

Abstract
Pseudomonas aeruginosa produces N-(3-oxo-dodecanoyl)-l-homoserine lactone (3OC12), a crucial signaling molecule that elicits diverse biological responses in host cells thought to subvert immune defenses. The mechanism mediating many of these responses remains unknown. The intracellular lactonase paraoxonase 2 (PON2) hydrolyzes and inactivates 3OC12 and is therefore considered a component of host cells that attenuates 3OC12-mediated responses. Here, we demonstrate in cell lines and in primary human bronchial epithelial cells that 3OC12 is rapidly hydrolyzed intracellularly by PON2 to 3OC12 acid, which becomes trapped and accumulates within the cells. Subcellularly, 3OC12 acid accumulated within the mitochondria, a compartment where PON2 is localized. Treatment with 3OC12 caused a rapid PON2-dependent cytosolic and mitochondrial pH decrease, calcium release, and phosphorylation of stress signaling kinases. The results indicate a novel, PON2-dependent intracellular acidification mechanism by which 3OC12 can mediate its biological effects. Thus, PON2 is a central regulator of host cell responses to 3OC12, acting to decrease the availability of 3OC12 for receptor-mediated effects and acting to promote effects, such as calcium release and stress signaling, via intracellular acidification.
INTRODUCTION
Pseudomonas aeruginosa is a common pathogen causing serious infections in immunocompromised and ill individuals due to the bacteria's ability to evade host immune responses and acquire antibiotic resistance (1). Many Gram-negative bacteria, including P. aeruginosa, produce acyl-homoserine lactone (AHL) signaling molecules which regulate the cell density-dependent expression of virulence factors in a process termed quorum sensing (QS) (1). The AHL N-(3-oxododecanoyl)-l-homoserine lactone (3OC12) is a key P. aeruginosa QS signal that has been shown to be necessary for biofilm maturation and full expression of virulence in P. aeruginosa animal infection models (2–5). Concentrations up to 600 μM 3OC12 have been measured in P. aeruginosa biofilms in vitro (6). Concentrations of over 6 μM 3OC12 have been detected in the sputum of individuals with pulmonary P. aeruginosa infections (7), suggesting active 3OC12 signaling in the human disease as well.
In addition to modulating bacterial gene expression, 3OC12 elicits a multitude of biological responses in diverse mammalian cell types (8). Depending upon the cell type and dose, 3OC12 (10 to 100 μM) can induce apoptosis, endoplasmic reticulum (ER) stress, chemotaxis, and proinflammatory gene expression (8–10). Conversely, 3OC12 inhibited lipopolysaccharide (LPS) induction of proinflammatory mediators in macrophages, fibroblasts, and epithelial cells and in vivo by repressing nuclear factor κ-light chain enhancer of activated B-cell (NF-κB) signaling (11). In antigen-stimulated T-lymphocytes, 3OC12 inhibits cell proliferation and production of gamma interferon and interleukin-4 (IL-4), critical regulators of immunity (8, 12). These diverse responses suggest that 3OC12 acts through multiple, and cell-type-dependent, mechanisms.
Delineating the role of 3OC12 in P. aeruginosa pathogenicity is difficult due to the multitude of often disparate effects it has on host cells, but also because the mechanisms by which 3OC12 mediates these effects are poorly understood. 3OC12 does not act through immune pattern recognition receptors such as Toll-like receptors and nucleotide binding or oligomerization domain-like receptors (13). In sinonasal epithelial cells the taste receptor 2 member 38 (T2R38) mediated a rapid Ca2+ and NO release by 3OC12; however, T2R38 likely only mediates responses in upper respiratory cell types (14). Due to its lipophilicity, 3OC12 rapidly enters mammalian cells (12). In Caco-2 intestinal epithelial cells, 3OC12 was found to alter cell migration, likely via interacting with the IQ-motif-containing GTPase activating protein (IQGAP1) and modulating its signaling (15). 3OC12 can interact with nuclear hormone peroxisome proliferator-activated receptor (PPAR) transcription factors, resulting in increased cytokine expression (16, 17). However, such effects are relatively slow, occurring at ≥6 h after 3OC12 treatment. Many effects of 3OC12, such as Ca2+ release and kinase activation, occur within 5 min of treatment (13, 18, 19), a timeline preceding any gene expression. The mechanism mediating these early effects of 3OC12 on host cells remains to be identified.
The paraoxonase (PON) family of mammalian esterases, PON1, PON2, and PON3, hydrolyze AHLs to their ring-opened biologically inactive carboxylic acid counterparts (20). PON2 is expressed intracellularly, is widely found in mammalian tissues and cell types, and efficiently hydrolyzes 3OC12 to 3OC12 acid (20–25). Independent of its hydrolytic activity, PON2 also has antioxidant activity and can protect cells from endoplasmic reticulum (ER) stress, including ER stress induced by 3OC12 (22, 23, 26). Such findings suggest that PON2 may be an important component of the innate defense by interfering with bacterial QS and attenuating 3OC12-mediated effects on host cells.
Recently, it was demonstrated that a relatively rapid, ≤2-h, induction of cytosolic Ca2+ and of markers of apoptosis in mouse embryonic fibroblasts by 3OC12 was dependent upon PON2 hydrolytic activity (27). Such findings were counterintuitive as PON2 was thought to inactive 3OC12, and the PON2-dependent mechanism mediating these bioeffects could not be explained. Here, we identify a unique mechanism by which PON2 can mediate biological effects of 3OC12. We demonstrate that 3OC12, which freely partitions into host cell membranes, is very rapidly hydrolyzed by the membrane-associated PON2 to its corresponding acid form which, in contrast to the lactone, accumulates in cells. Through this effect, the 3OC12 acid acidifies the cytosol and mitochondria within minutes and triggers Ca2+ liberation and p38 and elongation initiation factor 2 alpha (eIF2α) phosphorylation. Thus, PON2 both inactivates the lactone form of 3OC12 and promotes 3OC12-mediated intracellular acidification and the ensuing biological responses. Such findings suggest a central role for the enzyme in modulating bacterial QS and regulating host cell responses to bacterial homoserine lactone signaling molecules.
MATERIALS AND METHODS
Cells.
Generation and culturing of stable EA.hy 926 (EA.hy) cells overexpressing PON2 (EA.hy PON2) and the inactive PON2-H114Q mutant (EA.hy H114Q) and PON2-overexpressing HEK cells have been described previously (22, 28). Primary human bronchial epithelial cells (HBEC), human umbilical vein endothelial cells (HUVEC), and primary cell media and supplement mixes were from PromoCell, and cells were cultured as recommended by the supplier.
PON2 activity.
PON2 3OC12 (Sigma-Aldrich) hydrolytic activity was determined by high-performance liquid chromatography (HPLC) as previously described (20). Activity is expressed as units per milligram of lysate or purified protein. One unit equals 1 nmol of 3OC12 hydrolyzed per min. Recombinant human PON2 was purified as previously described (29).
Intracellular 3OC12 acid determination.
Cells were seeded in 24-well plates and the following day (at approximately 75% confluence) treated with 0.5 ml of medium containing 3OC12 and placed back in the cell culture incubator. At the times given in Fig. 1, the cells were rinsed with phosphate-buffered saline (PBS) and lysed with 100 μl of cold acetonitrile containing 25 μM N-dodecanoyl-l-homoserine lactone as the internal standard. Lysates were centrifuged for 1 min at 14,000 ×g and analyzed by HPLC as previously described (20). The concentration of 3OC12 acid in each sample was calculated from peak areas using a standard curve generated from 3OC12 acid standards. The 3OC12 acid was prepared by incubating 3OC12 in 5 mM NaOH for 2 h. Complete hydrolysis of 3OC12 to the 3OC12 acid was confirmed by HPLC analysis. Protein concentrations of cells were determined by lysis in radioimmunoprecipitation assay (RIPA) buffer, sonication, and analysis of lysates using the bicinchoninic acid (BCA) method (Thermo Scientific).
FIG 1.

Intracellular 3OC12 acid accumulation is PON2 dependent. (A and B) HEK and EA.hy cells were treated for the times shown with 25 μM 3OC12, rinsed, and lysed with acetonitrile, and lysates were analyzed for 3OC12 acid by HPLC. (C) HBEC were transfected with scrambled or PON2 siRNA for 3 days and then treated for 5 min with 25 μM 3OC12. Cells were then rinsed and lysed, and lysates were analyzed for 3OC12 acid. PON2 activity in scrambled and PON2 siRNA cell lysates was 16.8 ± 2.9 U/mg and 5.2 ± 0.9 U/mg, respectively (C). Data are the means of three separate experiments ± standard deviations. Differences between the results of the two groups were analyzed using a Student t test. *, P < 0.01; #, P < 0.05.
For determination of 3OC12 acid accumulation within the mitochondria, 5 × 106 cells were suspended in 10 ml of medium with 50 μM 3OC12 or with vehicle only for 30 min at 37°C. The cells were isolated and washed with PBS. An enriched mitochondrial fraction pellet was obtained as previously described (30). The mitochondrial pellet was lysed with 50 μl of acetonitrile, and 3OC12 acid was quantified by HPLC as described above. No peaks corresponding to the 3OC12 acid peak on the HPLC chromatogram from the control (vehicle-only) treated cells were present. To control for any 3OC12 acid potentially precipitating out of solution and/or associating with the mitochondrial fraction in a nonspecific manner, additional controls were analyzed in which 50 μM 3OC12 acid was added to the cell homogenate from untreated cells, followed by mitochondrial isolation. In these controls a small amount of 3OC12 acid was associated with the mitochondrial pellets. This background level of 3OC12 acid was subtracted from the amount of 3OC12 acid measured in the pellets isolated from 3OC12-treated cells. The mitochondrial protein, precipitated after acetonitrile lysis, was dissolved in 1 M NaOH overnight and quantified with the BCA method.
Cytosolic pH and Ca2+ measurements.
For determination of intracellular pH (pHi) and the cytosolic calcium concentration ([Ca2+]c) cells were seeded in black 96-well Costar plates so that they were 80 to 90% confluent at the time of assay. Treatments were performed at 37°C. For pHi determination, cells were loaded with 2 μM 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein-acetoxymethyl ester (BCECF-AM; Invitrogen) in 100 μl of medium for 40 min, washed, and incubated for 15 min with Hanks' balanced salt solution containing Ca2+ and Mg2+ (HBSS). Cells were washed again, treated with 3OC12 with or without triazolo[4,3-a]quinolone (TQ416; ChemDiv) or an equivalent volume of dimethyl sulfoxide (DMSO) as the control in 100 μl of HBSS, and fluorescence was measured using a Synergy HT fluorometric plate reader (Bio-Tek) with excitation wavelengths set at 485 nm and 360 nm and emission detected at 528 nm. The calibration of pHi was performed on cells using high-KCl buffers containing nigericin as previously described (31). Intracellular Ca2+ levels were determined as previously described with minor modifications (22). Plated cells were loaded with 4 μM Fluo-4 AM (Invitrogen) in medium containing 20 mM HEPES and 2.5 mM probenecid for 45 min and then washed three times in HBSS. Cells were treated with HBSS containing DMSO (controls), 3OC12, or 2,4,6-trimethyl-N-[3-(trifluoromethyl)phenyl]benzenesulfonamide (m-3M3FBS; EMD Millipore), and fluorescence was measured for 25 min with excitation and emission wavelengths of 480 nm and 530 nm, respectively.
Confocal microscopy.
EA.hy cells were seeded at 1×104 in four-well slide cover glass I chambers (Greiner Bio-One). The next day, cells were loaded with 5 μM SNARF-4F 5-(and-6)-carboxylic acid, acetoxymethyl ester, acetate (SNARF-4F; Invitrogen) and 1 μM Fluo-4 AM in Krebs buffer (Noxygen) at 37°C for 1 h. After brief washes, chambers were loaded on a 37°C, 5% CO2 incubator device mounted on a confocal Zeiss LSM710 laser scanning microscope. Upon exposure to 3OC12, cells were immediately imaged with an EC Plan-Neofluar 20×/0.50M27 objective, with 4.07 μs pixel dwell, 54-μm pinhole, and emission wavelengths of 493 to 516 nm (Fluo-4 AM); SNARF-4F was calculated as ratio of acidic (581 to 601 nm)/basic (650 to 738 nm) fluorescence units. Thus, an increased ratio indicates acidification. Using ZEN2009 software (Zeiss), the fluorescence intensities at every time point were recorded for approximately 50 to 100 cells per visible field and transferred to GraphPad Prism software (GraphPad Software, Inc.) for evaluation and data processing.
Mitochondrial pH (pHm) was determined as follows. HEK or EA.hy 926 cells were seeded in LabTek chambers (1.3 × 105 cells/chamber with four chambers per slide; Sarstedt). The next day, cells were gently washed with HBSS, stained with 5 μM 5-(and-6)-carboxy SNARF-1 acetoxymethyl ester, acetate (SNARF-1 AM; Invitrogen) in HBSS for 10 min at 37°C, and then kept in MICA buffer (145 mM KCl, 1.5 mM CaCl2, 1 mM MgSO4, 10 mM HEPES, 5.5 mM glucose, adjusted to pH 7.5 with Tris) for 3 h to allow mitochondrial targeting of SNARF-1. During a further 15-min incubation time, cells were stained in HBSS with 5 nM DiOC6 (3,3′-dihexyloxacarbocyanine iodide; Invitrogen), a cell-permeant, fluorescent dye that accumulates in respiratory-active mitochondria of live cells when used at low concentrations. We verified that 5 nM DiOC6 does not stain other organelles (data not shown). Then, chambers were mounted on the microscope for imaging. Cells were then treated with 50 μM 3OC12 in 500 μl of MICA buffer per chamber, accompanied by imaging on a Zeiss LSM710 (Plan/Apochromat 63×/1.4 oil differential interference contrast [DIC] objective; 1-min intervals for 15 min; z-stacking with 5 to 8 levels per image at 1 airy unit; excitation at 488 and 543 nm; emission at 500 to 520 nm for DiOC6, 580 to 595 nm for acidic SNARF-1, and 650 to 730 nm for basic SNARF-1). ImageJ software was used to evaluate SNARF-1 ratiometric changes in all single z-stacks for any time point using DiOC6 staining as a mitochondrial mask in order to collect only mitochondrial SNARF-1 signals. Changes in pHm were calculated after a preceding calibration with 10 μM nigericin-containing MICA buffers covering a pH range from 6.8 to 8.1. The treatment of EA.hy 926 cells differed from that of HEK cells in that the SNARF-1 incubation time was 90 min, which resulted in nearly complete overlap of SNARF-1 fluorescence with that of mitochondrial DiOC6.
Western blotting.
All lysates were produced in the presence of PhosphoStop phosphatase inhibitor (Roche) and HALT protease inhibitor (Thermo Scientific). The PON2 antibody and immunoblotting have been previously described (22). Antibodies against eIF2α/phospho-eIF2α (Ser51) and p38 (Thr180/Tyr182) were from Cell Signaling and used as recommended. Anti-glyceraldehyde-3-phosphate dehydrogenase ([GAPDH] clone 6C5; Santa Cruz) and mouse-anti-tubulin Ab2 (Dianova) were used at 1:5,000. Antibodies against cytochrome c oxidase IV (COX IV; Novus Biologicals) and histone 3 (Genetex) were used in accordance with the manufacturers' protocols. Horseradish peroxidase (HRP)-conjugated secondary antibodies were from Cell Signaling. Immunocomplexes were visualized and analyzed by Western blotting using a ChemiDoc XRS imaging system (Bio-Rad) equipped with QuantityOne, version 4.6.7, software. Phosphorylated proteins were normalized to total levels of the respective protein and/or to the level of tubulin or GAPDH.
RNA interference (RNAi).
Approximately 60% confluent cells were transfected with 50 nm PON2-specific or scrambled Stealth small interfering RNA (siRNA) (Invitrogen) using either SaintRed (Synvolux) or Lipofectamine RNAiMax (Invitrogen) transfection reagent according to the supplier's instructions. siRNA sequences and methods have been previously described (32). Stimulation with 3OC12 or LPS (Escherichia coli 0111:B4; Sigma) was performed 2.5 to 3 days after treatment since this was the time point of maximal knockdown. Efficiency was about 50 to 60% in HUVEC and ≥80% in all other cells (at protein and activity levels). Levels of phospho-p38 or phospho-eIF2α were normalized to total p38 or eIF2α levels, respectively.
Statistical analysis.
Curve-fitting and statistical analysis were performed with GraphPad Prism software.
RESULTS
PON2 mediates intracellular 3OC12 acid accumulation.
We along with others had previously observed that 3OC12 acid accumulated inside cells after treatment with 3OC12 (13). This suggested that the hydrophobic 3OC12 is hydrolyzed inside the cell to its corresponding ring-opened 3OC12 acid, which is much more polar and likely unable to readily cross cellular membranes. We therefore measured the rates of intracellular accumulation of 3OC12 acid in human embryonic kidney cells (HEK) and HEK PON2 cells stably transfected with a human PON2-green fluorescent protein (GFP) construct. The HEK cells express low basal levels of PON2, 2.2 ± 0.2 U/mg, while the stably transfected HEK PON2 cells expressed 70.4 ± 7.0 U/mg of PON2. When cells were treated with 3OC12, the rate of intracellular 3OC12 acid accumulation was much faster in the HEK PON2 cells and began to diminish rapidly, within about 4 min (Fig. 1A). This decrease in the rate of 3OC12 acid accumulation could at least partly be due to the resulting acidification, which would decrease the rate of pH-dependent lactone hydrolysis and the ability of 3OC12 to cause inactivation of PON2 activity (19, 33).
We also compared the rates of intracellular 3OC12 acid accumulation in a human endothelial cell line, EA.hy 926, stably transfected with human PON2 (EA.hy PON2) or an inactive PON2-H114Q mutant (EA.hy H114Q). The PON2 mutant retains its antioxidant and antiapoptotic functions but does not have 3OC12 hydrolytic activity (32). Thus, this mutant controls for effects due to increased protein expression and any effects of PON2 not associated with its hydrolytic activity. We have also previously established that PON2 is the only enzyme that hydrolyzes 3OC12 in EA.hy cells (20). The EA.hy H114Q cells have 6.8 ± 0.4 U/mg of PON2 activity, due to significant basal levels of PON2, while the EA.hy PON2 cells have 25.8 ± 2.5 U/mg of PON2 activity. The rate of accumulation of 3OC12 acid in the EA.hy PON2 cells was significantly higher than that in the EA.hy H114Q cells and also began to diminish rapidly (Fig. 1B).
The contribution of PON2 to 3OC12 acid accumulation was also evaluated in primary human bronchial epithelial cells (HBEC), cells that come into direct contact with P. aeruginosa during pulmonary infections. The cells were first transfected for 3 days with PON2 siRNA to decrease PON2 levels or with a scrambled siRNA as a control. Upon treatment with 3OC12, 3OC12 acid accumulated rapidly in the HBEC, and this accumulation was significantly diminished in the PON2 siRNA-treated cells (Fig. 1C). 3OC12 was not detected in any of the cells at any time point (detection limit, 0.5 nmol/mg of cell lysate). These results demonstrate that rapid intracellular accumulation 3OC12 acid largely depends on hydrolytically active PON2.
3OC12 causes a rapid PON2-dependent cytosolic acidification and calcium release.
The rapid intracellular accumulation of 3OC12 acid suggested a potential corresponding decrease in intracellular pH (pHi). Therefore, we directly visualized the change in pHi in naive (nontransfected) EA.hy cells after 3OC12 treatment by laser scanning confocal microscopy. Because decreased pHi can induce Ca2+ release and because increased cytosolic Ca2+ [(Ca2+)c] is a common response to 3OC12 in mammalian cells (8, 34), we also concomitantly measured [Ca2+]c fluxes. The cells were simultaneously loaded with the Ca2+ indicator Fluo-4 AM and the ratiometric pH indicator SNARF-4F and treated with 3OC12, and time-lapse images were acquired. As shown in Fig. 2A and B, 3OC12 caused a very rapid decrease in cytosolic pHi (as measured by an increase in SNARF-4F fluorescence) and an increase in [Ca2+]c. Interestingly, the pHi appeared to return to initial levels within 4 min but then decreased again before slowly returning to initial levels (Fig. 2B). The decrease in pHi appeared to just precede the increase in [Ca2+]c, consistent with a potential intracellular acidification mediating Ca2+ release into the cytosol.
FIG 2.

3OC12 mediates intracellular acidification and Ca2+ rise. (A and B) Naive EA.hy cells were loaded with Fluo-4 AM and SNARF-4F, and fluorescence intensities were recorded in confocal time-lapse images to monitor intracellular calcium and pH fluxes, respectively, in response to 50 μM 3OC12. In panel A, the green channel (Fluo-4 AM) reports the cytosolic Ca2+ rise; the intensity of the blue channel (ratio of acidic/basic SNARF-4F signals) is a measure of cytosolic acidification. Scale bar, 50 μm (A). Fluorescence intensities for all cells were individually measured, allowing simultaneous determination of changes in pH and Ca2+ within the same cell. In panel B, the SNARF-4F ratio before 3OC12 stimulation was set at zero. The graph shows the mean from four representative experiments. (C and D) HBEC loaded with BCECF-AM to detect cytosolic pH values (C) or Fluo-4 AM to detect cytosolic Ca2+ rise (D) were treated with increasing concentrations of 3OC12, and fluorescence was measured on a microplate reader. Each trace is the mean of three separate experiments in which the background (DMSO-treated samples) values were subtracted from the values of the 3OC12-treated samples. Dashed lines denote the standard deviations. Differences between the groups were analyzed by a two-way repeated-measures analysis of variance. The pH drop at 50 μM 3OC12 was significantly different from that at 25 μM 3OC12 (C), and the Ca2+ increases at 25 μM and 50 μM 3OC12 were significantly greater than the background value (P, < 0.01). PMT, photomultiplier tube; RFU, relative fluorescence units; a.u., arbitrary units.
The ability of 3OC12 to cause cytosolic acidification and calcium release was also evaluated with the pH indicator BCECF-AM and the Ca2+ indicator Fluo-4 AM, respectively, in the HBEC. 3OC12 caused a rapid and dose-dependent pHi decrease and [Ca2+]c increase (Fig. 2C and D). The pHi began to recover after 30 min (Fig. 2C) but at lower rate than in the EA.hy cells. The [Ca2+]c remained elevated in the HBEC even after 25 min (Fig. 2D). Thus, different cell types react with individual kinetics; however, the general responses to 3OC12 are consistent.
In mouse embryonic fibroblasts, a [Ca2+]c increase with 3OC12 treatment was shown to be dependent upon PON2 (27). We further explored the dependence of the [Ca2+]c increase on PON2 in our PON2-expressing cell lines as well as in the HBEC transfected with PON2 siRNA. In addition, we evaluated the 3OC12-mediated pHi changes. In the HEK cells 3OC12 caused no detectable change in pHi and a slow, minor rise in [Ca2+]c (Fig. 3A and D). Conversely, in the HEK PON2 cells 3OC12 caused a rapid, almost immediate, decrease in pHi with a concomitant pronounced rise in [Ca2+]c (Fig. 3A and D). Unlike levels in the HBEC, the pHi and [Ca2+]c returned to initial levels relatively quickly, by 10 to 15 min posttreatment. 3OC12 also caused a rapid decrease in pHi with a concomitant increase in [Ca2+]c in the EA.hy H114Q and EA.hy PON2 cells (Fig. 3B and E). However, the pHi decrease and the [Ca2+]c increase in EA.hy PON2 cells were significantly greater than changes in levels in EA.hy H114Q cells. Also, the increase in [Ca2+]c was delayed in EA.hy H114Q cells compared to that in EA.hy PON2 cells. Within each cell type the extent of intracellular acidification and [Ca2+]c flux corresponded closely with the time course of 3OC12 acid accumulation and cellular PON2 levels. The pHi and [Ca2+]c changes in the EA.hy H114Q and EA.hy PON2 cells were also transient, lasting 20 to 25 min.
FIG 3.

3OC12-mediated intracellular acidification and Ca2+ rise are PON2-dependent. Cells were loaded with BCECF-AM to detect cytosolic pH (A to C) or Fluo-4 AM to detect cytosolic Ca2+ rise (D to F); cells were then treated with test compounds, and fluorescence was measured on a microplate reader. HEK and EA.hy 926 cells were treated with 25 μM 3OC12. HBEC (C and F) were transfected with scrambled or PON2 siRNA and 3 days later treated with 50 μM 3OC12. For the experiment shown in panel F, cells were also treated with a 25 μM concentration of the phospholipase C activator m-3M3FBS. PON2 activity levels in scrambled and PON2 siRNA transfected cell lysates were 15.0 ± 2.9 U/mg and 1.6 ± 0.7 U/mg, respectively (C and F). Each trace is the mean of three separate experiments in which the background (DMSO-treated cells) values were subtracted from the values of the stimulus-treated cells. Dashed lines denote the standard deviations. Differences between the results for the groups were analyzed by a two-way repeated-measures analysis of variance. The pH drop was significantly lower in the HEK PON2 (P < 0.01), EA.hy H114Q PON2 (P < 0.05), and the scrambled siRNA (P < 0.01) groups than in the respective control groups in panels A, B and C, respectively. The Ca2+ rise was significantly different from that of the control group (D and E) and from that of the PON2 siRNA 3OC12-treated group (F) (P < 0.01).
Lowering PON2 levels by RNAi in the HBEC nearly eliminated the 3OC12-mediated acidification and [Ca2+]c rise (Fig. 3C and F). PON2 had no direct effect on calcium release as treatment of HBEC with the phospholipase C activator m-3M3FBS, which generates inositol phosphate and subsequent release of intracellular calcium stores, resulted in the same [Ca2+]c increases in both the PON2 and scrambled siRNA transfected cells (Fig. 3F). Thus, as with the cell lines, the 3OC12-mediated pHi and [Ca2+]c changes are dependent upon PON2 in the primary HBEC.
3OC12 causes PON2-dependent phosphorylation of mitogen-activated protein kinase (MAPK) p38 and eIF2α.
p38 and eIF2α are kinases that are activated in response to stressors. Activated eIF2α inhibits protein translation and is a marker of ER stress, and phosphorylation of both p38 and eIF2α is established as an immediate response to 3OC12 (11) (Fig. 4A). Furthermore, p38 is phosphorylated in response to intracellular acidification (35). Therefore, we hypothesized that the phosphorylation of these kinases by 3OC12 treatment would be dependent on PON2. Compared to naive EA.hy cells and EA.hy H114Q cells, the EA.hy PON2 cells exhibited increased p38 and eIF2α phosphorylation in response to 3OC12 (Fig. 4B to D). Phosphorylation of p38 by LPS was the same in the EA.hy naive, H114Q, and PON2 cells, demonstrating that PON2 affects only 3OC12-mediated p38 phosphorylation (Fig. 4B to D).
FIG 4.

3OC12-induced activation of p38 and eIF2α signaling is PON2 dependent. (A) Primary HUVEC were treated with indicated concentrations of 3OC12 for 10 min. Subsequently, levels of total and phosphorylated p38 and eIF2α were assessed by Western blotting. Tubulin served as a loading control. (B and C) EA.hy cells were treated with 3OC12 (or LPS) as indicated, followed by immunoblotting against phosphorylated p38 and eIF2α. WT, wild type. (D) Quantitative evaluation ± standard errors of the means of p38 and eIF2α phosphorylation from four individual experiments as described for panels B and C. (E) The experiment was similar to that described in panels B and C but used HUVEC that were untreated (naive) or treated with PON2-specific or a control siRNA. (F) Quantitative evaluation ± standard errors of the means of four individual experiments as described in panel E. Values for naive and control siRNA-treated cells were the same (data not shown). (G) HBEC treated with control or PON2-specific siRNA were incubated with 3OC12 as indicated, followed by immunoblotting for total/phosphorylated p38 and eIF2α, PON2, and α-tubulin as described before. (H) Quantification of the average ± the range of blots from two individual experiments as described for panel G. *, P < 0.05; **, P < 0.01.
To see if such effects were also dependent on PON2 in primary cells, phosphorylation of p38 and eIF2α was investigated in both primary human umbilical vein endothelial cells (HUVEC) and the HBEC. Decreasing PON2 levels by RNAi also diminished the 3OC12-mediated p38 and eIF2α phosphorylation in the HUVEC (Fig. 4E and F). Treatment of HBEC with 25 μM 3OC12 for 10 min did not induce p38 or eIF2α phosphorylation (Fig. 4G and H). p38 and eIF2α phosphorylation was induced by treating HBEC with 50 μM 3OC12 for 10 min and with 25 μM and 50 μM 3OC12 for 30 min (Fig. 4G and H). In all treatments that induced p38 and eIF2α phosphorylation, decreasing PON2 levels by RNAi significantly diminished phosphorylation (Fig. 4G and H). Collectively, the data demonstrate that the 3OC12-mediated phosphorylation of p38 and eIF2α is dependent upon PON2.
3OC12 acid accumulates within the mitochondria.
PON2 is predominantly localized to the inner mitochondrial membrane and likely facing the lumen in the ER (26, 36, 37). Therefore, we hypothesized (i) that it is within these organelles where 3OC12 acid is predominantly accumulating and (ii) that the pH drop measured in the cytosol is a reflection of a much greater pH drop within the ER and/or mitochondria. To demonstrate this principle, an enriched mitochondrial fraction was isolated from HEK PON2 cells after treatment with 3OC12 and analyzed for 3OC12 acid levels. Western analysis using organelle-specific markers demonstrated that the isolation method used resulted in preparations enriched in mitochondria (Fig. 5A). 3OC12 acid was present inside the mitochondrial fraction at 3.9 ± 0.6 nmol/mg protein after treatment of cells for 30 min with 50 μM 3OC12 (Fig. 5B). Visual inspection of the mitochondrial pellets indicated that they were very small, occupying less than about 0.5 μl in volume. Thus, the concentration of 3OC12 acid inside the enriched mitochondria can be estimated at greater than 220 μM, roughly 4-fold greater than the concentration of the 3OC12 used to treat the cells. No 3OC12 was detected in the enriched mitochondrial fraction.
FIG 5.
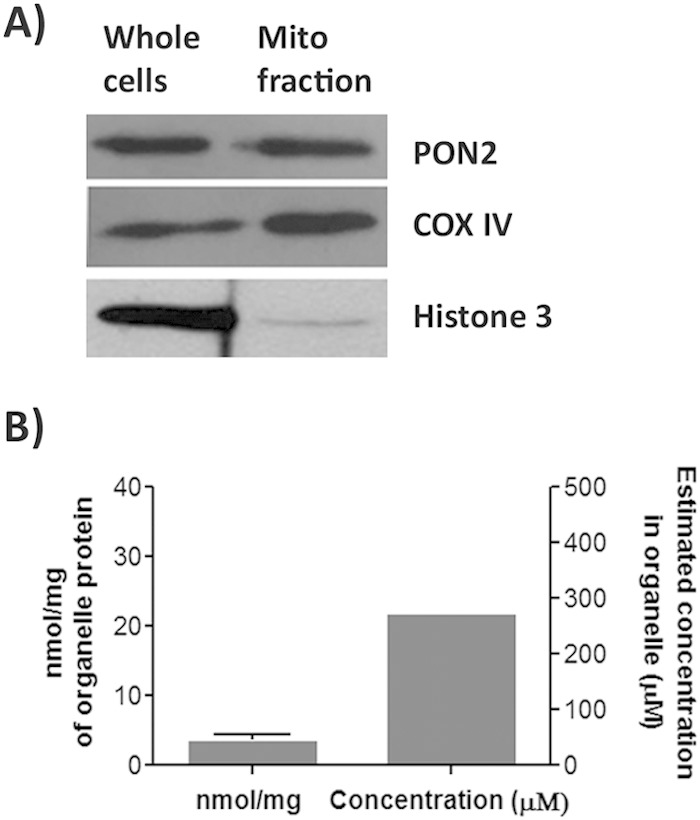
3OC12 acid accumulates within the mitochondrial fraction of 3OC12 treated cells. (A) Levels of PON2, COX IV (mitochondrial marker), and histone 3 (nuclear marker) in whole-cell lysates and isolated mitochondrial fractions from HEK PON2 cells were estimated by Western analysis. (B) Mitochondrial fractions from HEK PON2 cells treated with 50 μM 3OC12 were isolated, lysed, and analyzed for 3OC12 acid levels. The error bar represents the standard deviation of three separate experiments.
While the procedure used to isolate mitochondria results in a fraction highly enriched in mitochondria, it does not completely remove lysosomes, peroxisomes, or the Golgi complex (30). Therefore, to provide confirmatory evidence that 3OC12 acid is accumulating within the mitochondria, we determined if 3OC12 could decrease pH specifically within this organelle. EA.hy 926 cells stably overexpressing nonfluorescent PON2-hemagglutinin (HA) or the empty plasmid (pCDNA3-HA) (22) were loaded with SNARF-1, a ratiometric pH indicator targeting the mitochondria. pHm was visualized by three-dimensional (3D) multicolor time-lapse live-cell imaging. To account for mitochondrial movement and cell morphology changes during imaging, tracking of mitochondria through different image layers was established by concomitant cell loading with the mitochondrial dye DiOC6. The low concentration of DiOC6 (5 nM) did not stain ER membranes, and the dye perfectly localized with dyes such as MitoTracker-Orange (data not shown). Using the DiOC6 signal as a mask, SNARF-1 ratios at DiOC6-positive areas, i.e., mitochondria, were quantified. This enabled estimation of SNARF-1 ratios for all recorded z-layers at every time point. A calibration curve had been established by nigericin-containing buffers at different pH values. As shown in Fig. 6A and B, 3OC12 treatment of control EA.hy 926 cells resulted in an immediate decrease of pHm during the first 10 min. More importantly, upon overexpression of PON2, this acidification was significantly enhanced (Fig. 6C). Mitochondrial acidification in response to 3OC12 was also seen in HUVEC and, to a lower extent, in HEK cells (data not shown). Collectively, these data demonstrate a general mitochondrial acidification in response to 3OC12 exposure dependent on PON2-mediated acidification.
FIG 6.

3OC12 induces PON2-mediated mitochondrial acidification. (A) EA.hy 926 cells were loaded with the mitochondrial ratiometric pH indicator SNARF-1 (orange/red) and mitochondrial dye DiOC6 (green) as indicated in Materials and Methods. Cells were treated with 3OC12 (50 μM). pHm changes were observed in time-lapse 3D confocal imaging for the indicated duration (representative images for 1, 3, 5, 7, 9 min are shown). A change of SNARF-1 ratiometric fluorescence from orange/red to yellow/green indicates acidification. (B) Quantification of pHm changes during 3OC12 treatment, with a typical trace for EA.hy cells. (C) EA.hy 926 cells with PON2 overexpression (red curve) show an increased rate of 3OC12-induced pHm drop compared to that in control EA.hy (blue; empty plasmid) or solvent (0.1% methanol)-treated controls (black). Symbols show means ± standard errors of the means of 8 to 15 individual experiments demonstrating the SNARF-1 ratiometric pHm response; slopes of linear regression curves differ with a P of 0.004 for naive versus EA.hy PON2 cells. t, time.
A small-molecule inhibitor of 3OC12-mediated cellular responses also inhibits PON2.
The triazolo[4,3-a]quinolone compound TQ416 was recently identified in a high-throughput screen as a potent inhibitor of 3OC12 effects in cell cultures (38). At a concentration of 1 μM, TQ416 restored the 3OC12-mediated inhibition of LPS-induced NF-κB activation and prevented 3OC12-induced (Ca2+)c release, phosphorylation of p38, and caspase activation (38). This suggested to us that TQ416 may be preventing these effects of 3OC12 via inhibiting PON2 activity. Indeed, TQ416 was a potent inhibitor of PON2 3OC12 hydrolysis (Fig. 7A). TQ416 also potently inhibited 3OC12-mediated cytosolic acidification in EA.hy PON2 cells (Fig. 7B). These findings suggest that at least some of the inhibitory effects of TQ416 on 3OC12's biological actions are via inhibition of PON2-dependent 3OC12 intracellular acidification. Interestingly, we have thus identified a likely TQ416 target and the first inhibitor of PON2 activity.
FIG 7.

TQ416 inhibits PON2 and prevents intracellular acidification by 3OC12. Purified recombinant PON2 (A) and HEK PON2 (B) cell lysates were analyzed for PON2 activity in the presence of increasing concentrations of TQ416. Data are the means of three separate experiments ± standard deviations. Differences between the control (0 μM TQ416) and TQ416-treated groups were analyzed using a Student t test (*, P < 0.01). (C) EA.hy PON2 cells were loaded with BCECF-AM, and decreases in intracellular acidification were determined after treatment with 50 μM 3OC12 in the presence of increasing concentrations of TQ416 as described in Materials and Methods. Each trace is the mean of three separate experiments in which the background (DMSO-treated cells) values were subtracted from the values of treated cells. Error bars denote the standard deviations. Differences between the results of the groups were analyzed by a two-way repeated-measures analysis of variance. The pH drop in the TQ416-treated groups was significantly different from that of the group treated with 3OC12 only (P < 0.01).
DISCUSSION
3OC12 elicits a spectrum of biological effects in diverse host cell types, many of which are believed to favor P. aeruginosa persistence. While 3OC12 can activate the T2R38 receptor in sinonasal cells and modulate PPAR receptor activities, the mechanism mediating many of 3OC12's effects remains enigmatic (14, 16). Here, we demonstrate in both cell lines and primary HBEC that the [Ca2+]c increase and phosphorylation of p38 and eIF2α, common immediate effects of 3OC12, are dependent upon intracellular PON2-mediated hydrolysis of 3OC12 to 3OC12 acid. The time course of 3OC12 acid accumulation corresponds closely with the time course of cytosolic acidification and the measured biological responses. Thus, our data demonstrate a novel mechanism in which PON2 hydrolyzes 3OC12 to its carboxylic acid, which becomes trapped within the cell, causing an intracellular acidification triggering pH-dependent biological responses. These findings indicate that 3OC12 lactone hydrolysis is not always an inactivation pathway and reveals that, via both 3OC12 inactivation and 3OC12-mediated intracellular acidification, PON2 is likely a central regulator of 3OC12 biological functions and immunomodulatory responses.
We propose that intracellular acidification is the initial event mediating our observed biological responses to 3OC12. Phosphorylation of p38 by 3OC12, demonstrated to be dependent on PON2 in this study, is known to be induced by intracellular acidification (35). Many proteins that have critical roles in biological functions are very sensitive to minute changes in pH (39). Therefore, cells stringently regulate pHi by employing diverse families of proton extruders and pumps and a balance of intracellular weak acids and bases to increase buffering capacity (39). This propensity of cells to maintain pH homeostasis is exemplified in our cell lines, where cytosolic pHi decreases return to initial levels within 10 to 20 min after 3OC12 treatment (Fig. 3D and E).
As PON2 is localized to the mitochondria and ER, we hypothesized that 3OC12 acid accumulation and acidification would be predominantly occurring in these organelles. Indeed, we demonstrate that 3OC12 acid is concentrating in the mitochondrial fraction and that mitochondria undergo a rapid PON2-dependent acidification after 3OC12 treatment. Mitochondria rely on a proton gradient to drive ATP synthesis and to transport metabolites and ions across the inner mitochondrial membrane, making this organelle exquisitely sensitive to pH changes (40). Thus, we hypothesize that acidification within the mitochondria, and possibly the ER, is the primary event triggering the pH-dependent biological responses to 3OC12.
Because PON2 is widely expressed in cells and tissues (21, 25), most mammalian cells would be expected to hydrolyze 3OC12 intracellularly and potentially undergo subsequent acidification. However, due to the array of pH-regulating mechanisms within cells, the responses of different cell types to 3OC12 acid accumulation would not be expected to be the same (35, 39). We found that the rate of intracellular 3OC12 acid accumulation in the HEK PON2 cells was slightly greater than that in the EA.hy PON2 cells, yet the pHi drop was less extensive and had a shorter time course (Fig. 1 and 3). Interestingly, despite the less extensive pHi drop in the HEK PON2 cells, the maximal increase in [Ca2+]c was greater and occurred earlier than in the EA.hy PON2 cells (Fig. 3). Thus, compared to the EA.hy PON2 cells, the HEK PON2 cells appear to be resistant to cytosolic pHi changes but sensitive to pHi-mediated (Ca2+)c induction. Our findings suggest that, in addition to cellular PON2 levels, the ability of a cell type to regulate pHi and the sensitivity of its signaling pathways to pHi changes will likely be critical factors modulating the response to 3OC12.
3OC12 has been shown to mediate biological responses in host cells by several mechanisms. 3OC12 activates the G-protein-coupled receptor T2R38 to induce Ca2+ and nitric oxide synthase in sinusoidal cells and has been shown to interact with IQGAP1, affecting cell migration in Caco-2 epithelial cells (15). 3OC12 was shown to bind PPARγ receptors, resulting in modulation of cytokine expression (16, 17). Evidence has also been provided that direct interaction of 3OC12 with the plasma membrane may mediate some biological responses (41). PON2 will act to attenuate signaling through these pathways that are mediated by the lactone form of 3OC12. Conversely, PON2 will promote p38, eIF2α, and Ca2+ signaling via intracellular acidification in response to 3OC12 exposure. Apoptotic responses to 3OC12 in mouse embryo fibroblasts were recently shown to depend upon PON2 hydrolytic activity (27). Thus, it appears that 3OC12-mediated apoptosis is driven by PON2-dependent intracellular acidification as well although further studies are needed to determine the contribution of PON2 to 3OC12-mediated apoptosis in different cell types.
The overall cellular response to 3OC12 will depend upon multiple factors, including the signaling pathways present in the cell type, the sensitivity of the cell signaling pathways to 3OC12, and 3OC12 exposure duration and concentrations. Given the ubiquitous expression of PON2, simultaneous signaling via PON2-dependent acidification and other receptor-mediated pathways is likely. A diagram illustrating 3OC12 signaling via identified mechanisms and PON2's potential role in modulating the pathways is shown in Fig. 8. We have previously shown that 25 μM 3OC12 is below saturating concentrations for PON2 and that the rate of hydrolysis by the enzyme drops considerably below 25 μM 3OC12 (20). This, together with our demonstration that a PON2-dependent [Ca2+]c increase by 3OC12 is measurable only at 3OC12 concentrations above 10 μM (Fig. 2D), suggests that the PON2-dependent acidification effects may be appreciably activated only at 3OC12 concentrations above 10 μM. Thus, presuming that PON2 is expressed at significant levels, at higher 3OC12 concentrations, ≥25 μM, intracellular acidification effects may predominate and also modulate receptor-driven effects, whereas as at lower 3OC12 concentrations, ≤10 μM, the more sensitive receptor-driven effects will predominate. It is clear that the biological effects elicited by 3OC12 will depend greatly on the signaling pathways present in the cell type under evaluation.
FIG 8.

Scheme depicting the role of PON2 in modifying host cellular responses to 3OC12. 3OC12 concentrations of 25 μM are below saturating for PON2. Therefore, the rate of 3OC12 hydrolysis by PON2 drops significantly as the substrate falls below 25 μM. At concentrations below 10 μM, the lower rate of 3OC12 hydrolysis by PON2 will result in limited effects due to intracellular acidification. Also, below 10 μM 3OC12, PON2 would not be as effective at limiting the availability of 3OC12-lactone for receptor-mediated effects. At concentrations of 10 to 25 μM 3OC12, both receptor and intracellular acidification effects may occur, and acidification may significantly modulate receptor-mediated effects. Above 25 μM 3OC12, acidification effects would be expected to predominate due to a rapid and extensive intracellular acidification and reduction of the 3OC12-lactone bioavailability.
The concentration range of 3OC12 used in this study, 25 to 50 μM, is that which is typically used to evaluate 3OC12's biological effects. However, the levels of 3OC12 that cells are exposed to in vivo during P. aeruginosa infection are uncertain and a matter of debate. While concentrations of 3OC12 from 1 to 10 μM have been measured in planktonic cultures, concentrations of ∼600 μM have been measured in P. aeruginosa biofilms grown in vitro (6). Concentrations of up to 6.9 μM were recently measured in sputum samples from subjects with pulmonary P. aeruginosa infections (7). However, the authors of this study indicated that 3OC12 concentrations may be higher near sites of P. aeruginosa colonization due to differing environments within the lung and colonization densities. Our findings have significant implications with respect to quantifying the level of 3OC12 produced in vivo. 3OC12 has a very high partitioning coefficient of ∼1,000:1 in a lipophilic/aqueous system (6). Thus, consistent with our data and those of others (12), the 3OC12 produced would be expected to rapidly partition into host cells. Once in the cells, it will be rapidly hydrolyzed by PON2 to 3OC12 acid and become trapped, precluding detection of the metabolite extracellularly. Furthermore, P. aeruginosa produces N-acylases with high activity toward 3OC12 that will likely contribute to 3OC12 elimination (42). Therefore, due to intracellular sequestration and hydrolysis, metabolism via bacterial enzymes, and dilution into large sputum volumes, the low-micromolar concentrations of 3OC12 measured in sputum samples from infected subjects are likely a significant underestimation of concentrations to which host cells are exposed. The rapid partitioning of 3OC12 into host cells and intracellular entrapment of 3OC12 acid must be considered in future studies designed to determine the amount of 3OC12 produced in vivo during infection.
In summary, this study demonstrates a novel PON2-dependent mechanism by which 3OC12 elicits biological effects in mammalian cells. Additionally, we show that this mechanism is operative in all immortalized and primary cells analyzed in this study, suggesting that it is generally biologically relevant. The PON2-mediated intracellular acidification occurs rapidly and thus likely contributes to many of the previously reported, yet unexplained, immediate cellular responses to 3OC12. This finding should greatly accelerate the understanding of how P. aeruginosa, as well as other Gram-negative bacteria, utilize AHLs to modulate host immune responses. In addition, it may reveal novel therapeutic targets which may be exploited to limit the pathogenicity of P. aeruginosa.
ACKNOWLEDGMENTS
J.F.T. and S.H. were funded by a U.S. Department of Defense award (W81XWH-12-2-0091). S.H. was also funded by intramural funds from the University Medical Center Mainz, the German Research Foundation (DO1289/6-1), Gerhard and Martha Röttger Foundation, and the Center for Thrombosis and Hemostasis, Mainz, Germany (BMBF funding allocation ID 01E01003, project TRPA11i).
REFERENCES
- 1.Schuster M, Greenberg EP. 2006. A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa. Int J Med Microbiol 296:73–81. doi: 10.1016/j.ijmm.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 2.Hoffmann N, Rasmussen TB, Jensen PO, Stub C, Hentzer M, Molin S, Ciofu O, Givskov M, Johansen HK, Hoiby N. 2005. Novel mouse model of chronic Pseudomonas aeruginosa lung infection mimicking cystic fibrosis. Infect Immun 73:2504–2514. doi: 10.1128/IAI.73.4.2504-2514.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakagami G, Morohoshi T, Ikeda T, Ohta Y, Sagara H, Huang L, Nagase T, Sugama J, Sanada H. 2011. Contribution of quorum sensing to the virulence of Pseudomonas aeruginosa in pressure ulcer infection in rats. Wound Repair Regen 19:214–222. doi: 10.1111/j.1524-475X.2010.00653.x. [DOI] [PubMed] [Google Scholar]
- 4.Christensen LD, Moser C, Jensen PO, Rasmussen TB, Christophersen L, Kjelleberg S, Kumar N, Hoiby N, Givskov M, Bjarnsholt T. 2007. Impact of Pseudomonas aeruginosa quorum sensing on biofilm persistence in an in vivo intraperitoneal foreign-body infection model. Microbiology 153:2312–2320. doi: 10.1099/mic.0.2007/006122-0. [DOI] [PubMed] [Google Scholar]
- 5.Wu H, Song Z, Hentzer M, Andersen JB, Molin S, Givskov M, Hoiby N. 2004. Synthetic furanones inhibit quorum-sensing and enhance bacterial clearance in Pseudomonas aeruginosa lung infection in mice. J Antimicrob Chemother 53:1054–1061. doi: 10.1093/jac/dkh223. [DOI] [PubMed] [Google Scholar]
- 6.Charlton TS, de NR, Netting A, Kumar N, Hentzer M, Givskov M, Kjelleberg S. 2000. A novel and sensitive method for the quantification of N-3-oxoacyl homoserine lactones using gas chromatography-mass spectrometry: application to a model bacterial biofilm. Environ Microbiol 2:530–541. doi: 10.1046/j.1462-2920.2000.00136.x. [DOI] [PubMed] [Google Scholar]
- 7.Struss AK, Nunes A, Waalen J, Lowery CA, Pullanikat P, Denery JR, Conrad DJ, Kaufmann GF, Janda KD. 2013. Toward implementation of quorum sensing autoinducers as biomarkers for infectious disease states. Anal Chem 85:3355–3362. doi: 10.1021/ac400032a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teplitski M, Mathesius U, Rumbaugh KP. 2011. Perception and degradation of N-acyl homoserine lactone quorum sensing signals by mammalian and plant cells. Chem Rev 111:100–116. doi: 10.1021/cr100045m. [DOI] [PubMed] [Google Scholar]
- 9.Zimmermann S, Wagner C, Muller W, Brenner-Weiss G, Hug F, Prior B, Obst U, Hansch GM. 2006. Induction of neutrophil chemotaxis by the quorum-sensing molecule N-(3-oxododecanoyl)-l-homoserine lactone. Infect Immun 74:5687–5692. doi: 10.1128/IAI.01940-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tateda K, Ishii Y, Horikawa M, Matsumoto T, Miyairi S, Pechere JC, Standiford TJ, Ishiguro M, Yamaguchi K. 2003. The Pseudomonas aeruginosa autoinducer N-3-oxododecanoyl homoserine lactone accelerates apoptosis in macrophages and neutrophils. Infect Immun 71:5785–5793. doi: 10.1128/IAI.71.10.5785-5793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Grauer DC, Lehmann M, Meijler MM, Janda KD, Ulevitch RJ. 2008. Modulation of gene expression via disruption of NF-κB signaling by a bacterial small molecule. Science 321:259–263. doi: 10.1126/science.1156499. [DOI] [PubMed] [Google Scholar]
- 12.Ritchie AJ, Whittall C, Lazenby JJ, Chhabra SR, Pritchard DI, Cooley MA. 2007. The immunomodulatory Pseudomonas aeruginosa signalling molecule N-(3-oxododecanoyl)-l-homoserine lactone enters mammalian cells in an unregulated fashion. Immunol Cell Biol 85:596–602. doi: 10.1038/sj.icb.7100090. [DOI] [PubMed] [Google Scholar]
- 13.Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Wood MR, Brogan AP, Lehmann M, Mee JM, Iwata K, Pan Q, Fearns C, Knaus UG, Meijler MM, Janda KD, Ulevitch RJ. 2006. N-(3-oxo-acyl)homoserine lactones signal cell activation through a mechanism distinct from the canonical pathogen-associated molecular pattern recognition receptor pathways. J Biol Chem 281:28822–28830. doi: 10.1074/jbc.M606613200. [DOI] [PubMed] [Google Scholar]
- 14.Lee RJ, Xiong G, Kofonow JM, Chen B, Lysenko A, Jiang P, Abraham V, Doghramji L, Adappa ND, Palmer JN, Kennedy DW, Beauchamp GK, Doulias PT, Ischiropoulos H, Kreindler JL, Reed DR, Cohen NA. 2012. T2R38 taste receptor polymorphisms underlie susceptibility to upper respiratory infection. J Clin Invest 122:4145–4159. doi: 10.1172/JCI64240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karlsson T, Turkina MV, Yakymenko O, Magnusson KE, Vikstrom E. 2012. The Pseudomonas aeruginosa N-acylhomoserine lactone quorum sensing molecules target IQGAP1 and modulate epithelial cell migration. PLoS Pathog 8:e1002953. doi: 10.1371/journal.ppat.1002953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jahoor A, Patel R, Bryan A, Do C, Krier J, Watters C, Wahli W, Li G, Williams SC, Rumbaugh KP. 2008. Peroxisome proliferator-activated receptors mediate host cell proinflammatory responses to Pseudomonas aeruginosa autoinducer. J Bacteriol 190:4408–4415. doi: 10.1128/JB.01444-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooley MA, Whittall C, Rolph MS. 2010. Pseudomonas signal molecule 3-oxo-C12-homoserine lactone interferes with binding of rosiglitazone to human PPARγ. Microbe Infect 12:231–237. doi: 10.1016/j.micinf.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 18.Shiner EK, Terentyev D, Bryan A, Sennoune S, Martinez-Zaguilan R, Li G, Gyorke S, Williams SC, Rumbaugh KP. 2006. Pseudomonas aeruginosa autoinducer modulates host cell responses through calcium signalling. Cell Microbiol 8:1601–1610. doi: 10.1111/j.1462-5822.2006.00734.x. [DOI] [PubMed] [Google Scholar]
- 19.Horke S, Witte I, Altenhofer S, Wilgenbus P, Goldeck M, Forstermann U, Xiao J, Kramer GL, Haines DC, Chowdhary PK, Haley RW, Teiber JF. 2010. Paraoxonase 2 is down-regulated by the Pseudomonas aeruginosa quorum sensing signal N-(3-oxododecanoyl)-l-homoserine lactone and attenuates oxidative stress induced by pyocyanin. Biochem J 426:73–83. doi: 10.1042/BJ20091414. [DOI] [PubMed] [Google Scholar]
- 20.Teiber JF, Horke S, Haines DC, Chowdhary PK, Xiao J, Kramer GL, Haley RW, Draganov DI. 2008. Dominant role of paraoxonases in inactivation of the Pseudomonas aeruginosa quorum-sensing signal N-(3-oxododecanoyl)-l-homoserine lactone. Infect Immun 76:2512–2519. doi: 10.1128/IAI.01606-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackness B, Beltran-Debon R, Aragones G, Joven J, Camps J, Mackness M. 2010. Human tissue distribution of paraoxonases 1 and 2 mRNA. IUBMB Life 62:480–482. doi: 10.1002/iub.347. [DOI] [PubMed] [Google Scholar]
- 22.Horke S, Witte I, Wilgenbus P, Kruger M, Strand D, Forstermann U. 2007. Paraoxonase-2 reduces oxidative stress in vascular cells and decreases endoplasmic reticulum stress-induced caspase activation. Circulation 115:2055–2064. doi: 10.1161/CIRCULATIONAHA.106.681700. [DOI] [PubMed] [Google Scholar]
- 23.Kim JB, Xia YR, Romanoski CE, Lee S, Meng Y, Shi YS, Bourquard N, Gong KW, Port Z, Grijalva V, Reddy ST, Berliner JA, Lusis AJ, Shih DM. 2011. Paraoxonase-2 modulates stress response of endothelial cells to oxidized phospholipids and a bacterial quorum-sensing molecule. Arterioscler Thromb Vasc Biol 31:2624–2633. doi: 10.1161/ATVBAHA.111.232827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Devarajan A, Bourquard N, Grijalva VR, Gao F, Ganapathy E, Verma J, Reddy ST. 2013. Role of PON2 in innate immune response in an acute infection model. Mol Genet Metab 110:362–370. doi: 10.1016/j.ymgme.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marsillach J, Mackness B, Mackness M, Riu F, Beltran R, Joven J, Camps J. 2008. Immunohistochemical analysis of paraoxonases-1, 2, and 3 expression in normal mouse tissues. Free Radic Biol Med 45:146–157. doi: 10.1016/j.freeradbiomed.2008.03.023. [DOI] [PubMed] [Google Scholar]
- 26.Altenhofer S, Witte I, Teiber JF, Wilgenbus P, Pautz A, Li H, Daiber A, Witan H, Clement AM, Forstermann U, Horke S. 2010. One enzyme, two functions: PON2 prevents mitochondrial superoxide formation and apoptosis independent from its lactonase activity. J Biol Chem 285:24398–24403. doi: 10.1074/jbc.M110.118604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwarzer C, Fu Z, Morita T, Whitt AG, Neely AM, Li C, Machen TE. 2015. Paraoxonase 2 serves a proapopotic function in mouse and human cells in response to the Pseudomonas aeruginosa quorum-sensing molecule N-(3-oxododecanoyl)-homoserine lactone. J Biol Chem 290:7247–7258. doi: 10.1074/jbc.M114.620039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schweikert EM, Devarajan A, Witte I, Wilgenbus P, Amort J, Forstermann U, Shabazian A, Grijalva V, Shih DM, Farias-Eisner R, Teiber JF, Reddy ST, Horke S. 2012. PON3 is upregulated in cancer tissues and protects against mitochondrial superoxide-mediated cell death. Cell Death Differ 19:1549–1560. doi: 10.1038/cdd.2012.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Draganov DI, Teiber JF, Speelman A, Osawa Y, Sunahara R, La Du BN. 2005. Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlapping and distinct substrate specificities. J Lipid Res 46:1239–1247. doi: 10.1194/jlr.M400511-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Graham JM. 2001. Isolation of mitochondria from tissues and cells by differential centrifugation. Curr Protoc Cell Biol Chapter 3:Unit 3.3. doi: 10.1002/0471143030.cb0303s04. [DOI] [PubMed] [Google Scholar]
- 31.Grant RL, Acosta D. 1997. Ratiometric measurement of intracellular pH of cultured cells with BCECF in a fluorescence multi-well plate reader. In Vitro Cell Dev Biol Anim 33:256–260. doi: 10.1007/s11626-997-0044-z. [DOI] [PubMed] [Google Scholar]
- 32.Witte I, Altenhoefer S, Wilgenbus P, Amorat J, Clement AM, Pautz A, Li H, Foerstermann U, Horke S. 2011. Beyond reduction of atherosclerosis: PON2 provides apoptosis resistance and stabilizes tumor cells. Cell Death Dis 2:e112. doi: 10.1038/cddis.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teiber JF, Draganov DI, La Du BN. 2003. Lactonase and lactonizing activities of human serum paraoxonase (PON1) and rabbit serum PON3. Biochem Pharmacol 66:887–896. doi: 10.1016/S0006-2952(03)00401-5. [DOI] [PubMed] [Google Scholar]
- 34.Speake T, Yodozawa S, Elliott AC. 1998. Modulation of calcium signalling by intracellular pH in exocrine acinar cells. Eur J Morphol 36(Suppl):165–169. [PubMed] [Google Scholar]
- 35.Riemann A, Schneider B, Ihling A, Nowak M, Sauvant C, Thews O, Gekle M. 2011. Acidic environment leads to ROS-induced MAPK signaling in cancer cells. PLoS One 6:e22445. doi: 10.1371/journal.pone.0022445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hagmann H, Kuczkowski A, Ruehl M, Lamkemeyer T, Brodesser S, Horke S, Dryer S, Schermer B, Benzing T, Brinkkoetter PT. 2014. Breaking the chain at the membrane: paraoxonase 2 counteracts lipid peroxidation at the plasma membrane. FASEB J 28:1769–1779. doi: 10.1096/fj.13-240309. [DOI] [PubMed] [Google Scholar]
- 37.Devarajan A, Bourquard N, Hama S, Navab M, Grijalva VR, Morvardi S, Clarke CF, Vergnes L, Reue K, Teiber JF, Reddy ST. 2011. Paraoxonase 2 deficiency alters mitochondrial function and exacerbates the development of atherosclerosis. Antioxid Redox Signal 14:341–351. doi: 10.1089/ars.2010.3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valentine CD, Zhang H, Phuan PW, Nguyen J, Verkman AS, Haggie PM. 2014. Small molecule screen yields inhibitors of Pseudomonas homoserine lactone-induced host responses. Cell Microbiol 16:1–14. doi: 10.1111/cmi.12176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casey JR, Grinstein S, Orlowski J. 2010. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol 11:50–61. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- 40.Santo-Domingo J, Demaurex N. 2012. Perspectives on: SGP symposium on mitochondrial physiology and medicine: the renaissance of mitochondrial pH. J Gen Physiol 139:415–423. doi: 10.1085/jgp.201110767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis BM, Jensen R, Williams P, O'Shea P. 2010. The interaction of N-acylhomoserine lactone quorum sensing signaling molecules with biological membranes: implications for interkingdom signaling. PLoS One 5:e13522. doi: 10.1371/journal.pone.0013522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wahjudi M, Papaioannou E, Hendrawati O, van Assen AH, van Merkerk R, Cool RH, Poelarends GJ, Quax WJ. 2011. PA0305 of Pseudomonas aeruginosa is a quorum quenching acylhomoserine lactone acylase belonging to the Ntn hydrolase superfamily. Microbiology 157:2042–2055. doi: 10.1099/mic.0.043935-0. [DOI] [PubMed] [Google Scholar]