

Abstract
Transcriptional regulatory networks are biological network motifs that act in accordance with each other to play decisive roles in the pathological processes of cancer. One of the most common types, the feed-forward loop (FFL), has recently attracted interest. Three connected deregulated nodes, a transcription factor (TF), its downstream microRNA (miRNA) and their shared target gene can make up a class of cancer-involved FFLs as ≥1 of the 3 can act individually as a bona fide oncogene or a tumor suppressor. Numerous notable elements, such as p53, miR-17-92 cluster and cyclins, are proven members of their respective FFLs. Databases of interaction prediction, verification of experimental methods and confirmation of loops have been continually emerging during recent years. Development of TF-miRNA-target loops may help understand the mechanism of tumorgenesis at a higher level and explain the discovery and screening of the therapeutic target for drug exploitation.
Keywords: feed-forward loop, transcription factor, microRNA, cancer
1. Introduction
Although theoretical concepts and technological approaches have made significant progress, the molecular basis of carcinogenesis and progression of various types of cancer remains to be understood. This deficit in knowledge hinders the development of effective therapies and progress in the treatment of cancers remains slow. As a result, the curability of cancers is still poor (1).
To promote the discovery of oncogenic pathways, investigators have assessed biological networks, such as transcriptional regulatory networks (TRNs). TRNs (also known as gene regulatory networks) can offer the possibility to improve the understanding of the topology and function of gene regulation of the cellular responses to environmental changes at a system level. One important local property of biological networks is the ‘network motifs’, first described by Milo et al (2). They are patterns of interconnections occurring in complex networks and may reflect a framework in which particular functions are achieved efficiently. Much experimental study has been devoted to understanding network motifs in TRNs, as they define the core of the regulatory machinery of cellular life and are largely responsible for information processing and decision making (3).
The transcription network is a collection of DNA segments in a cell, which interacts with each other indirectly (through their RNA and protein expression products) and with other substances in the cell, thereby governing the expression levels of mRNA and proteins. In the network, a gene serves as the source of a direct regulatory edge by producing an RNA or protein molecule that functions as a transcriptional activator or inhibitor of the target gene. The network consists of network motifs, such as feed-forward loops (FFLs), feed-back loops (FBLs) and single-input modules. FFLs have been shown to be one of the most important and promising classes of transcriptional network motifs (2,4,5).
The FFL, a three-node motif pattern, is composed of two input elements, one of which regulates the other, both jointly regulating a target gene. Each of the three interactions in the FFL can be either activating or repressing (6,7).
As the research is being driven by its promising prospects, the importance of post-transcriptional processes have become more evident than previously expected in the regulation of gene expression. Among the various mechanisms of TRNs, transcription factors (TFs) and a class of small RNAs, known as microRNAs (miRNAs or miRs), are frequently observed in numerous TRN motifs, joining transcriptional and post-transcriptional regulatory interactions together, so as to play their prominent roles in regulation. Additionally, as the research regarding cancers expands, FFLs composed of a TF, an miRNA and their equivalent target gene are becoming apparent and the number of studies with such FFLs of different components reported is continuously increasing (8–12).
All the possible FFLs involving miRNA, TF and target gene are shown in Fig. 1. The TF-miRNA-target gene FFLs, as the network motif of typical TRNs that we will discuss in the present review, are depicted in Fig. 1.
Figure 1.
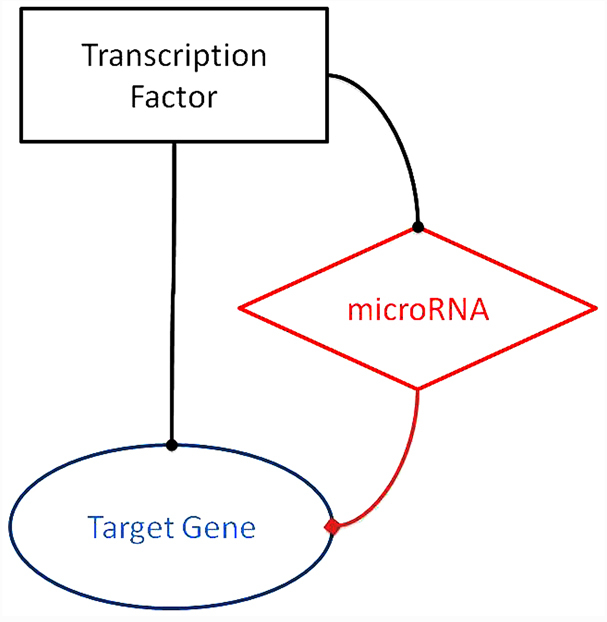
Typical feed-forward loops (FFLs). Representation of the FFL discussed in the present study. The square box represents the master transcription factor, the diamond-shaped box represents the microRNA involved in the circuit, and the oval box represents the downstream protein-coding target gene. Inside this circuit, the black line indicates transcriptional activation/repression, while the red line indicates post-transcriptional repression.
In molecular biology and genetics, a TF (sometimes known as a sequence-specific DNA-binding factor) is a protein that binds to specific DNA sequences, thereby controlling the flow (or transcription) of genetic information from DNA to messenger RNA (mRNA) (13). Characterized by containing one or more DNA-binding domains, which attach to specific sequences of DNA adjacent to the genes that they regulate, TFs are essential for the regulation of gene expression and are found in all living organisms. TFs can read and interpret the genetic ‘blueprint’ in the DNA. They bind to the DNA and help initiate a program of increased or decreased gene transcription. As such, they are vital for numerous important cellular processes (14–16).
The most well-known TF, p53, regulates genes, such as p21, cdc25c, bax and puma, which are elucidated to have an indispensable role in cell cycle arrest, apoptosis or cell senescence, and all these can eliminate or reverse the presence of progenitor cancer cells in the body (17). p53 function appears to be crucial in tumors: i) p53 limits the first steps of transformation by preventing the proliferation of cells with damaged genomes or dysregulated growth. ii) p53 may act as an emergency brake at later stages of tumor progression, by preventing cells from accumulating multiple mutations and developing an invasive phenotype. Taken together, these mechanisms explain the effects of TP53 mutation in numerous types of human cancer, detectable sometimes as an early event in precursor lesions or as a later event at the transition from in situ to invasive cancer (18). Cancer associated miRNAs, such as miR-34, miR-221 and miR-15/16, harbor p53 consensus binding sites and are already confirmed to be regulated by p53 and thus control downstream genes, including Bcl2, p27, E2F3 and CDK6, to carry out anti-tumorigenesis function (19). In 2008, Brosh et al (20) reported an FFL constructed by p53, E2F and miR-106b/93/25 polycistron, of which the target gene E2F was also a TF.
Numerous other TFs, such as WT1, TAL1/SCL and Myc, have also been proven to be associated with their regulating miRNA or target gene in cancer (21,22).
Increasing attention has focused on miRNAs as they have been indicated in various types of human cancer. miRNAs are small, evolutionarily conserved, endogenous non-coding RNAs of 18–25 nucleotides (nts) in length that have an important function in gene regulation by pairing to the mRNAs of protein-coding genes to direct their post-transcriptional repression. Their silencing effects are exerted by cleavage of their target mRNAs and by inhibition of their translation. Each miRNA can target a large number of genes (mRNAs) and each mRNA can be targeted by several miRNAs. It is generally observed that miRNAs only have a minor influence on the protein levels of their targets; however, miRNAs can have a profound influence on cell-fate determination. It can even change a phenotype by modulating a single miRNA. miRNAs are now known to repress thousands of target genes and coordinate normal processes, such as developmental timing, pattern formation, embryogenesis, differentiation, organogenesis, growth control and cell death. This discovery established a new paradigm of gene regulation (23–29). The alteration of miRNAs also contributes to a range of human pathologies, including cancer. Different associations between miRs and cancer have accumulated since the first evidence of an oncogene, KRAS, being targeted by an miRNA, the let-7 family, was reported in 2005 (30).
Beyond the impact of somatic, genetic and epigenetic lesions, the altered expression of miRNAs in cancer can arise through the aberrant activity of TFs that control their expression. Of note, the same TFs are often targets of miRNA-mediated repression, which gives rise to complex regulatory circuits (24).
Following this, as the understanding of miRNAs improved due to high-throughput miRNA expression profiling, bioinformatic prediction and other advanced technologies, their functions as regulators in signaling pathways and transcription networks have been revealed step by step. The maps of the networks have been completed gradually.
Directed by the theoretical discovery, research on signal flow inside the cell has increased. Researchers have carried out various experiments on the typical transcription network motifs. Subsequently, more hypotheses and their respective confirmative experiments were conducted and the TF-miRNA-target gene FFL motif theory was proven repeatedly, particularly in various types of human cancer (31–34).
2. Reported TF-miRNA-target gene FFLs in cancer
One of the best-characterized oncogenic miRNAs is miR-17-92, a polycistronic miRNA cluster also designated by He et al (35) as oncomiR-1 in 2005. This was the first time that the concept of an ‘oncogenic miRNA (oncomiR)’ became apparent. As more miRNAs have been identified to act as oncogenes, tumor suppressors and important modulators in cellular pathways have been divided into two classes: Increased activity of oncomiRs leads to inhibition of tumor-suppressor genes, facilitating cell proliferation and tumor progression. Decreased activity of tumor-suppressor miRNAs (tsmiRs) leads to increased oncogene translation, contributing to tumor formation (36,37).
Since the same TF can act as an activator or a repressor under different conditions, its directly-regulating downstream miRNA can be either an oncomiR or a tsmiR regardless of whether the TF is an oncogene or a tumor repressor. For instance, Myc, the c-Myc oncogenic TF, is known to directly upregulate a pro-tumorigenic group of miRNAs known as the miR-17-92 cluster, however, the predominant consequence of Myc activation is widespread repression of miRNA expression (38). The involved miRNAs, including miR-26a, miR-150 and miR-195/miR-497, whose tumor-suppressing properties are to be confirmed (39–41).
TF-miRNA-target gene FFL circuits with its TF as an oncogene in cancer
In 2005, O'Donnell et al (42) reported that the loop consisting of c-Myc, miR-17-5p and miR-20a cluster and E2F1 modulates cellular proliferation in P493-6 cells. c-Myc simultaneously activates E2F1 transcription and limits its translation by upregulating miR-17-5p and miR-20a, allowing a tightly controlled proliferative signal.
Burk et al (31) reported an FFL in 2008. In the FFL, zinc-finger E-box binding homeobox 1 (ZEB1) directly suppressed the transcription of miRNA-200 family members, miR-141 and miR-200c, which suppress target gene transforming growth factor-β2 (TGFβ2) and strongly activate epithelial differentiation so as to repress the epithelial-mesenchymal transition (EMT) in pancreatic, colorectal and breast cancer cells. As less-suppressed TGFβ2 in return upregulates ZEB1, ZEB1 triggers this miRNA-mediated FFL that stabilizes EMT and promotes invasion. TF ZEB1 itself is a crucial inducer of EMT in various human tumors and was shown to promote invasion and metastasis of tumor cells (43).
Following this, KRAS was proved to be a target for several miRNAs and KRAS activation indicated the repression of several miRNAs. For example, in pancreatic cancer with mutant KRAS, RAS-responsive element-binding protein 1 (RREB1) represses the miR-143 and miR-145 promoter and at the same time KRAS and RREB1 are targets of miR-143 and miR-145, revealing a feed-forward regulatory circuit that increases the effect of RAS signaling (44).
El Baroudi et al (45) made a summary of simple and mixed FFLs involving c-Myc in 2011. There are various complex circuits with c-Myc involved, such as the MYC/PTEN/miR-106b, miR-93, miR-25, miR-19a, miR-22, miR-26a, miR-193b and miR-23b circuit, acting as a noise-buffering circuit to guarantee a steady level of the PTEN protein as a tumor-suppressor gene. The MYC/retinoblastoma 1 (RB1)/miR-106a, miR-106b and miR-17 circuit has a critical role in the pathogenesis of solid cancer by repressing the transcription and translation of tumor-suppressor gene RB1. The MYC/vascular endothelial growth factor (VEGF)/miR-106b, miR-106a, miR-93, miR-34a, miR-20a, miR-17, miR-16 and miR-15a circuit, can be classified as a coherent or incoherent loop, depending on the different functional roles of VEGF ranging from cell migration to apoptosis.
In the year 2013, Polioudakis et al (46) confirmed that miR-22, activated by the TF Myc when quiescent cells enter proliferation, could inhibit the Myc transcriptional repressor MXD4, mediating an FFL to elevate Myc expression levels in HeLa cells and human foreskin fibroblasts.
Also in 2013, Zhao et al (41) published a study that identified a MYC-miRNAs-EZH2 FFL linking overexpression of MYC, EZH2 and miR-26a repression in aggressive B cell lymphomas.
TF-miRNA-target gene FFL circuits with its TF as a tumor repressor in cancer
He et al (47) reported that the loop formed by p53 and miR-34a-c promotes cell cycle arrest and inhibits inappropriate cell proliferation in 2007. They proved direct regulation of the association between p53 and miR-34a, miR-34a and its target genes CDK4 and MET; however, in 2011, Hwang et al (32) established the exact p53-regulated FFL: Regulation of cancer-invasion-promoting gene MET by wild-type p53 consists of miR-34-dependent and -independent mechanisms. p53 activates miR-34, which represses MET and p53 can repress MET itself.
Untypical or uncertain reported FFLs are associated with cancer
In 2008, Lin et al (48) showed that TF c-Myc directly activates transcription of the 3 subunits of eIF4F (eIF4E, eIF4AI and eIF4GI), which is thought to be the rate-limiting phase of translation. Increased eIF4F levels result in stimulation of c-Myc mRNA translation specifically. This FFL involving c-Myc and eIF4F that serves to link transcription and translation could contribute to the effects of c-Myc on cell proliferation and neoplastic growth. The following year, the investigators published another study confirming the FFL association and highlighted that the regulators of the transcription and translation that affect Myc function (such as Mad1 or antisense approaches) or eIF4F activity (such as mammalian target of rapamycin) are expected to act as rheostats during normal growth and development to fine-tune the outcomes of the Myc/eIF4F FFL, representing promising targets for cancer therapy (49).
By studying head and neck squamous cell carcinoma in 2009, Cohen and Rosner (50) and Cohen et al (51) identified an FFL in cell cycle regulation involving protein kinase Cα (PKCα) that activates mitogen-activated protein kinase (MAPK), as well as cyclin E translation via inhibition of miR-15a. Of note, while one arm of the network entails classic transcriptional regulation of cyclins by the MAPK pathway, the other arm involves regulation of miR15a inhibiting cyclin E translation. The FFL is constitutively driven by PKCα activation, leading to the unabated proliferation inherent to cancer cells. Although the specific elements may differ, their results suggest that FFL networks could play a fundamental role in controlling DNA synthesis and cell cycle progression in tumor cells. In this FFL, PKCα is not typically a TF, the mechanism of how it downregulates miR-15a remains to be elucidated.
Using a coculture model system, Rokavec et al (52) showed a feed-forward inflammatory signaling circuit in breast cancer in 2012. The circuit was composed of miR-200c, p65, c-Jun N-terminal kinase 2 (JNK2), heat-shock factor 1 (HSF1) and interleukin 6 (IL-6). Suppression of miR-200c by IL-6 constitutively activates p65/RelA and JNK2, and the latter phosphorylates and activates HSF1. In turn, HSF1 triggers demethylation of the IL-6 promoter that facilitates the binding of p65 and c-Jun, which together drive constitutive IL-6 transcription, promoting transformation in human cancer cells and in a mouse model of ErbB2-driven breast cancer.
3. Finding and confirming a specific TF-miRNA-target gene FFL
Research has accumulated in this field of the typical TF-miRNA-target protein coding gene formed FFL involved in cancers. During the past decades, investigators have developed an accession of procedure to predict, investigate and verify the interaction of the 3 elements of a special FFL circuit. A series of databases appeared with respectively different algorithms to offer bioinformatic support of predicted connections.
miRNAs repress the translation of target genes by binding, in a Watson-Crick complementary manner, to 7-nt long sequences present at the 3′-untranslated region (3′-UTR) of the regulated genes. The binding usually involves 2–8 nts of the miRNA, known as the ‘seed’. The large amount of research associated with the discovery of TF binding sites suggest that transcriptional and post-transcriptional regulatory interactions could be predicted in silico by searching over-represented short sequences of nts present in promoters or 3′-UTRs and by filtering the results with suitable evolutionary or functional constraints.
Independent computational evidence for the regulatory interactions of the TF-miRNA-target gene FFL can be extracted from the ECRbase, miRBase, PicTar and TargetScan databases, with relevance to cancer of the TF-miRNA-target gene FFL as deduced from their intersection with the oncomiR and cancer gene census databases. In addition, Gene Ontology enrichment provides detailed information regarding the joint targets of the loop (5).
Establishment of the miRNA-target gene association
Significant progress has been made in computational algorithms for miRNA target prediction during the last decade. Currently, there are databases such as TargetScan, miRanda, PicTar, RNA22 and DIANA for target gene prediction (53–55) (Table I). When miRNA and its potential target gene emerge from bioinformatics, gain- and loss-of-function methods, most commonly transfection, are ready to be applied to evaluate the effect of the miRNA in respective human cell lines. Immunology or molecular biology experiments, such as western blot analysis, quantitative polymerase chain reaction (qPCR), and miRNA and mRNA microarray expression profiling, are also involved to verify the linking dynamic expression changes of the miRNA, or its mimics and its target genes' mRNA and protein. For microarray analysis, the stand-alone software tool CoExpress (freely available at www.bioinformatics.lu/CoExpress), could be chosen to perform interactive detection of correlated profiles in large expression data sets. Finally, a luciferase reporter assay will confirm associations between the miRNA and its targets (56). Publications of validated miR-target correlations are recorded in TarBase.
Table I.
Examples of common databases for miRNA-TG and TF-TG interactions.
| Databases | URL |
|---|---|
| miRNA target prediction databases | |
| TargetScan | http://www.targetscan.org/ |
| miRanda | http://www.microrna.org/ |
| PicTar | http://pictar.mdc-berlin.de/ |
| RNA22 | https://cm.jefferson.edu/rna22/Interactive/ |
| DIANA | http://diana.imis.athena-innovation.gr/DianaTools/index.php |
| Databases on validated microRNA targets | |
| TarBase | http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=tarbase/index |
| miRWalk | http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/mirnapredictedtarget.php |
| miRBase | http://www.mirbase.org/ |
| Motif search database for TF binding site | |
| TRANSFAC | http://www.gene-regulation.com/ |
| JASPAR | http://jaspar.genereg.net/ |
| TRED | http://rulai.cshl.edu/cgi-bin/TRED/tred.cgi?process=home |
| DBTSS | http://dbtss.hgc.jp |
| TRRD | http://wwwmgs.bionet.nsc.ru/mgs/gnw/trrd/ |
miRNA, microRNA; TG, target gene; TF, trancription factor.
Establishment of the TF-target gene association
In an effort to further dissect the molecular pathways regulated by a particular TF, several genome-wide screens have been used to identify its transcriptional targets. The transcription regulation databases include TRANSFAC, JASPAR, TRED, DBTSS and TRRD (57–61). Among these databases mentioned, TRANSFAC is the most commonly used (Table I). First, researchers use bioinformatics to select consensus DNA elements situated in the 5′ regulatory region of genes and subsequently they measure TF binding to those sequences in vivo by, most commonly, quantitative chromatin immunoprecipitation (ChIP). Other experimental methods include using the luciferase reporter gene, electrophoretic mobility shift assays and DNase footprinting. Recently, as the technology of microarray and new generation sequencing develop, high-throughput methods based on ChIP are emerging. These advanced approaches are ChIP-chip and ChIP-seq, which are expensive but extremely promising. Classic ChIP results from electrophoretic analysis of PCR amplification products, and this method can only observe some specific target genes. However, the emergence of ChIP-chip and ChIP-seq technology has made the observation on the whole genome of protein and DNA combination possible (62–66).
Establishment of the TF-miRNA association
Based on bioinformatic prediction, using a miRNA microarray containing different miRNAs and a set of miRNA qPCR assays to validate the microarray results, the correct miRNAs can be identified that are induced by a special TF in vitro, whether upregulated or downregulated with the TF amplification. The TransmiR database can also be used, which is a TF-miRNA regulatory database built by researchers from Peking University (Beijing China) who manually surveyed ~5,000 reports in the literature and identified 243 TF-miRNA regulatory associations, which were supported experimentally from 86 studies (67).
In 2012, Yan et al (68) described a novel method for integrating gene and miRNA expression profiles in cancer using FFLs consisting of TFs, miRNAs and their common target genes. This was the dChip-GemiNi (Gene and miRNA Network-based Integration) method, available at www.canevolve.org/dChip-GemiNi/usergemini.php. It statistically ranks computationally predicted TF-miRNA-target gene FFLs by their explanatory power to account for differential gene and miRNA expression between two biological conditions, such as normal and cancer. Compared to existing approaches, GemiNI also computationally derives information regarding TF-target gene and miRNA-mRNA interactions. The integrated modeling of expression data and FFLs better identifies cancer-related TFs and miRNAs.
All the connections of these TF-miRNA-target gene FFLs were based on experimentally validated interactions referenced in the Ingenuity Knowledge Base, based on which the powerful software Ingenuity Pathway Analysis (IPA) was built.
4. Conclusions
As the concepts of transcription network motifs and TF-miRNA-target gene FFLs emerge, our understanding of the molecular deregulatory mechanism of the cancer step delves further into the unknown field. Researchers could study the complicated deregulation system involving numerous elements at a higher level. Associated TF-miRNA-target gene FFLs can be confirmed and collected to form a regulatory network, similar to a jigsaw puzzle. Key nodes may be used therapeutically as a target for drugs or as the drug itself. More experimental analyses in vivo and more accurate network constituent data analyses may lead to the discovery of crucial principles of cancer, which indicates curability and hope of overcoming malignancy.
However, since miRNAs can also regulate other non-coding RNAs (for example, long non-coding RNAs), which have a role in cancer development and vice versa (69), and TFs are also involved in the regulation of other cancer-associated non-coding RNAs. Each member as a node of a special FFL can be another motifs' indispensable element, which indicates that the overlaying of TF-miRNA-target gene FFLs should put other types of motifs (such as FBL) into consideration. These two elements make the profile of transcription networks more complex and have a larger role than expected, which is currently unknown. New strategies to identify and characterize the entire targets of individual miRNAs and TFs, with an improved high-throughput, and to determine how they function in combination to regulate specific targets, will be required to understand their action on cancer pathology.
Acknowledgements
The present study was supported by grants from the National Natural Science Foundation of China (nos. 81101824 and 81302112) and from the Outstanding Youth Science Foundation of Tongji Hospital (no. YXQN005).
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U. Network motifs: Simple building blocks of complex networks. Science. 2002;298:824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- 3.Widder S, Solé R, Macía J. Evolvability of feed-forward loop architecture biases its abundance in transcription networks. BMC Syst Biol. 2012;6:7. doi: 10.1186/1752-0509-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen-Orr SS, Milo R, Mangan S, Alon U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat Genet. 2002;31:64–68. doi: 10.1038/ng881. [DOI] [PubMed] [Google Scholar]
- 5.Re A, Corá D, Taverna D, Caselle M. Genome-wide survey of microRNA-transcription factor feed-forward regulatory circuits in human. Mol Biosyst. 2009;5:854–867. doi: 10.1039/b900177h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma HW, Kumar B, Ditges U, Gunzer F, Buer J, Zeng AP. An extended transcriptional regulatory network of Escherichia coli and analysis of its hierarchical structure and network motifs. Nucleic Acids Res. 2004;32:6643–6649. doi: 10.1093/nar/gkh1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mangan S, Alon U. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci USA. 2003;100:11980–11985. doi: 10.1073/pnas.2133841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He XX, Guo AY, Xu CR, et al. Bioinformatics analysis identifies miR-221 as a core regulator in hepatocellular carcinoma and its silencing suppresses tumor properties. Oncol Rep. 2014;32:1200–1210. doi: 10.3892/or.2014.3306. [DOI] [PubMed] [Google Scholar]
- 9.Yan JW, Lin JS, He XX. The emerging role of miR-375 in cancer. Int J Cancer. 2014;135:1011–1018. doi: 10.1002/ijc.28563. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Liang C, Easterbrook S, Luo J, Zhang Z. Investigating the functional implications of reinforcing feedback loops in transcriptional regulatory networks. Mol Biosys. 2014;10:3238–3248. doi: 10.1039/C4MB00526K. [DOI] [PubMed] [Google Scholar]
- 11.Fujita Y, Komatsu N, Matsuda M, Aoki K. Fluorescence resonance energy transfer based quantitative analysis of feedforward and feedback loops in epidermal growth factor receptor signaling and the sensitivity to molecular targeting drugs. FEBS J. 2014;281:3177–3192. doi: 10.1111/febs.12852. [DOI] [PubMed] [Google Scholar]
- 12.Hsieh WT, Tzeng KR, Ciou JS, Tsai JJ, Kurubanjerdjit N, Huang CH, et al. Transcription factor and microRNA-regulated network motifs for cancer and signal transduction networks. BMC Syst Biol. 2015;9:S5. doi: 10.1186/1752-0509-9-S1-S5. (Suppl 1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Latchman DS. Transcription factors: An overview. Int J Biochem Cell Biol. 1997;29:1305–1312. doi: 10.1016/S1357-2725(97)00085-X. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell PJ, Tjian R. Transcriptional regulation in mammalian cells by sequence-specific DNA binding proteins. Science. 1989;245:371–378. doi: 10.1126/science.2667136. [DOI] [PubMed] [Google Scholar]
- 15.Ptashne M, Gann A. Transcriptional activation by recruitment. Nature. 1997;386:569–577. doi: 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]
- 16.van Nimwegen E. Scaling laws in the functional content of genomes. Trends Genet. 2003;19:479–484. doi: 10.1016/S0168-9525(03)00203-8. [DOI] [PubMed] [Google Scholar]
- 17.Lane D, Levine A. p53 Research: The past thirty years and the next thirty years. Cold Spring Harb Perspect Biol. 2010;2:a000893. doi: 10.1101/cshperspect.a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hainaut P, Wiman KG. 30 years and a long way into p53 research. Lancet Oncol. 2009;10:913–919. doi: 10.1016/S1470-2045(09)70198-6. [DOI] [PubMed] [Google Scholar]
- 19.Hermeking H. p53 enters the microRNA world. Cancer Cell. 2007;12:414–418. doi: 10.1016/j.ccr.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 20.Brosh R, Shalgi R, Liran A, Landan G, Korotayev K, Nguyen GH, Enerly E, Johnsen H, Buganim Y, Solomon H, et al. p53-Repressed miRNAs are involved with E2F in a feed-forward loop promoting proliferation. Mol Syst Biol. 2008;4:229. doi: 10.1038/msb.2008.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altshuler ML, Severin SE, Glukhov AI. The tumor cell and telomerase. Biochemistry (Mosc) 2003;68:1275–1283. doi: 10.1023/B:BIRY.0000011648.51641.04. [DOI] [PubMed] [Google Scholar]
- 22.Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, Ma W, Tatarek J, Ahn Y, Kelliher MA, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell. 2012;22:209–221. doi: 10.1016/j.ccr.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartel DP. MicroRNAs: Target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lujambio A, Lowe SW. The microcosmos of cancer. Nature. 2012;482:347–355. doi: 10.1038/nature10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calin GA, Croce CM. MicroRNA-cancer connection: The beginning of a new tale. Cancer Res. 2006;66:7390–7394. doi: 10.1158/0008-5472.CAN-06-0800. [DOI] [PubMed] [Google Scholar]
- 26.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 27.Eiring AM, Harb JG, Neviani P, Garton C, Oaks JJ, Spizzo R, Liu S, Schwind S, Santhanam R, Hickey CJ, et al. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell. 2010;140:652–665. doi: 10.1016/j.cell.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moretti F, Thermann R, Hentze MW. Mechanism of translational regulation by miR-2 from sites in the 5′ untranslated region or the open reading frame. RNA. 2010;16:2493–2502. doi: 10.1261/rna.2384610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shalgi R, Brosh R, Oren M, Pilpel Y, Rotter V. Coupling transcriptional and post-transcriptional miRNA regulation in the control of cell fate. Aging (Albany NY) 2009;1:762–770. doi: 10.18632/aging.100085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 31.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9:582–589. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwang CI, Choi J, Zhou Z, Flesken-Nikitin A, Tarakhovsky A, Nikitin AY. MET-dependent cancer invasion may be preprogrammed by early alterations of p53-regulated feedforward loop and triggered by stromal cell-derived HGF. Cell Cycle. 2011;10:3834–3840. doi: 10.4161/cc.10.22.18294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Avraham R, Yarden Y. Regulation of signalling by microRNAs. Biochem Soc Trans. 2012;40:26–30. doi: 10.1042/BST20110623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobayashi K, Sakurai K, Hiramatsu H, Inada K, Shiogama K, Nakamura S, et al. The miR-199a/Brm/EGR1 axis is a determinant of anchorage-independent growth in epithelial tumor cell lines. Sci Rep. 2015;5:8428. doi: 10.1038/srep08428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9:775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song S, Ajani JA. The role of microRNAs in cancers of the upper gastrointestinal tract. Nat Rev Gastroenterol Hepatol. 2013;10:109–118. doi: 10.1038/nrgastro.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang X, Huang H, Li Z, Li Y, Wang X, Gurbuxani S, et al. Blockade of miR-150 maturation by MLL-fusion/MYC/LIN-28 is required for MLL-associated leukemia. Cancer Cell. 2012;22:524–535. doi: 10.1016/j.ccr.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Musilova K, Mraz M. MicroRNAs in B-cell lymphomas: How a complex biology gets more complex. Leukemia. 2015;29:1004–1017. doi: 10.1038/leu.2014.351. [DOI] [PubMed] [Google Scholar]
- 41.Zhao X, Lwin T, Zhang X, Huang A, Wang J, Marquez VE, Chen-Kiang S, Dalton WS, Sotomayor E, Tao J. Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell lymphoma survival and clonogenicity. Leukemia. 2013;27:2341–2350. doi: 10.1038/leu.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 43.Ning Z, Wang A, Liang J, et al. USP22 promotes epithelial-mesenchymal transition via the FAK pathway in pancreatic cancer cells. Oncol Rep. 2014;32:1451–1458. doi: 10.3892/or.2014.3354. [DOI] [PubMed] [Google Scholar]
- 44.Kent OA, Chivukula RR, Mullendore M, Wentzel EA, Feldmann G, Lee KH, Liu S, Leach SD, Maitra A, Mendell JT. Repression of the miR-143/145 cluster by oncogenic Ras initiates a tumor-promoting feed-forward pathway. Genes Dev. 2010;24:2754–2759. doi: 10.1101/gad.1950610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.El Baroudi M, Corà D, Bosia C, Osella M, Caselle M. A curated database of miRNA mediated feed-forward loops involving MYC as master regulator. PLoS One. 2011;6:e14742. doi: 10.1371/journal.pone.0014742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Polioudakis D, Bhinge AA, Killion PJ, Lee BK, Abell NS, Iyer VR. A Myc-microRNA network promotes exit from quiescence by suppressing the interferon response and cell-cycle arrest genes. Nucleic Acids Res. 2013;41:2239–2254. doi: 10.1093/nar/gks1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–1134. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin CJ, Cencic R, Mills JR, Robert F, Pelletier J. c-Myc and eIF4F are components of a feedforward loop that links transcription and translation. Cancer Res. 2008;68:5326–5334. doi: 10.1158/0008-5472.CAN-07-5876. [DOI] [PubMed] [Google Scholar]
- 49.Lin CJ, Malina A, Pelletier J. c-Myc and eIF4F constitute a feedforward loop that regulates cell growth: Implications for anticancer therapy. Cancer Res. 2009;69:7491–7494. doi: 10.1158/0008-5472.CAN-09-0813. [DOI] [PubMed] [Google Scholar]
- 50.Cohen EE, Rosner MR. MicroRNA-regulated feed forward loop network. Cell Cycle. 2009;8:2477–2478. doi: 10.4161/cc.8.16.9271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cohen EE, Zhu H, Lingen MW, Martin LE, Kuo WL, Choi EA, Kocherginsky M, Parker JS, Chung CH, Rosner MR. A feed-forward loop involving protein kinase Calpha and microRNAs regulates tumor cell cycle. Cancer Res. 2009;69:65–74. doi: 10.1158/0008-5472.CAN-08-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rokavec M, Wu W, Luo J-L. IL6-mediated suppression of miR-200c directs constitutive activation of inflammatory signaling circuit driving transformation and tumorigenesis. Mol Cell. 2012;45:777–789. doi: 10.1016/j.molcel.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alexiou P, Maragkakis M, Papadopoulos GL, Reczko M, Hatzigeorgiou AG. Lost in translation: An assessment and perspective for computational microRNA target identification. Bioinformatics. 2009;25:3049–3055. doi: 10.1093/bioinformatics/btp565. [DOI] [PubMed] [Google Scholar]
- 54.Reyes-Herrera PH, Ficarra E. One decade of development and evolution of microRNA target prediction algorithms. Genomics Proteomics Bioinformatics. 2012;10:254–263. doi: 10.1016/j.gpb.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mendes ND, Freitas AT, Sagot MF. Current tools for the identification of miRNA genes and their targets. Nucleic Acids Res. 2009;37:2419–2433. doi: 10.1093/nar/gkp145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nazarov PV, Reinsbach SE, Muller A, Nicot N, Philippidou D, Vallar L, Kreis S. Interplay of microRNAs, transcription factors and target genes: Linking dynamic expression changes to function. Nucleic Acids Res. 2013;41:2817–2831. doi: 10.1093/nar/gks1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dubchak I, Munoz M, Poliakov A, Salomonis N, Minovitsky S, Bodmer R, Zambon AC. Whole-Genome rVISTA: A tool to determine enrichment of transcription factor binding sites in gene promoters from transcriptomic data. Bioinformatics. 2013;29:2059–2061. doi: 10.1093/bioinformatics/btt318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krystkowiak I, Lenart J, Debski K, Kuterba P, Petas M, Kaminska B, Dabrowski M. Nencki Genomics Database - Ensembl funcgen enhanced with intersections, user data and genome-wide TFBS motifs. Database (Oxford) 2013;2013:bat069. doi: 10.1093/database/bat069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang C, Xuan Z, Zhao F, Zhang MQ. TRED: A transcriptional regulatory element database, new entries and other development. Nucleic Acids Res. 2007;35:D137–D140. doi: 10.1093/nar/gkl1041. (Database) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamashita R, Sugano S, Suzuki Y, Nakai K. DBTSS: DataBase of Transcriptional Start Sites progress report in 2012. Nucleic Acids Res. 2012;40:D150–D154. doi: 10.1093/nar/gkr1005. (D1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kolchanov NA, Podkolodnaia OA, Anan'ko EA, Ignat'eva EV, Podkolodnyĭ NL, Merkulov VM, Stepanenko IL, Pozdniakov MA, Belova OE, Grigorovich DA, et al. Regulation of eukaryotic gene transcription: Description in the TRRD database. Mol Biol (Mosk) 2001;35:934–942. (In Russian) [PubMed] [Google Scholar]
- 62.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 63.Perez-Pinera P, Ousterout DG, Brunger JM, Farin AM, Glass KA, Guilak F, Crawford GE, Hartemink AJ, Gersbach CA. Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat Methods. 2013;10:239–242. doi: 10.1038/nmeth.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009;6:283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc. 2007;2:1849–1861. doi: 10.1038/nprot.2007.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115–1129. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang J, Lu M, Qiu C, Cui Q. TransmiR: A transcription factor-microRNA regulation database. Nucleic Acids Res. 2010;38:D119–D122. doi: 10.1093/nar/gkp803. (Database) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yan Z, Shah PK, Amin SB, Samur MK, Huang N, Wang X, Misra V, Ji H, Gabuzda D, Li C. Integrative analysis of gene and miRNA expression profiles with transcription factor-miRNA feed-forward loops identifies regulators in human cancers. Nucleic Acids Res. 2012;40:e135. doi: 10.1093/nar/gks395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–1038. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]