

Abstract
Focal adhesion kinase (FAK) is strongly activated by integrins and growth factors and is essential for embryonic development. We previously showed that the C terminus of FAK is expressed as a separate protein termed FAK-related nonkinase (FRNK) in a smooth muscle cell–selective fashion and that FRNK functions to buffer FAK-dependent signals. We now show that FRNK is also transiently expressed in the neonatal myocardium, with peak levels occurring 5 to 7 days postnatal, just before cell cycle withdrawal. Using novel mouse models, we demonstrate that cardiac-selective expression of FRNK (leading to inhibition of FAK) starting at embryonic day 10.5 leads to a severe ventricular noncompaction defect associated with reduced cardiomyocyte proliferation. Remarkably, postnatal expression of nearly identical levels of FRNK is well tolerated and does not affect viability or anabolic cardiac growth. Nonetheless, FRNK expression in the adult heart does attenuate pathological cardiac hypertrophy following aortic banding, confirming and extending our previous data that this compensatory response is blunted in FAK null hearts. Our mechanistic studies in cultured neonatal cardiomyocytes reveal that FRNK expression induces p38/p27kip-dependent cell cycle withdrawal and attenuates extracellular signal-regulated kinase–dependent hypertrophic growth. These findings indicate that dynamic expression of FRNK in the neonatal heart may function to promote cardiomyocyte quiescence in an environment that is particularly rich in growth factors and growth promoting extracellular matrices.
Keywords: cardiomyocyte, cell cycle, focal adhesion kinase, ventricular noncompaction
Robust, locally restricted myocyte proliferation is necessary for several stages of cardiac morphogenesis, and, although it has long been known that cardiomyocytes undergo terminal differentiation and exit from the cell cycle shortly after birth, the mechanisms that regulate fetal myocyte cell cycle progression and those that promote cell cycle withdrawal are not well understood.
The nonreceptor tyrosine kinase focal adhesion kinase (FAK) is strongly activated by both integrins and growth factors and is a likely candidate to integrate signals from these diverse pathways during growth and development.1 Germline deletion of FAK results in general mesodermal defects and embryonic lethality between embryonic day (E)8.5 to E10, and examination of the malformed hearts from these embryos indicated a putative function for FAK in cardiomyocyte maturation.2 However, because FAK can serve both structural and signaling roles within focal adhesions, it has been difficult to determine whether FAK activity per se is essential for regulating cardiomyocyte growth. Interestingly, our laboratory recently showed that FAK activity can be regulated by the endogenous expression of a separate protein comprising the carboxyl terminus of FAK, termed FRNK (FAK-related nonkinase) that acts as a dominant interfering mutant for FAK.3 FRNK transcription results from the utilization of an alternative start site within the FAK gene, and FRNK expression is independently regulated by a distinct promoter embedded within FAK intronic sequences.4,5 Whereas FAK expression is ubiquitous relatively constantly during development, we previously found that FRNK protein was particularly high in the vasculature when smooth muscle cells (SMCs) are transitioning from a synthetic to contractile phenotype.4
We now present evidence that FRNK is also transiently expressed in the neonatal myocardium before cell cycle withdrawal. By conditionally expressing FRNK in the fetal and neonatal myocardium in the mouse, we show that FAK activity mediates both hyperplastic and pathological hypertrophic growth but not anabolic growth of the heart. Our findings lead us to propose that dynamic expression of FRNK in the neonatal heart may function to dampen cardiomyocyte cell cycle progression (and to facilitate cell cycle withdrawal) in an environment that is particularly rich in growth factors and growth-promoting extracellular matrix (ECM).
Materials and Methods
Transgenic mice were generated and backcrossed to the C57black6 background. Mice were housed in the University of North Carolina Animal Care Facility, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International, and all experimental procedures were approved by the University of North Carolina Animal Care and Use Committee. See the expanded Materials and Methods section in the online data supplement at http://circres.ahajournals.org for a complete description of the reagents, DNA constructs, transgenic mouse generation, and general methods used for these studies.
Statistical Analysis
All quantitative data represent at least 3 separate experiments and are presented as means±SEM. Means were compared by 2-tailed Student's t test. P<0.05 was considered statistically significant, as indicated by an asterisk. All other data including, Western analysis, are representative of at least 3 individual experiments.
Results
Transient Expression of Cardiac FRNK
We previously reported that FRNK is dynamically and selectively expressed in SMCs during neonatal development and following vascular injury in adult rodents.4 However, during the course of our previous examination of FAK signaling in the developing myocardium,6 we made the somewhat surprising observation that FRNK is also transiently expressed in the neonatal myocardium. As shown in Figure 1a, peak FRNK protein levels were observed between postnatal day (P)5 and P7, whereas high levels of FAK were observed in the myocardium from E10.5 to P7. During heart development, 2 forms of FRNK (with apparent molecular masses of 41 and 43 kDa) appear to be present, consistent with our previous findings in SMCs.4 (See the online data supplement for further discussion.) We next isolated cardiomyocytes from neonatal rat hearts and performed Western analysis to ensure that FRNK was selectively expressed in these cells. As shown in Figure 1b, expression of FRNK in purified cardiomyocytes mirrored the levels found in the intact myocardium, with high levels observed in isolated P7 relative to P0 cardiomyocytes. The temporal expression of FRNK in neonatal cardiomyocytes highlights the possibility that precise control of FAK signaling might be necessary for proper growth of the heart.
Figure 1.
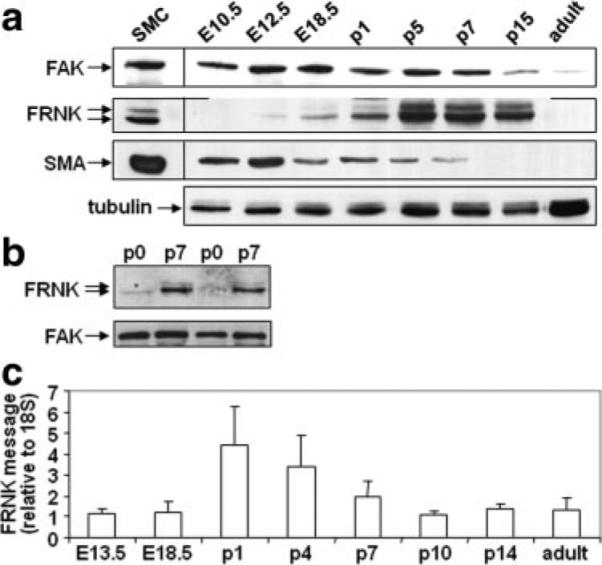
Endogenous cardiac FRNK expression during heart development. a and b, Western blots of cardiac lysate from indicated embryonic (E), postnatal day (p), or adult C57black6 mice (a) or cardiomyocytes isolated from P0 or P7 neonatal rat hearts (b). Western blots are representative of at least 3 separate experiments. c, Quantitative RT-PCR for FRNK message in C57black6 hearts. FRNK levels were normalized to 18S and presented as fold change over message levels detected at E13.5±SEM (n=at least 4 for each time point).
FRNK transcription results from the utilization of an alternative promoter embedded between exons encoding the FAK-kinase domain and the FAK carboxyl-terminal protein-binding region,5 and we showed that FRNK-specific mRNAs are initiated within a unique 5′ noncoding exon in vascular SMCs and smooth muscle–containing tissues.4 However, data also indicate that FRNK-like peptides can be generated in cells by protease degradation of FAK.7,8 To discriminate between these 2 possibilities, we subjected ventricular myocardial RNAs (isolated from the apex of the heart) to quantitative RT-PCR using probes and primers directed against the FRNK noncoding exon. As shown in Figure 1c, FRNK mRNA was apparent in ventricular myocardial tissue, and the levels peaked between P1 and P4, correlating well with FRNK protein patterns detected by immunoblot analysis. This transient expression of FRNK in the early postnatal heart coincides with the window in which cardiomyocytes transition to terminally differentiated cells. Because our previous data indicated that FRNK functions to buffer of FAK-dependent growth signals in SMCs,9 we reasoned that FRNK might function to decrease DNA synthesis and possibly promote cell cycle withdrawal in cardiomyocytes.
Generation of FRNKloxP Mice
To carefully examine the function of FRNK in the myocardium in vivo, we generated a Cre-dependent transgenic mouse model in which FRNK can be overexpressed in a time-and tissue-restricted fashion (Figure 2A; see the expanded Results section in the online data supplement for description). Western blot analysis revealed moderate levels of green fluorescent protein (GFP) expression in all tissues examined from 2 independent transgenic lines of these so-named FRNKloxp mice, whereas myc-FRNK was undetectable in these lysates (Figure I in the online data supplement). Immunohistochemistry for GFP revealed uniform expression throughout the heart (Figure 2d) and several other tissues as expected (data not shown).
Figure 2.

Generation of transgenic mice with temporal and myocardial-restricted FRNK expression. a, Schematic of construct used to generate FRNKLoxP transgenic mice. b and c, Western blot analysis of myc-FRNK expression in tissues from FRNKMlc2v double transgenic mice. d, Immunohistochemical analysis of GFP expression levels in left ventricle from 8-week-old FRNKLoxP or FRNKMlc2v mice. e, Western analysis for active, phospho-Y397 FAK, or total FAK in ventricular lysate from 1-week FRNKLoxP or FRNKMlc2v mice. f, Viable FRNKNkx2.5 or FRNKMlc2v offspring at indicated time points. Dashed line denotes expected Mendelian ratio of 25%. See Online Table I for number of animals per time point. g, Western blot analysis of myc-FRNK expression in hearts of FRNKMlc2v and FRNKNkx2.5 double transgenic mice at the indicated ages. Western blots are representative of at least 3 separate experiments.
Targeted Expression of FRNK in the Myocardium
By crossing the FRNKloxP mice to well-established Nkx2.5Cre and MLC2vCre lines, we were able to explore the functional consequences of FRNK expression early and late during cardiac development.10,11 FRNKloxP/MLC2vCre double transgenic mice (hereafter referred to as FRNKMlc2v) were born in the expected Mendelian ratios, and Western blot analysis revealed cardiac-restricted expression of myc-FRNK from P1 onwards (Figure 2b and 2c and Online Figure II). Moreover, only a few GFP-positive cells were detected by immunohistochemistry in the ventricles of the FRNKMlc2v hearts, indicating nearly complete transgene recombination (Figure 2d). Importantly, hearts from FRNKMlc2v mice exhibited decreased FAK activity relative to hearts from control FRNKloxP mice, as determined by Western blotting with an anti–pFAK Y-397 antibody (Figure 2e). Nonetheless, FRNKMlc2v mice did not exhibit any overt abnormalities and had normal life expectancies (Figure 2f and Online Table I).
In stark contrast, crossing FRNKloxP mice with the Nkx2.5Cre line yielded no viable double transgenics, and subsequent studies revealed that embryonic lethality in FRNKNkx2.5 mice occurred between E14.5 and E16.5 (Figure 2f and Online Table I). Western analysis revealed detectable levels of myc-FRNK as early as E10.5 in FRNKNkx2.5 hearts (data not shown), with maximal levels observed by E13.5 (Figure 2g). Importantly, although the timing of myc-FRNK expression was different between the 2 models, the maximal levels of myc-FRNK protein expressed in the hearts of FRNKNkx2.5 and the FRNKMlc2v mice were remarkably similar and modestly (3-fold) higher than levels of endogenous FRNK expressed in control hearts at P7 (see Online Figure II for quantification). Collectively, these data indicate that embryonic cardiac development is exquisitely sensitive to FRNK expression (and a reduction in FAK activity), whereas further postnatal development is FAK-independent.
Histological and Morphological Analysis of FRNKNkx2.5 Mice
Gross morphology of FRNKNkx2.5 embryos at E14.5 revealed extreme hydrops fetalis and blood pooling, likely attributable to cardiac failure in these embryos (Figure 3a). Histological examination of coronal sections through the FRNKNkx2.5 hearts revealed similar heart size and ventricular wall thickness at E10.5 (the onset of myc-FRNK expression). However, as development progressed from E10.5 to E14.5, little growth was observed in the FRNKNkx2.5 hearts in comparison to genetic controls, which underwent dramatic expansion of the ventricular walls and growth of the interventricular septum (Figure 3b). Whereas trabeculae were reasonably well formed in the FRNKNkx2.5 hearts, compaction of the ventricular chambers was significantly impaired. Indeed, quantification of E14.5 coronal heart sections revealed a 48% reduction in cell density in the FRNKNkx2.5 hearts compared to age-matched controls (Figure 3c). There were no overall gross or histological differences observed in other tissues derived from nkx2.5-expressing cells compared to genetic controls at E14 (Online Figure IV and Online Results), indicating that lethality in FRNKNkx2.5 embryos was likely attributable to heart failure at approximately E15.5.
Figure 3.
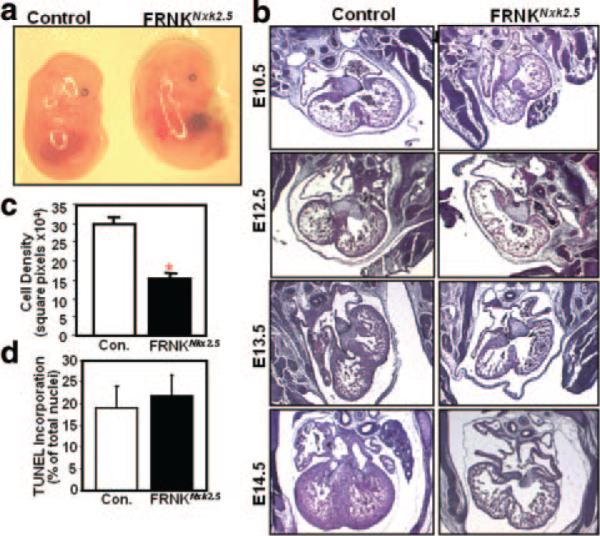
Histological and morphological analysis of FRNKNkx2.5 mice. a, Gross morphology of genetic control and FRNKNkx2.5 embryos at E14.5. b, Hematoxylin/eosin-stained sections of hearts from the indicated embryonic day. c, Cellular density was quantified in serial sections from E14.5 genetic control and FRNKNkx2.5 hearts. d, Ventricle sections from E13.5 genetic control and FRNKNkx2.5 mice were costained with FragEL and cardiac troponin T to identify apoptotic cardiomyocytes. Graphical data are expressed as means±SEM (n=6 for each condition; *P<0.01).
myc-FRNK Expression Attenuates Myocyte Proliferation
Because no significant differences were observed in the cardiac differentiation program or the extent of apoptosis between E13.5 FRNKNkx2.5 and genetic control hearts (Figure 3d and Online Results), we next sought to determine whether FRNK expression led to a proliferation defect. Indeed the number of 5-bromodeoxyuridine (BrdUrd)-labeled cells in the ventricles of E13.5 FRNKNkx2.5 hearts was significantly reduced when compared to genetic control hearts (especially within the compact zone; Figure 4). We did not observe significant differences in BrdUrd incorporation in any other tissues examined (data not shown). We next turned to cultured primary cardiomyocytes to determine whether FRNK expression could directly impair proliferation of neonatal cardiomyocytes. To this end, we infected P0 cardiomyocytes with GFP or GFP-FRNK adenovirus and examined the rate of proliferation of these cells in serum-containing media. Under these conditions the rate of BrdUrd incorporation in GFP-infected cells was ≈ 18%, whereas only 4% of the FRNK-infected cells were BrdUrd-positive (Figure 5a and 5f). In addition to this striking reduction in BrdUrd incorporation, the FRNK-infected cells exhibit a blunted hypertrophic response to serum, as assessed by reduced cell size and sarcomeric actin organization (as assessed by cardiac troponin T staining; see Online Figure VIII, a, for higher-power images), consistent with our previous findings.12 Collectively, these data indicate that FRNK expression negatively regulates embryonic and neonatal cardiomyocyte growth but does not affect cell survival during postnatal heart development.
Figure 4.

FRNKNkx2.5 hearts exhibit a noncompaction defect associated with reduced myocyte proliferation. a, High power hematoxylin/eosin-stained images of left ventricular compact zone from E14.5 FRNKloxP and FRNKNkx2.5 embryos. b, BrdUrd incorporation in E13.5 FRNKloxP and FRNKNkx2.5 ventricles. c, Quantification of BrdUrd-positive nuclei in heart (means±SEM; n≥6; *P<0.01).
Figure 5.

FRNK expression attenuates DNA synthesis and promotes cell cycle withdrawal in a p38-dependent fashion. a, BrdUrd incorporation in isolated cardiomyocytes (P0) infected with GFP- or GFP-FRNK adenovirus (10 multiplicities of infection). Cells were maintained in serum-containing medium and treated with vehicle or SB203580 (10 μmol/L) as indicated. Costaining with anti–cardiac troponin T (cTnT) was performed to identify cardiomyocytes. b, Western blot analysis in cardiac lysates from E13.5 FRNKloxP and FRNKNkx2.5 embryos. c and d, Rat neonatal cardiomyocytes (P0) plated on fibronectin were infected with GFP- or GFP-FRNK adenovirus (10 multiplicities of infection), replaced with serum-free media, and treated with FGF-2 (c) or maintained in serum-containing conditions (d) for the indicated times before immunoblotting. See Online Figure VI (a through c) for densitometric quantification of 3 separate experiments. e, Isolated cardiomyocytes, treated as described for a, were processed by immunocytochemistry for p27kip (myocytes are identified by striated actin visualized by phalloidin). f and g, Quantification of BrdUrd- and p27kip-positive cardiomyocytes (means±SEM; n=3; minimum of 300 cells/condition). Additional representative images are provided in Online Figure VIII.
myc-FRNK Expression Regulates Mitogen-Activated Protein Kinase Signaling
Previous studies have revealed that the mitogen-activated protein kinases (MAPKs) extracellular signal-regulated kinase (ERK) and p38 play positive and negative roles, respectively, in regulating cardiomyocyte cell cycle progression.13–17 Interestingly, we found that ERK activity (Figure 5b) and expression of the ERK-dependent immediate early gene c-fos (Online Figure V, b) were each significantly reduced in E13.5 FRNKNkx2.5 hearts compared to littermate controls at this time point (the peak of myocyte proliferation). In contrast, p38 activity was significantly elevated in FRNKNkx2.5 hearts (Figure 5b). To confirm that regulation of these MAPKs was attributable to a primary change in FRNK expression, we examined activation of these pathways in cultured cardiomyocytes. Indeed, ectopic expression of FRNK significantly enhanced p38 activity in a time-dependent fashion but attenuated growth factor (fibroblast growth factor [FGF]-2 and insulin-like growth factor [IGF]-1)–induced ERK activity in these cells (Figures 5c and 6b; see Online Figure VI for quantification).
Figure 6.

Anabolic cardiac growth and AKT signaling are not affected by FRNK expression. a, Low- and high-power images of cross-sections of 8-week-old FRNKloxp and FRNKMlc2v hearts stained with trichrome (blue corresponds to collagen) (top and middle) or with an anti–cardiac troponin T antibody (bottom). b and c, Rat neonatal cardiomyocytes were infected as described above or plated on fibronectin (FN) or collagen (COL) (10 μg/mL) and treated with IGF-1 or FGF-2 for the indicated times. Lysates were immunoblotted for phospho- and total ERK and AKT. Western data are representative of at least 3 separate experiments. See Online Figure VI for quantification.
To determine the functional importance of these pathways with respect to FRNK-dependent inhibition of myocyte proliferation, we explored whether inhibition of p38 or activation of ERK would restore myocyte proliferation in FRNK-expressing cardiomyocytes. As shown in Figure 5a and 5f, treatment with the p38 inhibitor SB203580 (10 μmol/L) enhanced BrdUrd incorporation in GFP-expressing cells (by 2-fold) and even more markedly enhanced BrdUrd incorporation in FRNK-expressing cells by 5-fold (to 25%; surpassing the level observed in GFP control cells). With respect to ERK activation, we found that treatment of GFP-expressing cardiomyocytes with the MEK1 inhibitor UO927 reduced BrdUrd incorporation by ≈50%, indicating that ERK activation is necessary for maximal serum-stimulated BrdUrd incorporation. However, adenoviral-mediated expression of constitutively active MEK1, which led to sustained elevated ERK activity (see Online Figure VIII, b), induced only a modest increase in BrdUrd incorporation in both GFP- and FRNK-infected cells, and no synergy was found on treatment with constitutively active MEK and the p38 inhibitor (Figure 5f; see Online Figure VIII, a, for representative images). Importantly, FRNK was expressed in comparable levels under all treatment groups, as assessed by Western analysis (Online Figure VIII, b).
We next evaluated the expression levels of the cell cycle inhibitor p27kip (a key p38-dependent cell cycle regulator) in GFP- and FRNK-expressing cardiomyocytes. As shown in Figure 5e, expression of p27kip was markedly upregulated in FRNK-expressing cells in comparison to the GFP-expressing controls. Notably, treatment of the FRNK-expressing cells with SB203580 restored expression of p27 kip to control levels. p27kip levels were also significantly elevated in E13.5 FRNKNkx2.5 hearts in comparison to genetic controls (Figure 5b). Collectively, these data indicate that FRNK expression attenuates cardiomyocyte cell cycle progression and induces cell cycle withdrawal, in large part, by a mechanism that involves enhanced activation of p38, an important negative regulator of cardiomyocyte cytokinesis.16,17 Interestingly, we also observed upregulation of p38 activity in FAK-null hearts (see Online Results and Online Figure VII), indicating that FRNK expression likely dampens cardiomyocyte proliferation by relieving FAK-dependent repression of p38.
FRNKMlc2v Mice Exhibit Normal Anabolic Growth but a Diminished Response to Pressure Overload
Because FAK activity was clearly necessary for growth of the fetal heart, we carefully assessed the affect of FRNK expression on postnatal myocardial growth. After extensive measurements of heart size, heart weight, cardiomyocyte cross-sectional area, myofibrillar organization, collagen deposition, and physiological function (by echocardiography), we concluded that there were no significant differences in hearts from FRNKLoxP and FRNKMlc2v mice up to 8 months of age (Figure 6a and Online Table II). These data reveal that FRNK expression is well tolerated in terminally differentiated myocytes and imply that FAK activity is not necessary for anabolic growth of the heart.
Postnatal anabolic (physiological) hypertrophic growth is known to be controlled primarily by IGF-1– and growth hormone–dependent stimulation of the phosphatidylinositol 3-kinase/AKT/p70S6K protein synthesis pathway.18–23 Interestingly, we found that whereas IGF-1–stimulated (and FGF-stimulated) ERK activation was attenuated by FRNK expression in neonatal cardiomyocytes, IGF-1–stimulated activation of AKT was not (Figure 6b). Moreover, IGF-1 stimulation led to a comparable induction of AKT in cardiomyocytes plated on either fibronectin (which promotes β1 integrin–dependent FAK activation) or on type I collagen (which does not promote FAK activation; Figure 6c), whereas IGF-stimulated (and FGF-stimulated) ERK activity required fibronectin (Figure 6d). Collectively, these data reveal that FRNK/FAK signaling modulates a subset of myocyte growth responses, effecting signals that regulate cell cycle progression but not anabolic growth.
Although FAK protein levels and basal activity are relatively low in the adult myocardium, several reports have indicated that FAK activity is markedly induced following mechanical overload, and we recently published a report showing that myocyte-restricted FAK deletion attenuates compensatory hypertrophic remodeling following pressure overload.24 However, questions remain as to whether FAK plays a structural role or signaling role in this setting. Thus, we next strove to determine whether specific inhibition of FAK activity in vivo in the FRNKMlc2v line was sufficient to blunt pressure overload–induced hypertrophic remodeling. To this end, we subjected FRNKMlc2v and wild-type littermate control mice to a minimally invasive aortic banding procedure that provides an approximate 50% reduction in the lumen of the ascending aorta.25 Following chronic aortic constriction, we examined the mice for a variety of hypertrophic changes. As shown in Figure 7a, FRNKMlc2v mice had significantly lower heart weight versus body weight in comparison to genetic controls following banding. In addition, myocyte cross-sectional area was significantly lower in postbanded FRNKMlc2v hearts compared to postbanded genetic controls (Figure 7b). Echocardiographic analysis of genetic control mice also revealed significant differences in the extent of remodeling following banding. Notably, the pressure overload–induced increase in thickness of the left ventricular wall and intraventricular septum were both significantly reduced in the FRNKMlc2v hearts when compared to the genetic controls (Figure 7c). Hearts from FRNKMlc2v and control mice were also analyzed for hypertrophic marker gene expression by quantitative RT-PCR before and after banding. Importantly, the fold increase in the canonical hypertrophic marker gene, atrial natriuretic factor was significantly attenuated in the FRNKMlc2v hearts in comparison to the genetic controls (control, 66±10.5-fold increase in banded over sham; FRNK, 3.8±1.2-fold increase in banded over sham; P<0.05; n=6 per genotype).
Figure 7.

FRNKMlc2v mice exhibit attenuated pressure overload–induced hypertrophy. a, Left ventricular (LVW) and body (BW) weight measurements were taken from control and FRNKMlc2v animals at baseline and following 14 days of transaortic constriction (TAC). Data are presented as the means±SEM (n=4/time point per genotype; *P<0.05). b, Cross-sectional myocyte area in sham and banded control and FRNKMlc2v hearts (n=at least 130 cells from 4 mice/condition; *P<0.05 compared to control banded). c, M-mode echocardiographic measurements of the left ventricle from baseline and 14-day banded control and FRNKMlc2v mice during diastole (d) and systole (s). Data are expressed as percentage of change from pre- to post-TAC. #P<0.05 when compared with percentage of change in control mice. d, Control (C) and FRNKMlc2v (F) mice were injected IP with vehicle or adrenergic cocktail for 7 minutes containing phenylephrine and isoproterenol (30 mg/kg each), and ventricular lysate was processed for Western blot analysis.
As expected, adrenergic agonist-induced ERK activation was also dampened in FRNKMlc2v hearts in comparison to those from genetic controls (Figure 7d). Interestingly, we noted in our proliferation studies that ectopic expression of Mek1 (but not inhibition of p38) reversed the hypertrophic growth defect in FRNK-expressing cells (see Figure 5a and Online Figure VIII, a). Subsequent studies revealed that expression of constitutively active Mek1 induced a 2-fold increase in cell area in FRNK-expressing cardiomyocytes and enhanced sarcomeric actin organization in these cells (a hallmark of pathological hypertrophic growth; Online Figure VIII, c and d). Collectively, these data indicate that promotion of p38 activity and attenuation of ERK activity are likely the major mechanisms by which FRNK expression represses proliferation and pathological hypertrophic growth, respectively.
Discussion
It is clear that coordinated signaling through integrins and growth factors plays an important role in regulating both cardiac morphogenesis and the progression of cardiac disease, but how these processes are fine-tuned during the different phases of heart growth and development is unknown. Herein, we show that FRNK, the endogenous inhibitor of FAK, is transiently expressed in heart, with peak levels occurring at P5 to P7, when cardiomyocytes are transitioning to terminally differentiated cells. Using novel mouse models, we demonstrated that cardiac-selective expression of FRNK (leading to inhibition of FAK) starting at E10.5 led to a severe ventricular noncompaction defect associated with impaired cardiomyocyte proliferation. Remarkably, postnatal expression of nearly identical levels of FRNK was well tolerated and did not affect viability or anabolic cardiac growth but did attenuate pressure overload–induced hypertrophic remodeling. Collectively, our mechanistic studies indicate that FAK regulates p38-dependent myocyte proliferation in the developing myocardium and ERK-dependent promotion of pathological hypertrophy following biomechanical stress but that FAK activity is dispensable for AKT-dependent physiological, anabolic growth of the heart.
Our recent demonstration that FAK activity is enhanced in tissues and cells derived from FRNK−/− mice strongly supports the hypothesis that the dynamic regulation of FRNK expression can impart specific spatial and temporal control of FAK activity in vivo.26 Additionally, a very recent publication revealed that myocyte-specific deletion of FAK in mid-gestation (using the Mlc2aCre line) phenocopies the defects we observed in the FRNKNkx2.5 embryos including embryonic lethality, global edema, thin ventricle wall, and reduced rates of myocyte proliferation, although the underlying signaling mechanisms were not explored.27 Thus, mice with cardiac-restricted expression of FRNK exhibit remarkably similar developmental and hypertrophic growth defects as mice with similar temporal deletion of FAK. Collectively, these studies strongly support the notion that the effects of FRNK are mediated by inhibition of FAK-dependent signals and that FAK activity is necessary to promote myocyte proliferation in a cell-autonomous fashion.
Proliferation of cardiomyocytes within the compact zone at mid-gestation is necessary to support the increasing hemodynamic load of the embryo, and a large body of evidence indicates that FGF-, IGF-, and transforming growth factor/bone morphogenetic protein–induced signals appear to play a critical role in this process because genetic deletion of these ligands or their receptors lead to compaction defects.15 In terms of signaling, many of these receptors are linked to the MAPK signaling pathways terminating in activation of ERK, c-Jun N-terminal kinase, and p38. Several recent studies indicate that p38 may function at a G2/M checkpoint to block cardiomyocyte cell cycle progression,16,17 including the findings that (1) p38α activity is inversely correlated with cardiac growth during development; (2) activation of p38 in vivo (by forced expression of MKK3) reduced fetal cardiomyocyte proliferation; and (3) cardiac-restricted deletion of p38α significantly enhanced myocyte proliferation. Our studies reveal that expression of FRNK limits cardiomyocyte proliferation and induces cell cycle arrest in a p38-dependent fashion. Indeed, we found that expression of FRNK (or inactivation of FAK) promotes p38 activity and regulates expression of the p38-dependent cell cycle modifier, p27kip and that pharmacological inhibition of p38 restores DNA synthesis in FRNK-expressing cells. Thus, it is tempting to speculate that basal FAK activity may act to repress p38, a necessary function for tyrosine kinase–dependent growth, in addition to growth induced by equally important parallel pathways such as the bone morpho-genetic protein/Smad4 signaling cascade.28,29
Our studies reveal that the timing of FRNK expression in the postnatal heart corresponds to the window during which cardiomyocytes exit the cell cycle. (See the online data supplement for further discussion of FRNK expression in the myocardium.) Because misexpression of FRNK can attenuate proliferation of embryonic cardiomyocytes, we speculate that transient expression of endogenous FRNK in the perinatal heart is necessary to achieve quiescence in this growth-promoting environment. Several studies indicate that the cardiomyocyte growth state in the developing heart correlates with regulated shifts in expression of ECM and integrin receptors and the ability of these matrices to support myocyte growth in vitro.30,31 Thus, we speculate that FRNK may not be necessary in the adult myocardium, because although growth factors such as FGF-2 and IGF-1 continue to be highly expressed, the ECM composition in the adult heart (mainly type I collagen) is not growth-permissive. Interestingly, periostin (a component of the ECM that activates β1, 3, and 5 integrins) is also highly expressed in the developing myocardium and is re-expressed in the diseased adult heart and a recent high profile study demonstrated that recombinant periostin was sufficient to induce cell-cycle re-entry in adult cardiomyocytes both in vitro and in vivo.32 These data underscore the importance of integrin signaling in the regulation of cardiomyocyte cell cycle and support the notion that FAK activity may play a critical role in this response.
Our new studies also reveal that FAK does not merely have a cyto-architectural role in the adult myocardium, but has a key enzymatic function to transmit pressure-induced compensatory progrowth signals. Interestingly, most known genetic mutations leading to familial hypertrophic cardiomyopathy (HCM, which resembles concentric compensatory hypertrophy) are attributable to point mutations in sarcomeric proteins,33 and because these structural molecules are involved in the generation of force that is ultimately transmitted to the cell membrane and ECM, it will be interesting to determine whether FAK can also act to sense the altered load imparted by these mutant muscle fibers. We speculate that FAK might be aberrantly activated in the hearts of mice carrying hypertrophic cardiomyopathy mutations and that FRNK expression might attenuate the extent of hypertrophic remodeling observed. (See the online data supplement for further discussion regarding the necessity of FAK activity for pathological but not anabolic growth of the heart.)
In summary, we show that FRNK expression increases in the myocardium at birth and that FRNK has the capacity to dampen cardiomyocyte proliferation by enhancing activity of the cell cycle inhibitor p38. FRNK−/− mice were recently generated, and these mice were reportedly born in the expected Mendelian frequency and showed no gross phenotype.34 However, our studies indicate that a specific evaluation of cardiac growth in these mice is warranted. Additionally, because we provide evidence that FAK provides cues to neutralize signals responsible for cell cycle restriction, it will also be of interest to determine to whether targeted FAK activation can induce post-mitotic cardiomyocyte cell cycle reentry.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported, in part, by National Heart, Lung, and Blood Institute grants HL-081844 (to J.M.T.), HL-071054 (to J.M.T.), and HL070953 (to C.P.M.) and American Heart Association Grants 0355776U (to J.M.T.) and 0555476U (to C.P.M.).
Footnotes
Disclosures
None.
References
- 1.Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 2.Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 3.Richardson A, Parsons T. A mechanism for regulation of the adhesion-associated proteintyrosine kinase pp125FAK. Nature. 1996;380:538–540. doi: 10.1038/380538a0. [DOI] [PubMed] [Google Scholar]
- 4.Taylor JM, Mack CP, Nolan K, Regan CP, Owens GK, Parsons JT. Selective expression of an endogenous inhibitor of FAK regulates proliferation and migration of vascular smooth muscle cells. Mol Cell Biol. 2001;21:1565–1572. doi: 10.1128/MCB.21.5.1565-1572.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nolan K, Lacoste J, Parsons JT. Regulated expression of focal adhesion kinase-related nonkinase, the autonomously expressed C-terminal domain of focal adhesion kinase. Mol Cell Biol. 1999;19:6120–6129. doi: 10.1128/mcb.19.9.6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hakim ZS, Dimichele LA, Doherty JT, Homeister JW, Beggs HE, Reichardt LF, Schwartz RJ, Brackhan J, Smithies O, Mack CP, Taylor JM. Conditional deletion of focal adhesion kinase leads to defects in ventricular septation and outflow tract alignment. Mol Cell Biol. 2007;27:5352–5364. doi: 10.1128/MCB.00068-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carragher NO, Levkau B, Ross R, Raines EW. Degraded collagen fragments promote rapid disassembly of smooth muscle focal adhesions that correlates with cleavage of pp125(FAK), paxillin, and talin. J Cell Biol. 1999;147:619–630. doi: 10.1083/jcb.147.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen LP, Fahrni JA, Troie S, Guan JL, Orth K, Rosen GD. Cleavage of focal adhesion kinase by caspases during apoptosis. J Biol Chem. 1997;272:26056–26061. doi: 10.1074/jbc.272.41.26056. [DOI] [PubMed] [Google Scholar]
- 9.Sundberg LJ, Galante LM, Bill HM, Mack CP, Taylor JM. An endogenous inhibitor of focal adhesion kinase blocks Rac1/JNK but not Ras/ERK-dependent signaling in vascular smooth muscle cells. J Biol Chem. 2003;278:29783–29791. doi: 10.1074/jbc.M303771200. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Kubalak SW, Chien KR. Ventricular muscle-restricted targeting of the RXRalpha gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development. 1998;125:1943–1949. doi: 10.1242/dev.125.10.1943. [DOI] [PubMed] [Google Scholar]
- 11.Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis. 2001;31:176–180. doi: 10.1002/gene.10022. [DOI] [PubMed] [Google Scholar]
- 12.Taylor JM, Rovin JD, Parsons JT. A role for focal adhesion kinase in phenylephrine-induced hypertrophy of rat ventricular cardiomyocytes. J Biol Chem. 2000;275:19250–19257. doi: 10.1074/jbc.M909099199. [DOI] [PubMed] [Google Scholar]
- 13.Moorman AF, Christoffels VM. Cardiac chamber formation: development, genes, and evolution. Physiol Rev. 2003;83:1223–1267. doi: 10.1152/physrev.00006.2003. [DOI] [PubMed] [Google Scholar]
- 14.Lavine KJ, Yu K, White AC, Zhang X, Smith C, Partanen J, Ornitz DM. Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev Cell. 2005;8:85–95. doi: 10.1016/j.devcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev. 2007;87:521–544. doi: 10.1152/physrev.00032.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, Jiang H, Wang Y, Keating MT. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–1187. doi: 10.1101/gad.1306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Engel FB, Hsieh PC, Lee RT, Keating MT. FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction. Proc Natl Acad Sci U S A. 2006;103:15546–15551. doi: 10.1073/pnas.0607382103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shioi T, Kang PM, Douglas PS, Hampe J, Yballe CM, Lawitts J, Cantley LC, Izumo S. The conserved phosphoinositide 3-kinase pathway determines heart size in mice. EMBO J. 2000;19:2537–2548. doi: 10.1093/emboj/19.11.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, Cantley LC, Izumo S. Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799–2809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Kang PM, Izumo S. Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci U S A. 2003;100:12355–12360. doi: 10.1073/pnas.1934654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McMullen JR, Shioi T, Huang WY, Zhang L, Tarnavski O, Bisping E, Schinke M, Kong S, Sherwood MC, Brown J, Riggi L, Kang PM, Izumo S. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110alpha) pathway. J Biol Chem. 2004;279:4782–4793. doi: 10.1074/jbc.M310405200. [DOI] [PubMed] [Google Scholar]
- 22.Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie-Sasaki J, Sah R, Cheng HY, Rybin VO, Lembo G, Fratta L, Oliveira-dos-Santos AJ, Benovic JL, Kahn CR, Izumo S, Steinberg SF, Wymann MP, Backx PH, Penninger JM. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell. 2002;110:737–749. doi: 10.1016/s0092-8674(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 23.Condorelli G, Drusco A, Stassi G, Bellacosa A, Roncarati R, Iaccarino G, Russo MA, Gu Y, Dalton N, Chung C, Latronico MV, Napoli C, Sadoshima J, Croce CM, Ross J., Jr. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:12333–12338. doi: 10.1073/pnas.172376399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiMichele LA, Doherty JT, Rojas M, Beggs HE, Reichardt LF, Mack CP, Taylor JM. Myocyte-restricted focal adhesion kinase deletion attenuates pressure overload-induced hypertrophy. Circ Res. 2006;99:636–645. doi: 10.1161/01.RES.0000240498.44752.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu P, Zhang D, Swenson L, Chakrabarti G, Abel ED, Litwin SE. Minimally invasive aortic banding in mice: effects of altered cardiomyocyte insulin signaling during pressure overload. Am J Physiol Heart Circ Physiol. 2003;285:H1261–H1269. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- 26.Sayers RL, Sundberg-Smith LJ, Rojas M, Hayasaka H, Parsons JT, Mack CP, Taylor JM. FRNK expression promotes smooth muscle cell maturation during vascular development and after vascular injury. Arterioscler Thromb Vasc Biol. 2008;28:2115–2122. doi: 10.1161/ATVBAHA.108.175455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng X, Wu X, Druso JE, Wei H, Park AY, Kraus MS, Alcaraz A, Chen J, Chien S, Cerione RA, Guan JL. Cardiac developmental defects and eccentric right ventricular hypertrophy in cardiomyocyte focal adhesion kinase (FAK) conditional knockout mice. Proc Natl Acad Sci U S A. 2008;105:6638–6643. doi: 10.1073/pnas.0802319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qi X, Yang G, Yang L, Lan Y, Weng T, Wang J, Wu Z, Xu J, Gao X, Yang X. Essential role of Smad4 in maintaining cardiomyocyte proliferation during murine embryonic heart development. Dev Biol. 2007;311:136–146. doi: 10.1016/j.ydbio.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 29.Song L, Yan W, Chen X, Deng CX, Wang Q, Jiao K. Myocardial smad4 is essential for cardiogenesis in mouse embryos. Circ Res. 2007;101:277–285. doi: 10.1161/CIRCRESAHA.107.155630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brancaccio M, Hirsch E, Notte A, Selvetella G, Lembo G, Tarone G. Integrin signalling: the tug-of-war in heart hypertrophy. Cardiovasc Res. 2006;70:422–433. doi: 10.1016/j.cardiores.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 31.Hescheler J, Fleischmann BK. Integrins and cell structure: powerful determinants of heart development and heart function. Cardiovasc Res. 2000;47:645–647. doi: 10.1016/s0008-6363(00)00164-4. [DOI] [PubMed] [Google Scholar]
- 32.Kuhn B, del Monte F, Hajjar RJ, Chang YS, Lebeche D, Arab S, Keating MT. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat Med. 2007;13:962–969. doi: 10.1038/nm1619. [DOI] [PubMed] [Google Scholar]
- 33.Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Annu Rev Genomics Hum Genet. 2005;6:185–216. doi: 10.1146/annurev.genom.6.080604.162132. [DOI] [PubMed] [Google Scholar]
- 34.Hayasaka H, Martin KH, Hershey ED, Parsons JT. Disruption of FRNK expression by gene targeting of the intronic promoter within the focal adhesion kinase gene. J Cell Biochem. 2007;102:947–954. doi: 10.1002/jcb.21329. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.