.S., nonsignificant; *, P < 0.02; **, P < 0.005.
DISCUSSION
In this study, we found that that GltT-GltM-dependent meningococcal internalization into HBMEC might be induced by the reduction of the environmental glutamate concentration upon infection. In addition, we showed that glutamate uptake via GltT-GltM is crucial for glutathione synthesis in N. meningitidis for HBMEC infection, which resulted in increased meningococcal survival in HBMEC. Considering these findings and the previous report that l-glutamate uptake via GltT-GltM is the source of the nutrient in human cells (27), we speculated that l-glutamate uptake via GltT-GltM performs multiple functions in meningococcal internalization into host cells.
Meningococcal GltT-GltM-dependent invasion and the accompanying ezrin accumulation frequently occurred under physiological glutamate conditions, under which N. meningitidis formed large colonies on HBMEC (Fig. 3 and 4), and defects in GltT-GltM-dependent invasion could be suppressed by a reduction of the environmental glutamate concentration (Fig. 1 and 3). In addition, glutamate uptake via GltT-GltM and the resultant glutathione synthesis were enhanced only upon meningococcal infection (Fig. 5 and 9). Since glutamate uptake upon infection was largely dependent on GltT-GltM, but not on GltS, which functions only under extracellular conditions (Fig. 5) (27, 29, 30), we speculated that the local environment between the meningococcal colony and HBMEC was different from the extracellular conditions. Moreover, the reduction of environmental glutamate itself did not affect ezrin accumulation (Fig. 6B, b), suggesting that ezrin accumulation and the concomitant meningococcal internalization were required for reduction in the local glutamate concentration to less than 100 μM by the meningococcal GltT-GltM. Based on these indirect findings, we propose that N. meningitidis GltT-GltM reduces the local glutamate concentration beneath the colony on HBMEC upon infection to enhance meningococcal internalization into HBMEC (Fig. 10). According to this model, the fact that some carriers are susceptible to meningococcal diseases while others are not might be partly explained by variations in the glutamate concentrations in the blood, which are affected by many factors (45, 50–52). However, reduction of the glutamate concentrations beneath meningococcal colonies on HBMEC could not be directly proven, and further examinations should be performed when an in vivo molecular probe for glutamate becomes available.
FIG 10.

Schematic representation of meningococcal infection of human cultured endothelial cells. During the course of the meningococcal invasion of human cells, N. meningitidis adheres to and forms large colonies on the endothelial cells. (Step 1) The glutamate concentrations around (possibly beneath) the colonies may be reduced by glutamate uptake via meningococcal GltT-GltM, which might stimulate host cell cytoskeleton rearrangements to enhance meningococcal internalization into the host cells. (Step 2) During adhesion to and invasion of HBMEC, l-glutamate obtained via GltT-GltM is also converted to glutathione in N. meningitidis. After internalization, the accumulated glutathione in N. meningitidis plays a role as an antioxidant in survival in the host cells. (Step 3) Moreover, N. meningitidis also utilizes intracellular glutamate as a nutrient by uptake via GltT-GltM. Thus, the meningococcal GltT-GltM performs multiple functions in internalization into host cells.
Ezrin accumulation beneath the ΔgltT ΔgltM mutant, HT1414, in AM was impaired (Fig. 6A, h). Certain environmental conditions, such as the presence of antibiotics and the oxygen concentration, affect neisserial piliation (53, 54), and the meningococcal pili, including the minor components PilV and PilX, are important for ezrin accumulation (42, 44, 55, 56). However, we confirmed that the expression of PilE, the major component of pili, and that of PilV, as well as the aggregate formation that was linked to meningococcal virulence (43) in HT1414, were similar to those in the wild-type strain, HT1125 (see Fig. S1 in the supplemental material). Considering these facts, it is unlikely that the defect in ezrin accumulation was due to the impairment of the expression and function of pili in HT1414. However, we could not eliminate the possibility that the GltT-GltM defect might indirectly affect some host cell infectivity properties, since the adhesion activity and the propensity of HT1414 to infect HBMEC were somewhat different from those of HT1125 (Fig. 1A, 3, and 6A).
The N. meningitidis capsule prevents meningococcal adhesion to and invasion of cultured human cells (57–60). Conversely, the meningococcal capsule plays important roles in intracellular survival (61) by its antibactericidal (62) (63) and antiphagocytic (64) effects. As larger amounts of capsular polysaccharide have been detected in clinical isolates from patients (65) and the production is phase variable (59, 66), unencapsulated bacteria within the nasopharynx may invade the epithelial and endothelial cells and then revert to the encapsulated state by the reversible capsule expression switch for dissemination in humans (59, 64, 66, 67). In this study, we used unencapsulated N. meningitidis to observe efficient internalization by eliminating the multiple effects of the capsule on meningococcal infection. As discussed above, the meningococcal capsule may affect intracellular survival ability (Fig. 8). However, since the majority of internalized bacteria are considered to remain unencapsulated until phase variation occurs, the intracellular survival of the unencapsulated state may also be important for the N. meningitidis infectious cycle.
Finally, our findings illustrate the diverse functions of glutamate uptake via GltT-GltM in N. meningitidis internalization into HBMEC (Fig. 10). N. meningitidis adheres to endothelial cells and forms colonies. In the environment around (beneath) the meningococcal colony, which is likely to have a low Na+ concentration, environmental glutamate is assimilated by the adhered N. meningitidis via the meningococcal GltT-GltM transporter. The glutamate concentration around the colony on HBMEC might thus be decreased and contribute to meningococcal internalization by host cells. Following internalization into the host cells, N. meningitidis synthesizes glutathione from glutamate as an antioxidant for meningococcal intracellular survival. N. meningitidis also acquires intracellular glutamate via GltT-GltM as a nutrient to grow within host cells (27). Thus, the uptake of l-glutamate via GltT-GltM performs multiple functions for meningococcal internalization into host cells. In Helicobacter pylori, the metabolism of environmental glutathione, glutamine (Gln), asparagine (Asn), glutamate (Glu), and aspartate (Asp) by γ-glutamyl aminopeptidase and arginase also performs multiple functions for (i) forming Glu and Asp as nutrients, (ii) producing ammonia, and (iii) depleting Gln/Glu and Asn/Asp to injure the host cells (68, 69). It seems logical that many functions performed by a single molecule are biologically more economical than one function accomplished by one molecule. These kinds of molecules seem to resemble the “moonlighting proteins,” molecules that perform multiple jobs, found in eukaryotes (70), virulent bacteria (70, 71), and N. meningitidis (72, 73). One of the reasons why the mechanism of meningococcal virulence has not been clearly elucidated might be the presence of unidentified moonlighting proteins in N. meningitidis and the contributions of such moonlighting (or multifunctional) proteins to meningococcal virulence. The multiple functions of GltT-GltM in meningococcal internalization into host cells shed light on new aspects of N. meningitidis virulence factors and pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (grant no. 24590545 and 15K08485) and by the Platform for Drug Discovery, Informatics, and Structural Life Science, funded by the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00654-15.
REFERENCES
- 1.Takahashi H, Kuroki T, Watanabe Y, Tanaka H, Inouye H, Yamai S, Watanabe H. 2004. Characterization of Neisseria meningitidis isolates collected from 1974 to 2003 in Japan by multilocus sequence typing. J Med Microbiol 53:657–662. doi: 10.1099/jmm.0.45541-0. [DOI] [PubMed] [Google Scholar]
- 2.Chang Q, Tzeng YL, Stephens DS. 2012. Meningococcal disease: changes in epidemiology and prevention. Clin Epidemiol 4:237–245. doi: 10.2147/CLEP.S28410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brouwer MC, Read RC, van de Beek D. 2010. Host genetics and outcome in meningococcal disease: a systematic review and meta-analysis. Lancet Infect Dis 10:262–274. doi: 10.1016/S1473-3099(10)70045-1. [DOI] [PubMed] [Google Scholar]
- 4.Davila S, Wright VJ, Khor CC, Sim KS, Binder A, Breunis WB, Inwald D, Nadel S, Betts H, Carrol ED, de Groot R, Hermans PW, Hazelzet J, Emonts M, Lim CC, Kuijpers TW, Martinon-Torres F, Salas A, Zenz W, Levin M, Hibberd ML. 2010. Genome-wide association study identifies variants in the CFH region associated with host susceptibility to meningococcal disease. Nat Genet 42:772–776. doi: 10.1038/ng.640. [DOI] [PubMed] [Google Scholar]
- 5.Caesar JJ, Lavender H, Ward PN, Exley RM, Eaton J, Chittock E, Malik TH, De Goiecoechea JE, Pickering MC, Tang CM, Lea SM. 2014. Competition between antagonistic complement factors for a single protein on N. meningitidis rules disease susceptibility. eLife 3:e04008. doi: 10.7554/eLife.04008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murray RL, Britton J, Leonardi-Bee J. 2012. Second hand smoke exposure and the risk of invasive meningococcal disease in children: systematic review and meta-analysis. BMC Public Health 12:1062. doi: 10.1186/1471-2458-12-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rashid H, Booy R. 2012. Passive smoking, invasive meningococcal disease and preventive measures: a commentary. BMC Med 10:160. doi: 10.1186/1741-7015-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Codjoe SN, Nabie VA. 2014. Climate change and cerebrospinal meningitis in the Ghanaian meningitis belt. Int J Environ Res Public Health 11:6923–6939. doi: 10.3390/ijerph110706923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Virji M. 2009. Pathogenic neisseriae: surface modulation, pathogenesis and infection control. Nat Rev Microbiol 7:274–286. doi: 10.1038/nrmicro2097. [DOI] [PubMed] [Google Scholar]
- 10.Hill DJ, Virji M. 2012. Meningococcal ligands and molecular targets of the host. Methods Mol Biol 799:143–152. doi: 10.1007/978-1-61779-346-2_9. [DOI] [PubMed] [Google Scholar]
- 11.Plant L, Sundqvist J, Zughaier S, Lovkvist L, Stephens DS, Jonsson AB. 2006. Lipooligosaccharide structure contributes to multiple steps in the virulence of Neisseria meningitidis. Infect Immun 74:1360–1367. doi: 10.1128/IAI.74.2.1360-1367.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis LA, Ram S. 2014. Meningococcal disease and the complement system. Virulence 5:98–126. doi: 10.4161/viru.26515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serruto D, Adu-Bobie J, Scarselli M, Veggi D, Pizza M, Rappuoli R, Arico B. 2003. Neisseria meningitidis App, a new adhesin with autocatalytic serine protease activity. Mol Microbiol 48:323–334. doi: 10.1046/j.1365-2958.2003.03420.x. [DOI] [PubMed] [Google Scholar]
- 14.Scarselli M, Serruto D, Montanari P, Capecchi B, Adu-Bobie J, Veggi D, Rappuoli R, Pizza M, Arico B. 2006. Neisseria meningitidis NhhA is a multifunctional trimeric autotransporter adhesin. Mol Microbiol 61:631–644. doi: 10.1111/j.1365-2958.2006.05261.x. [DOI] [PubMed] [Google Scholar]
- 15.Turner DP, Marietou AG, Johnston L, Ho KK, Rogers AJ, Wooldridge KG, Ala'Aldeen DA. 2006. Characterization of MspA, an immunogenic autotransporter protein that mediates adhesion to epithelial and endothelial cells in Neisseria meningitidis. Infect Immun 74:2957–2964. doi: 10.1128/IAI.74.5.2957-2964.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Comanducci M, Bambini S, Brunelli B, Adu-Bobie J, Arico B, Capecchi B, Giuliani MM, Masignani V, Santini L, Savino S, Granoff DM, Caugant DA, Pizza M, Rappuoli R, Mora M. 2002. NadA, a novel vaccine candidate of Neisseria meningitidis. J Exp Med 195:1445–1454. doi: 10.1084/jem.20020407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Merrell DS, Falkow S. 2004. Frontal and stealth attack strategies in microbial pathogenesis. Nature 430:250–256. doi: 10.1038/nature02760. [DOI] [PubMed] [Google Scholar]
- 18.Fraser C, Hanage WP, Spratt BG. 2005. Neutral microepidemic evolution of bacterial pathogens. Proc Natl Acad Sci U S A 102:1968–1973. doi: 10.1073/pnas.0406993102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snyder LA, Saunders NJ. 2006. The majority of genes in the pathogenic Neisseria species are present in non-pathogenic Neisseria lactamica, including those designated as ‘virulence genes'. BMC Genomics 7:128. doi: 10.1186/1471-2164-7-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marri PR, Paniscus M, Weyand NJ, Rendon MA, Calton CM, Hernandez DR, Higashi DL, Sodergren E, Weinstock GM, Rounsley SD, So M. 2010. Genome sequencing reveals widespread virulence gene exchange among human Neisseria species. PLoS One 5:e11835. doi: 10.1371/journal.pone.0011835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schoen C, Blom J, Claus H, Schramm-Gluck A, Brandt P, Muller T, Goesmann A, Joseph B, Konietzny S, Kurzai O, Schmitt C, Friedrich T, Linke B, Vogel U, Frosch M. 2008. Whole-genome comparison of disease and carriage strains provides insights into virulence evolution in Neisseria meningitidis. Proc Natl Acad Sci U S A 105:3473–3478. doi: 10.1073/pnas.0800151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Ulsen P, Tommassen J. 2006. Protein secretion and secreted proteins in pathogenic Neisseriaceae. FEMS Microbiol Rev 30:292–319. doi: 10.1111/j.1574-6976.2006.00013.x. [DOI] [PubMed] [Google Scholar]
- 23.Vandenberg RJ, Ryan RM. 2013. Mechanisms of glutamate transport. Physiol Rev 93:1621–1657. doi: 10.1152/physrev.00007.2013. [DOI] [PubMed] [Google Scholar]
- 24.van Heeswijk WC, Westerhoff HV, Boogerd FC. 2013. Nitrogen assimilation in Escherichia coli: putting molecular data into a systems perspective. Microbiol Mol Biol Rev 77:628–695. doi: 10.1128/MMBR.00025-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amon J, Titgemeyer F, Burkovski A. 2010. Common patterns—unique features: nitrogen metabolism and regulation in Gram-positive bacteria. FEMS Microbiol Rev 34:588–605. doi: 10.1111/j.1574-6976.2010.00216.x. [DOI] [PubMed] [Google Scholar]
- 26.Catlin BW. 1973. Nutritional profiles of Neisseria gonorrhoeae, Neisseria meningitidis, and Neisseria lactamica in chemically defined media and the use of growth requirements for gonococcal typing. J Infect Dis 128:178–194. doi: 10.1093/infdis/128.2.178. [DOI] [PubMed] [Google Scholar]
- 27.Monaco C, Tala A, Spinosa MR, Progida C, De Nitto E, Gaballo A, Bruni CB, Bucci C, Alifano P. 2006. Identification of a meningococcal L-glutamate ABC transporter operon essential for growth in low-sodium environments. Infect Immun 74:1725–1740. doi: 10.1128/IAI.74.3.1725-1740.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colicchio R, Ricci S, Lamberti F, Pagliarulo C, Pagliuca C, Braione V, Braccini T, Tala A, Montanaro D, Tripodi S, Cintorino M, Troncone G, Bucci C, Pozzi G, Bruni CB, Alifano P, Salvatore P. 2009. The meningococcal ABC-type L-glutamate transporter GltT is necessary for the development of experimental meningitis in mice. Infect Immun 77:3578–3587. doi: 10.1128/IAI.01424-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tala A, Monaco C, Nagorska K, Exley RM, Corbett A, Zychlinsky A, Alifano P, Tang CM. 2011. Glutamate utilization promotes meningococcal survival in vivo through avoidance of the neutrophil oxidative burst. Mol Microbiol 81:1330–1342. doi: 10.1111/j.1365-2958.2011.07766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahashi H, Kim KS, Watanabe H. 2011. Meningococcal internalization into human endothelial and epithelial cells is triggered by the influx of extracellular L-glutamate via GltT L-glutamate ABC transporter in Neisseria meningitidis. Infect Immun 79:380–392. doi: 10.1128/IAI.00497-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bottero V, Chakraborty S, Chandran B. 2013. Reactive oxygen species are induced by Kaposi's sarcoma-associated herpesvirus early during primary infection of endothelial cells to promote virus entry. J Virol 87:1733–1749. doi: 10.1128/JVI.02958-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alam J, Baek KJ, Choi YS, Kim YC, Choi Y. 2014. N-acetylcysteine and the human serum components that inhibit bacterial invasion of gingival epithelial cells prevent experimental periodontitis in mice. J Periodontal Implant Sci 44:266–273. doi: 10.5051/jpis.2014.44.6.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He L, Zhang Y, Fang Y, Liang W, Lin J, Cheng M. 2014. Classical swine fever virus induces oxidative stress in swine umbilical vein endothelial cells. BMC Vet Res 10:279. doi: 10.1186/s12917-014-0279-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Pietro M, Filardo S, De Santis F, Mastromarino P, Sessa R. 2015. Chlamydia pneumoniae and oxidative stress in cardiovascular disease: state of the art and prevention strategies. Int J Mol Sci 16:724–735. doi: 10.3390/ijms16010724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seib KL, Wu HJ, Kidd SP, Apicella MA, Jennings MP, McEwan AG. 2006. Defenses against oxidative stress in Neisseria gonorrhoeae: a system tailored for a challenging environment. Microbiol Mol Biol Rev 70:344–361. doi: 10.1128/MMBR.00044-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seib KL, Tseng HJ, McEwan AG, Apicella MA, Jennings MP. 2004. Defenses against oxidative stress in Neisseria gonorrhoeae and Neisseria meningitidis: distinctive systems for different lifestyles. J Infect Dis 190:136–147. doi: 10.1086/421299. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi H, Watanabe H. 2002. A broad-host-range vector of incompatibility group Q can work as a plasmid vector in Neisseria meningitidis: a new genetical tool. Microbiology 148:229–236. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi H, Kim KS, Watanabe H. 2008. Differential in vitro infectious abilities of two common Japan-specific sequence-type (ST) clones of disease-associated ST-2032 and carrier-associated ST-2046 Neisseria meningitidis strains in human endothelial and epithelial cell lines. FEMS Immunol Med Microbiol 52:36–46. doi: 10.1111/j.1574-695X.2007.00342.x. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi H, Carlson RW, Muszynski A, Choudhury B, Kim KS, Stephens DS, Watanabe H. 2008. Modification of lipooligosaccharide with phosphoethanolamine by LptA in Neisseria meningitidis enhances meningococcal adhesion to human endothelial and epithelial cells. Infect Immun 76:5777–5789. doi: 10.1128/IAI.00676-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sa E Cunha C, Griffiths NJ, Murillo I, Virji M. 2009. Neisseria meningitidis Opc invasin binds to the cytoskeletal protein α-actinin. Cell Microbiol 11:389–405. doi: 10.1111/j.1462-5822.2008.01262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sa E Cunha C, Griffiths NJ, Virji M. 2010. Neisseria meningitidis Opc invasin binds to the sulphated tyrosines of activated vitronectin to attach to and invade human brain endothelial cells. PLoS Pathog 6:e1000911. doi: 10.1371/journal.ppat.1000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takahashi H, Yanagisawa T, Kim KS, Yokoyama S, Ohnishi M. 2012. Meningococcal PilV potentiates Neisseria meningitidis type IV pilus-mediated internalization into human endothelial and epithelial cells. Infect Immun 80:4154–4166. doi: 10.1128/IAI.00423-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown DR, Helaine S, Carbonnelle E, Pelicic V. 2010. Systematic functional analysis reveals that a set of seven genes is involved in fine-tuning of the multiple functions mediated by type IV pili in Neisseria meningitidis. Infect Immun 78:3053–3063. doi: 10.1128/IAI.00099-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brissac T, Mikaty G, Dumenil G, Coureuil M, Nassif X. 2012. The meningococcal minor pilin PilX is responsible for type IV pilus conformational changes associated with signaling to endothelial cells. Infect Immun 80:3297–3306. doi: 10.1128/IAI.00369-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leibowitz A, Boyko M, Shapira Y, Zlotnik A. 2012. Blood glutamate scavenging: insight into neuroprotection. Int J Mol Sci 13:10041–10066. doi: 10.3390/ijms130810041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsukita S, Yonemura S, Tsukita S. 1997. ERM proteins: head-to-tail regulation of actin-plasma membrane interaction. Trends Biochem Sci 22:53–58. doi: 10.1016/S0968-0004(96)10071-2. [DOI] [PubMed] [Google Scholar]
- 47.Eugene E, Hoffmann I, Pujol C, Couraud PO, Bourdoulous S, Nassif X. 2002. Microvilli-like structures are associated with the internalization of virulent capsulated Neisseria meningitidis into vascular endothelial cells. J Cell Sci 115:1231–1241. [DOI] [PubMed] [Google Scholar]
- 48.Masip L, Veeravalli K, Georgiou G. 2006. The many faces of glutathione in bacteria. Antioxid Redox Signal 8:753–762. doi: 10.1089/ars.2006.8.753. [DOI] [PubMed] [Google Scholar]
- 49.Lu SC. 2013. Glutathione synthesis. Biochim Biophys Acta 1830:3143–3153. doi: 10.1016/j.bbagen.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernstrom JD, Wurtman RJ, Hammarstrom-Wiklund B, Rand WM, Munro HN, Davidson CS. 1979. Diurnal variations in plasma concentrations of tryptophan, tryosine, and other neutral amino acids: effect of dietary protein intake. Am J Clin Nutr 32:1912–1922. [DOI] [PubMed] [Google Scholar]
- 51.Tsai PJ, Huang PC. 1999. Circadian variations in plasma and erythrocyte concentrations of glutamate, glutamine, and alanine in men on a diet without and with added monosodium glutamate. Metabolism 48:1455–1460. doi: 10.1016/S0026-0495(99)90159-2. [DOI] [PubMed] [Google Scholar]
- 52.Song J, Viggiano A, Monda M, De Luca V. 2014. Peripheral glutamate levels in schizophrenia: evidence from a meta-analysis. Neuropsychobiology 70:133–141. doi: 10.1159/000364828. [DOI] [PubMed] [Google Scholar]
- 53.Stephens DS, Krebs JW, McGee ZA. 1984. Loss of pili and decreased attachment to human cells by Neisseria meningitidis and Neisseria gonorrhoeae exposed to subinhibitory concentrations of antibiotics. Infect Immun 46:507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dewenter L, Volkmann TE, Maier B. 20 April 2015. Oxygen governs gonococcal microcolony stability by enhancing the interaction force between type IV pili. Integr Biol doi: 10.1039/C5IB00018A. [DOI] [PubMed] [Google Scholar]
- 55.Coureuil M, Lecuyer H, Scott MG, Boularan C, Enslen H, Soyer M, Mikaty G, Bourdoulous S, Nassif X, Marullo S. 2010. Meningococcus hijacks a β2-adrenoceptor/β-Arrestin pathway to cross brain microvasculature endothelium. Cell 143:1149–1160. doi: 10.1016/j.cell.2010.11.035. [DOI] [PubMed] [Google Scholar]
- 56.Mikaty G, Soyer M, Mairey E, Henry N, Dyer D, Forest KT, Morand P, Guadagnini S, Prevost MC, Nassif X, Dumenil G. 2009. Extracellular bacterial pathogen induces host cell surface reorganization to resist shear stress. PLoS Pathog 5:e1000314. doi: 10.1371/journal.ppat.1000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Virji M, Makepeace K, Ferguson DJ, Achtman M, Sarkari J, Moxon ER. 1992. Expression of the Opc protein correlates with invasion of epithelial and endothelial cells by Neisseria meningitidis. Mol Microbiol 6:2785–2795. doi: 10.1111/j.1365-2958.1992.tb01458.x. [DOI] [PubMed] [Google Scholar]
- 58.Stephens DS, Spellman PA, Swartley JS. 1993. Effect of the (α2→8)-linked polysialic acid capsule on adherence of Neisseria meningitidis to human mucosal cells. J Infect Dis 167:475–479. doi: 10.1093/infdis/167.2.475. [DOI] [PubMed] [Google Scholar]
- 59.Hammerschmidt S, Muller A, Sillmann H, Muhlenhoff M, Borrow R, Fox A, van Putten J, Zollinger WD, Gerardy-Schahn R, Frosch M. 1996. Capsule phase variation in Neisseria meningitidis serogroup B by slipped-strand mispairing in the polysialyltransferase gene (siaD): correlation with bacterial invasion and the outbreak of meningococcal disease. Mol Microbiol 20:1211–1220. doi: 10.1111/j.1365-2958.1996.tb02641.x. [DOI] [PubMed] [Google Scholar]
- 60.Hardy SJ, Christodoulides M, Weller RO, Heckels JE. 2000. Interactions of Neisseria meningitidis with cells of the human meninges. Mol Microbiol 36:817–829. doi: 10.1046/j.1365-2958.2000.01923.x. [DOI] [PubMed] [Google Scholar]
- 61.Spinosa MR, Progida C, Tala A, Cogli L, Alifano P, Bucci C. 2007. The Neisseria meningitidis capsule is important for intracellular survival in human cells. Infect Immun 75:3594–3603. doi: 10.1128/IAI.01945-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hammerschmidt S, Birkholz C, Zahringer U, Robertson BD, van Putten J, Ebeling O, Frosch M. 1994. Contribution of genes from the capsule gene complex (cps) to lipooligosaccharide biosynthesis and serum resistance in Neisseria meningitidis. Mol Microbiol 11:885–896. doi: 10.1111/j.1365-2958.1994.tb00367.x. [DOI] [PubMed] [Google Scholar]
- 63.Vogel U, Frosch M. 1999. Mechanisms of neisserial serum resistance. Mol Microbiol 32:1133–1139. doi: 10.1046/j.1365-2958.1999.01469.x. [DOI] [PubMed] [Google Scholar]
- 64.Tzeng YL, Stephens DS. 2000. Epidemiology and pathogenesis of Neisseria meningitidis. Microbes Infect 2:687–700. doi: 10.1016/S1286-4579(00)00356-7. [DOI] [PubMed] [Google Scholar]
- 65.Cartwright KA, Stuart JM, Jones DM, Noah ND. 1987. The Stonehouse survey: nasopharyngeal carriage of meningococci and Neisseria lactamica. Epidemiol Infect 99:591–601. doi: 10.1017/S0950268800066449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hammerschmidt S, Hilse R, van Putten JP, Gerardy-Schahn R, Unkmeir A, Frosch M. 1996. Modulation of cell surface sialic acid expression in Neisseria meningitidis via a transposable genetic element. EMBO J 15:192–198. [PMC free article] [PubMed] [Google Scholar]
- 67.Yazdankhah SP, Caugant DA. 2004. Neisseria meningitidis: an overview of the carriage state. J Med Microbiol 53:821–832. doi: 10.1099/jmm.0.45529-0. [DOI] [PubMed] [Google Scholar]
- 68.Leduc D, Gallaud J, Stingl K, de Reuse H. 2010. Coupled amino acid deamidase-transport systems essential for Helicobacter pylori colonization. Infect Immun 78:2782–2792. doi: 10.1128/IAI.00149-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ricci V, Giannouli M, Romano M, Zarrilli R. 2014. Helicobacter pylori γ-glutamyl transpeptidase and its pathogenic role. World J Gastroenterol 20:630–638. doi: 10.3748/wjg.v20.i3.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Henderson B, Martin A. 2013. Bacterial moonlighting proteins and bacterial virulence. Curr Top Microbiol Immunol 358:155–213. doi: 10.1007/82_2011_188. [DOI] [PubMed] [Google Scholar]
- 71.Henderson B, Martin A. 2011. Bacterial virulence in the moonlight: multitasking bacterial moonlighting proteins are virulence determinants in infectious disease. Infect Immun 79:3476–3491. doi: 10.1128/IAI.00179-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Knaust A, Weber MV, Hammerschmidt S, Bergmann S, Frosch M, Kurzai O. 2007. Cytosolic proteins contribute to surface plasminogen recruitment of Neisseria meningitidis. J Bacteriol 189:3246–3255. doi: 10.1128/JB.01966-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tunio SA, Oldfield NJ, Berry A, Ala'Aldeen DA, Wooldridge KG, Turner DP. 2010. The moonlighting protein fructose-1,6-bisphosphate aldolase of Neisseria meningitidis: surface localization and role in host cell adhesion. Mol Microbiol 76:605–615. doi: 10.1111/j.1365-2958.2010.07098.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.