

Abstract
Further chemical optimization of the halopemide-derived family of dual PLD1/2 inhibitors afforded ML395 (VU0468809), a potent, >80-fold PLD2 selective allosteric inhibitor (cellular PLD1, IC50 >30,000 nM, cellular PLD2, IC50 = 360 nM). Moreover, ML395 possesses an attractive in vitro DMPK profile, improved physiochemical properties, ancillary pharmacology (Eurofins Panel) cleaner than any other reported PLD inhibitor, and has been found to possess interesting activity as an antiviral agent in cellular assays against a range of influenza strains (H1, H3, H5 and H7).
Keywords: phospholipase D, PLD2, inhibitor, antiviral, lipid
Phospholipase D (PLD) is a phospholipase that catalyzes the production of phosphatidic acid, an important lipid second messenger involved in a myriad of critical signaling and metabolic pathways.[1–3] In mammals there are two isoforms of PLD, coined PLD1 and PLD2, which are differentially regulated and perform distinct physiological roles. Data from biochemical and genetic studies have implicated aberrant PLD function and/or overexpression in cancer, viral infection and central nervious systems (CNS) disorders; however, due to a lack of highly isoform selective small molecule PLD inhibitors, the therapeutic potential of modulating PLD function has remained elusive.[1–4]
In 2007, halopemide 1, an atypical antipsychotic agent, was reported to inhibit PLD.[5] This report triggered a resurgence of interest in the therapeutic relevance of PLD inhibition since (i) no potent chemical inhibitors of PLD were previously known and (ii) 1 had been involved in clinical trials where PLD1 and PLD2 were inhibited without adverse events.[6] Therefore, our lab quickly initiated a diversity-oriented synthesis campaign around 1 (Figure 1), and developed the first direct, isoform selective PLD inhibitors represented by 2 (1,700-fold PLD1 selective) and 3 (75-fold PLD2 selective).[2] While these first generation tools were important in defining the individual contributions of PLD1 and PLD2 in various systems and diseases, neither 2 nor 3 possessed ideal physiochemical or DMPK properties for rigorous in vivo evaluation.[7–9] Subsequent optimization efforts provided the PLD2 selective probe ML298 (4) and the potent dual PLD1/2 inhibitor ML299 (5), with significantly improved ancillary pharmacology; however, the DMPK profiles and physiochemical properties were still lacking.[10] Therefore, we launched an optimization effort aimed at developing a third generation PLD2 selective inhibitor devoid of cytotoxicity, that displays improved DMPK and ancillary pharmacology profiles coupled with good CNS exposure.
Figure 1.
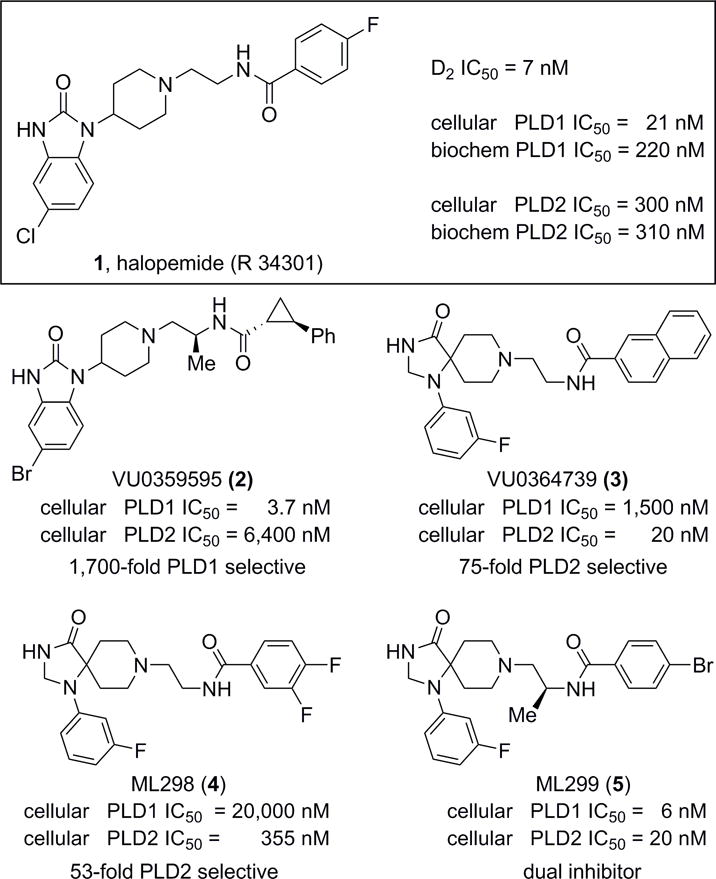
Structures of recently reported PLD inhibitors (1–5). Halopemide (1), an atypical antipsychotic agent was shown to be a dual PLD1/2 inhibitor, which spawned optimization campaigns that afforded isoform selective (either PLD1 (2) or PLD2 (3 and 4)) inhibitors, as well as a highly potent dual PLD1/2 inhibitor (5).
Thus far, all triazaspirone-based PLD inhibitors, such as 3–5, bore an N-aryl moiety (only naked phenyl or halogen substituted phenyl were evaluated), which left a great deal to explore in terms of SAR for PLD2 inhibition as well as in enhancement of physiochemical and DMPK properties.[8–10] Further probe optimization efforts were focused on surveying alternative, non-N-aryl moieties in the triazaspirone core (Figure 2) and to evaluate diverse analogs 6. To enable this effort, a synthetic route was devised to access advanced intermediate 11 (Scheme 1).[11] Beginning with N-benzyl protected piperidinone 7, a Strecker reaction with ammonia followed by sulfuric acid mediated hydrolysis, delivered carboxamide 8. Condensation with formamide followed by reduction with sodium borohydrideprovided the triazaspirone core 9 in 25% yield. Subsequent Boc protection of the secondary amine, deprotection of the primary benzyl amine, and reductive amination with tert-butyl (2-oxoethyl)carbamate furnished the bis-Boc 10 in 62% yield. Finally, global deprotection with HCl and selective acylation of the primary amine afforded advanced intermediate 11, with the free secondary amine to participate in alkylations, reductive aminations, and acylations to survey non-N-aryl moieties in analogues 6.
Figure 2.
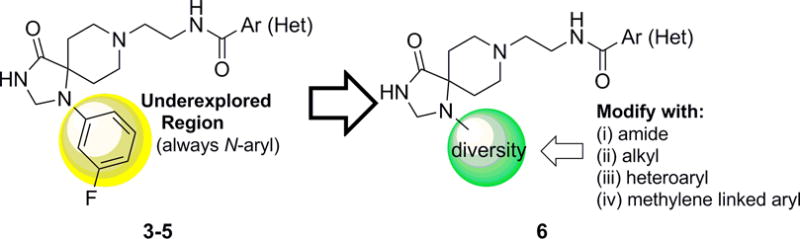
Optimization plan for 3–5 to produce analogues 6 with non-N-aryl diversity to improve activity, physiochemical properties and DMPK profile.
Scheme 1.
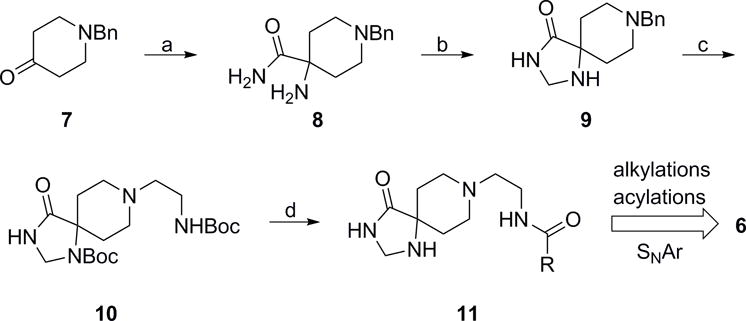
Preparation of analogues 6. Reagents and conditions: a) i. NaCN, NH4Cl, 7 M NH3/MeOH, rt, 4 h, ii. H2SO4, DCM, 62%; b) i) formamide, H2SO4, 175 °C, 16h ii) NaBH4, MeOH, 25% c) i. Boc2O, DIEA, 0.5 equiv. DMAP, THF, ii. H2, 10% Pd/C, MeOH, rt, iii. tert-butyl (2-bromoethyl)carbamate, K2CO3, DMF, rt, 62% over three steps; d) i. HCl, dioxanes, rt, ii. RCOCl, DCM, DIEA, rt, 51–84%.
From 11, multiple iterative libraries were synthesized (totally 80 novel analogues) that were evaluated for inhibitory activity against PLD1 and PLD2 in our standard cell-based assay. In the first iteration, the 2-naphthylamide moiety was held constant (a preferred group) and alterative N-substituents were surveyed in analogues 6. Table 1 highlights selected SAR for this series. Robust SAR was noted. The unsubstituted secondary amine 6a was inactive, while simple cycloalkyl amides, such as cyclopropyl 6b and cyclobutyl 7c were potent PLD2 inhibitors with IC50 values of 440 nM and 320 nM, respectively; however, both were modestly selective versus PLD1 (7- to 12-fold). Benzamide congeners, 6d–f, were uniformly inactive at both PLD1 and PLD2, while certain benzyl amines proved to be potent PLD2 inhibitors, such as the 2-fluorobenzyl analogue, 6h, displaying 22-fold selectivity over PLD1 and a PLD2 IC50 of 205 nM. The most interesting SAR concerned regioisomeric methylene linked pyridyl congeners.
Table 1.
Structures and PLD inhibitory activities of analogues 6.
| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R | PLD1 IC50 (nM)[a] | PLD2 IC50 (nM)[a] | Fold PLD2 selective |
| 6a | H | >30,000 | >30,000 | – |
| 6b |
|
3,100 | 440 | 7 |
| 6c |
|
4,000 | 320 | 12.5 |
| 6d |
|
19,000 | 2,050 | 9 |
| 6e |
|
8,760 | 2,750 | ~3 |
| 6f |
|
>20,000 | 2,540 | ~10 |
| 6g |
|
8,260 | 890 | 9.2 |
| 6h |
|
4,590 | 205 | 22 |
| 6i |
|
8,100 | 1,780 | 4.5 |
| 6j |
|
3,500 | 7,200 | – |
| 6k |
|
2,650 | 80 | 30 |
| 6l |
|
>30,000 | 360 | >80 |
| 6m |
|
24,000 | 850 | 28 |
IC50 values were determined in triplicate.
In this case, the methylene linked 2-pyridyl analogue 6k was 30-fold selective versus PLD1, the 3-pyridyl analogue 6l was >80-fold and very potent (PLD2 IC50 = 360 nM), and the 4-pyridyl methyl analogue 6m was 28-fold selective versus PLD1 (PLD2 IC50 = 850 nM). Thus, the 3-pyridyl methyl would be retained in future analogues 12 while surveying alternative amides (Table 2), employing a variation of the route depicted in Scheme 1. In this instance, SAR was shallow, with functionalized benzamides and heteroaryl amides affording inactive compounds. However, indole-based amides, 12a–12e, were the exception from this library, though SAR was still shallow. Notably, 12a, a 2-indolyl amide was a potent PLD2 inhibitor (PLD2 IC50 = 52 nM) with ~69-fold selectivity versus PLD1 (PLD1 IC50 = 3,600 nM). The 5-fluoro congener, 8b, suffered a moderate loss in potency (PLD2 IC50 = 120 nM) and selectivity (26-fold), but other substitution patterns were inactive. The N-methyl analogue of 12a, 12c, was uniformly inactive as were aza derivatives, such as 12d. Lastly, the 3-indolyl amide 8e was also devoid of PLD activity inhibition.
Table 2.
Structures and PLD inhibitory activities of analogues 12.
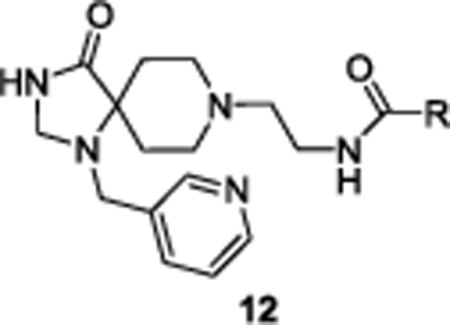
| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R | PLD1 IC50 (nM)[a] | PLD2 IC50 (nM)[a] | Fold PLD2 selective |
| 12a |
|
3,600 | 52 | 69 |
| 12b |
|
3,100 | 120 | 26 |
| 12c |
|
>20,000 | >20,000 | – |
| 12d |
|
>20,000 | >20,000 | – |
| 12e |
|
>20,000 | >20,000 | – |
IC50 values were determined in triplicate.
Since the methylene linked 3-pyridyl moiety was still optimal, this group was attached to the nitrogen of the triazaspirone and N-pyridyl congeners evaluated. SNAr chemistry afforded rapid access to a set of 12 analogues with either a 3-pyridyl 13 or 3-pyridyl-5-fluoro ring 14, the latter of which to combine the optimal moieties in 3–5 and the 3-pyridyl moiety (Figure 3). Interestingly, these analogues were all uniformly inactive. Thus, 3-pyridyl analogue 6l, a potent, direct inhibitor of PLD2 (cellular PLD2 IC50 = 360 nM, exogenous biochemical assay with purified PLD2 IC50 = 8.7 μM) with no measureable activity at PLD1 up to 30 μM (>80-fold selective versus PLD1) was designated ML395, an MLPCN probe molecule to be further characterized. To further evaluate ML395 (6l), the physiochemical and DMPK profile of this highly PLD2 selective inhibitor was assessed.[11] ML395 conforms to Lipinski’s rules (MW = 443, H-bond donors = 2) with favorable lipophilicity (cLogP = 2.74) and an acceptable TPSA of 77. Moreover, ML395 was found be both soluble in phosphate buffered saline (PBS), (95 μM at pH = 7.4 at 23 °C) and stable (~100% parent remaining at 48 hours in PBS at 23 °C). Moreover, ML395 displayed a favourable Cytochrome P450 profile (CYP3A4 IC50 = 3.9 μM, CYP2D6 IC50 = 16.4 μM, CYP1A2 IC50 > 30 μM CYP2C9 IC50 > 30 μM). In plasma protein binding experiments, the compound exhibited exceptionally high fraction unbound (fuplasma) in rat (26.3%) and moderately high fuplasma in human (8.8%), which prompted investigation into the compound’s stability due to the presence of amides. ML395 was stable over time (4 hours; 37 °C) in plasma from both species, revealing that the observed fuplasma values were accurate. In rat and human hepatic microsomes, ML395 exhibited moderate-to-high intrinsic clearance (rat CLint: 82.1 mL/min/kg, human CLint: 43 mL/min/kg) with predicted hepatic clearance values (rat CLhep: 64.3 mL/min/kg, human CLhep: 17 mL/min/kg) near the respective rates of hepatic blood flow in each species (Table 3). In order to determine the relevant biotransformation pathway(s) contributing to the modest metabolic stability observed in vitro (CLINT and CLHep), metabolite identification (Met ID) experiments were performed using the rat hepatic S9 fraction (Figure 4). This analysis revealed NADPH-dependent oxidation of the triazaspirone core consistent with common biotransformation pathways of piperidines where an initial oxygenation is followed by subsequent alcohol oxidation or dehydration. Another NADPH-dependent mono-oxidation pathway identified in rat S9 was N-dealkylation at the amide (M1; Figure 4).[11]
Figure 3.
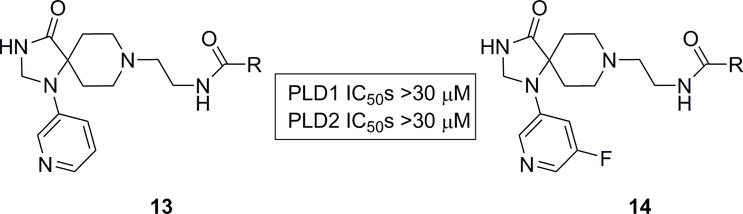
Direct N-pyridyl analogue libraries 13 and 14 were all uniformly inactive at both PLD1 and PLD2 (IC50s >30 μM).
Table 3.
DMPK profile of ML395.
| Species | Property | Value1 |
|---|---|---|
| Sprague Dawley Rat | Hepatic Microsomal CLint | 82.1 mL/min/kg |
| Predicted CLhep | 64.3 mL/min/kg | |
| fuplasma; fubrain | 0.26; 0.12 | |
| Cbrain:Cplasma (Kp) | 1.482 | |
|
| ||
| Human | Hepatic Microsomal CLint | 43 mL/min/kg |
| Predicted CLhep | 17 mL/min/kg | |
| fuplasma | 0.088 | |
| P450 1A2 IC50 | > 30 μM | |
| P450 2C9 IC50 | >30 μM | |
| P450 2D6 IC50 | 16.2 μM | |
| P450 3A4 IC50 | 3.9 μM | |
values represent means of at least two replicates with similar results
Kp determined at 0.25 hr following a 0.2 mg/kg IV dose (n = 2)
Figure 4.
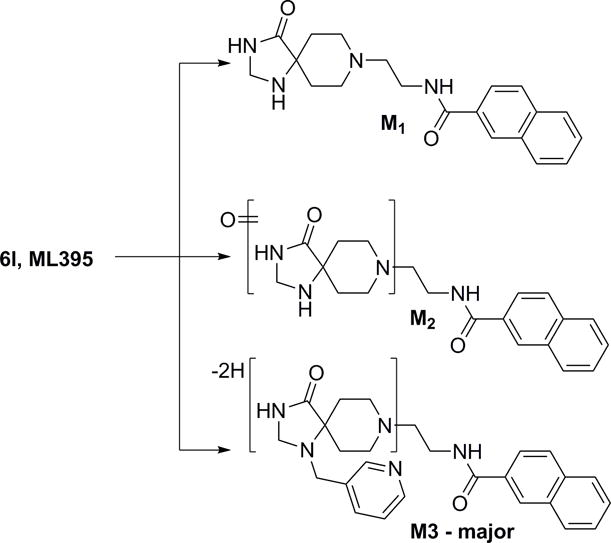
Metabolite identification for 7l, ML395 (VU0468809), in rat S9 with and without NADPH. Three routes of metabolism (M1–M3) were identified in the presence of NADPH, and none in the absence of NADPH.
In order to gauge distribution to the central nervous system (CNS), concentrations of ML395 in whole brain and plasma at a single time point (0.25 hr) were measured following a single intravenous (IV) administration (0.2 mg/kg) to male, Sprague Dawley rats (n = 2).[11] This study revealed a brain:plasma partition coefficient (Kp) of 1.48, indicating excellent distribution to the CNS (Table 1), and in a bidirectional MDCK-MDR1 transwell assay, ML395 (5 μM) exhibited an efflux ratio (ER) of 1.4, suggesting an absence of P-glycoprotein (P-gp)-mediated active efflux liabilities at the blood-brain barrier. Moreover, ML395 was screened in a Eurofins radioligand binding panel of 68 GPCRs, ion channels and transporters at a concentration of 10 μM,[12] and no significant activity was noted (no inhibition >50% at 10 μM) including hERG. Thus, in addition to unprecedented selectivity versus PLD1, ML395 displayed clean ancillary pharmacology against a diverse array of discrete molecular targets, and notably eliminated biogenic amine activity that persisted with 1–5.[3–10] Together, these findings suggest that ML395 possesses acceptable CNS compound exposure properties for pharmacodynamic studies in rodent species.
Based on the pronounced cytotoxicity of our standard selective PLD2 tool compound 3, we needed to assess if ML395 provided an improvement prior to advancing into key studies. As shown in Figure 5, ML395 was devoid of cytotoxicity at concentrations up to 50 μM, as opposed to 3 which displays significant cyototoxicity at concentrations above 10 μM.[11] Therefore, ML395 emerged as a preferred tool compound to assess selective PLD2 inhibition in cell-based assays, and represented a major advance over 3.
Figure 5.
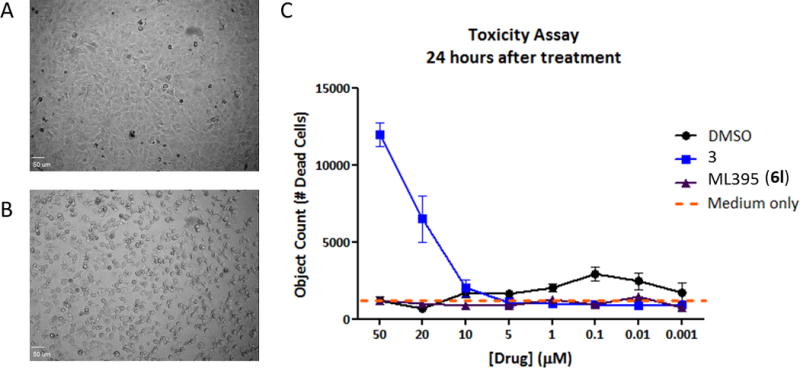
Assessing the cytotoxicity of PLD2 inhibitor 3 versus ML395 (6l). A) microscopic image of A549 cells treated with ML395 (6l) showing no cellular toxicity after 24 hours, bar = 50 μm. B) microscopic image of A549 cells treated with 3 showing significant cellular toxicity after 24 hours, bar = 50 μm. C) A dose response of 3 and ML395 in a 24 hour cytotoxicity in A549 cells. PLD2 inhibitor 3 displays significant toxicity at concentrations above 10 μM, whereas ML395 is comparable to DMSO at concentrations up to 50 μM.
Recently, we reported that influenza virus stimulates host cell PLD activity, and PLD co-localizes with influenza during infection.[13] By decreasing PLD2 activity using chemical inhibition, via 3, or through RNA interference, delayed viral entry and reduced viral titers in vitro were observed. In vivo, PLD2 inhibition with 3 reduced viral titer, increased survival, and correlated with significant increases in transcription of innate antiviral effectors.[13] However, due to the cytotoxicity of 3, and the modest activity toward PLD1 (IC50 = 1.5 μM), evaluation of the antiviral activity of ML395 (6l) due to its improved properties and lack of both cytotoxicity and activity at PLD1 was essential.
Employing the traditional TCID50 assay to assess viral reproduction in vitro,[13] A549 cells were treated with 10 μM 3 or ML395 (6l) for 1 hour before infection (Figure 6), and the cells were then infected with 1 multiplicity of infection (MOI) with four distinct strains of influenza (A/California/04/2009 (H1N1), A/Brisbane/10/2007 (H3N2), rg-A/Vietnam/1203/2004 (H5N1) and A/Anhui/01/2013 (H7N9)). At 24 hours post infection, infectious supernatant was removed from the A549 cells and titrated on MDCK cells to measure viral reproduction.[11,13] Across all four strains, both 3 and ML395 significantly lowered viral reproduction. The improved physiochemical properties of ML395 translated into comparable efficacy to 3, despite an ~20-fold reduction in PLD2 potency. Additionally, since this assay was performed at 10 μM, the effect of ML395 can be directly linked to PLD2 inhibition while a 10 μM concentration of 3 undoubtedly inhibits both PLD1 and PLD2. Notably, the PLD inhibitors displayed pan-anti-influenza activity across both seasonal influenza (H1N1), a low pathogenicity strain of influenza (H3N2), a highly pathogenic avian influenza (H5N1), and a recently emergent virus with pandemic potential (H7N9). Importantly, the apparent ease of resistance to both adamantane drugs and Tamiflu with H7N9 viruses is concerning, and thus small molecule PLD inhibition represents an exciting new mechanism with which to combat pathogenic influenza and provide new tools to assist in overcoming evolution of antiviral drug resistance.[14]
Figure 6.
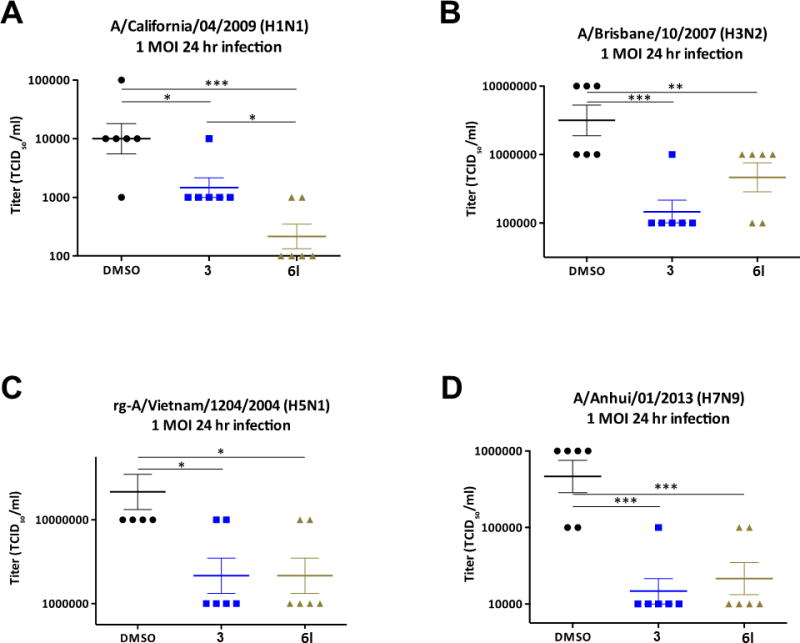
Influenza replication is significanlty reduced when PLD2 is inhibited by either VU034739 (3) or ML395 (6l). A549 cells were pretreated with either DMSO or 10 μM VU0364739 or 10 μM ML395 (7l) for 1 hour, then infected with either: A. 1 MOI H1N1 influenza B. 1 MOI H3N2 influenza C. 1 MOI H5N1 influenza D. 1 MOI H7N9 influenza. After 24 hours post-infection, the infectious supernatant containing the virus was removed and titrated on MDCK cells to assess viral production. Under PLD2 inhibitor treatment, poor viral replication was observed in all influenza strains tested by 24 hours post-infection, and the viral output defect was noted as early as 12 hours post-infection in the case of H3N2 and H7N9. Differences were assessed using a two-way ANOVA and Bonferroni’s post-test, where * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
In summary, a potent (PLD2 IC50 = 360 nM) and >80-fold selective (PLD1 IC50 >30,000 nM) PLD2 inhibitor was developed 6l (also known as ML395 or VU0468809), with improved physiochemical properties, no cytotoxicity, high CNS penetration and a favorable DMPK profile. SAR was steep for this series, with subtle modifications leading to complete loss of PLD inhibitory activity. In cell-based antiviral assays, ML395 was shown to dramatically inhibit infection of A549 cells by multiple strains of influenza. Importantly, inhibition of PLD2 has now emerged as a powerful new mechanism to treat not only classical H1N1, but also the highly virulent and treatment resistant H5N1 and H7N9 strains. Studies are underway to further explore the breadth of antiviral activity of PLD2 inhibition, as well as in vivo survival studies with ML395. ML395 is an MLPCN probe and is freely available upon request.
Supplementary Material
Acknowledgments
This work was generously supported by the NIH/MLPCN grant U54 MH084659 (C.W.L.). Dr. Lindsley acknowledges the Warren Family and Foundation for funding the William K. Warren, Jr. Chair in Medicine. M.C.O. acknowledges funding from a Predoctoral ACS Medicinal Chemistry Fellowship (2011–2012).
Footnotes
Experimental Section
Experimental procedures for medicinal chemistry, pharmacology and drug metabolism as well as characterization of compounds are provided in the Supporting information, available at http://dx.doi.org/10.1002/cmdc.20xxxxxxx.
References
- 1.Selvy PE, Lavieri R, Lindsley CW, Brown HA. Chem Rev. 2011;111:6064–6119. doi: 10.1021/cr200296t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scott SA, Selvy PE, Buck JR, Cho HP, Criswell TL, Thomas AL, Armstrong MD, Arteaga CL, Lindsley CW, Brown HA. Nat Chem Biol. 2009;5:108–117. doi: 10.1038/nchembio.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown HA, Henage LG, Preininger AM, Xiang Y, Exton JH. Methods Enzymol. 2007;434:49–87. doi: 10.1016/S0076-6879(07)34004-4. [DOI] [PubMed] [Google Scholar]
- 4.Scott SA, Matthews TP, Ivanova PT, Lindsley CW, Brown HA. Biochimica et Biophysica Acta – Molecular and Cell Biology of Lipids. in press. [Google Scholar]
- 5.Monovich L, Mugrage B, Quadros E, Toscano K, Tommasi R, LaVoie S, Liu E, Du ZM, LaSala D, Boyar W, Steed P. Bioorg Med Chem Lett. 2007;17:2310–2311. doi: 10.1016/j.bmcl.2007.01.059. [DOI] [PubMed] [Google Scholar]
- 6.De Cuyper H, van Praag HM, Verstraeten D. Neuropsychobiology. 1984;12:211–223. doi: 10.1159/000118141. [DOI] [PubMed] [Google Scholar]
- 7.Lewis JA, Scott SA, Lavieri R, Buck JR, Selvy PE, Stoops SL, Armstrong MD, Brown HA, Lindsley CW. Bioorg Med Chem Lett. 2009;19:1916–1919. doi: 10.1016/j.bmcl.2009.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lavieri R, Lewis JA, Scott SA, Selvy PE, Buck J, Armstrong MD, Brown HA, Lindsley CW. Bioorg Med Chem Lett. 2009;19:2240–2244. doi: 10.1016/j.bmcl.2009.02.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lavieri R, Scott SA, Selvy PE, Brown HA, Lindsley CW. J Med Chem. 2010;53:6706–6719. doi: 10.1021/jm100814g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Reilly MC, Scott SA, Brown KA, Oguin TH, III, Thomas PG, Daniels JS, Morrison R, Brown HA, Lindsley CW. J Med Chem. 2013;56:2695–2699. doi: 10.1021/jm301782e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For experimental details of the molecular pharmacology, DMPK, antiviral assays, chemical synthesis and characterization, see the Supporting Information.
- 12.For informaiton on the Eurofins Lead Profiling Screen, see: http://www.eurofins.com
- 13.Oguin T, Sharma S, Stuart AD, Jones CK, Daniels JS, Lindsley CW, Thomas PG, Brown HA. J Biol Chem. doi: 10.1074/jbc.M114.558817. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu Y, Lu S, Wang W, Hao P, Li J, Yen HL, Shi B, Li T, Guan W, Zu L, Liu Y, Zhang X, Tian D, Zhu Z, He J, Huang K, Chen H, Zheng L, Li X, Ping J, Kang B, Xi X, Zha L, Zhang Z, Peiris M, Yuan Z. Lancet. 2013;381:2273–2279. doi: 10.1016/S0140-6736(13)61125-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.