

Abstract
This letter describes the continued optimization of the MLPCN probe ML375, a highly selective M5 negative allosteric modulator (NAM), through a combination of matrix libraries and iterative parallel synthesis. True to certain allosteric ligands, SAR was shallow, and the matrix library approach highlighted the challenges with M5 NAM SAR within in this chemotype. Once again, enantiospecific activity was noted, and potency at rat and human M5 were improved over ML375, along with slight enhancement in physiochemical properties, certain in vitro DMPK parameters and CNS distribution. Attempts to further enhance pharmacokinetics with deuterium incorporation afforded mixed results, but pretreatment with a pan-P450 inhibitor (1-aminobenzotriazole; ABT) provided increased plasma exposure.
Keywords: M5, Muscarinic receptor, Negative allosteric modulator, Matrix library, Pharmacokinetics
Of the five muscarinic acetylcholinereceptors (mAChR subtypes M1–M5), far less in known about the neurobiological roles of M5 due both to limited CNS expression (< 2% of all mAChR protein in rat brain and found exclusively in the ventral tegmental area [VTA] on dopamine transporter [DAT]-expressing neurons and in the substantia nigra pars compacta [SNc]) and the lack of highly selective, in vivo probe molecules.1–10 Insight into the therapeutic potential of M5 comes largely from genetic studies in M5-KO mice, which exhibit reduced sensitivity to the rewarding effects of cocaine and opiates.11–13 Recently, an association between an M5 SNP and an addictive phenotype was observed in man, directly linking M5 to drug abuse and reward.14 To advance the M5 research field, small molecule probes are required to recapitulate the genetic data.
Previously, we have reported on the devleopment of several potent and selective M5 positive allosteric modualtor (PAM) chemotypes,15–18 as well as the first highly M5 selctive orthosteric antagonist;19 however, DMPK properties were generally poor and these efforts failed to produce in vivo probes. Last year we disclosed results from an M5 functional high-throughput screen that provided 1-(4-flurobenzoyl)-9b-phenyl-2,3-dihydro-1H-imidazo[2,1-a]isoindol-5(9bH)-one as an M5 negative allosteric modulator (NAM) hit, 1 (Fig. 1).20 A limited chemical optimization effort afforded ML375 (2), the first M5-selective NAM with favorable CNS exposure (brain:plasma Kp = 1.8), moderate PK, high plasma protein binding (rat fu = 0.029, human fu = 0.013, rat brain fu = 0.003) and enantiospecific activity (only the (S)-enantiomer of the 9b p-Cl phenyl wasactive).20 Despite a major advance in the field, due to weak potency at rat M5, coupled with high plasma protein and brain homogenate binding, ML375 lacked the requisite free drug exposure to serve as an in vivo tool compound.20 In this Letter, we report on the continued optimization of our first-in-class M5 NAM, and detail key tactics and noteworthy challenges en route to an M5 NAM in vivo probe.
Figure 1.

Structures and mAChR activities of M5 NAM HTS hit 1, and the optimized MLPCN probe ML375 (2). Inset, optimization plan for ML375 to improve rat potency and physiochemical/disposition properties. Potency values determined via a functional calcium mobilization assay in the presence of a fixed acetylcholine EC80 in recombinant cells.20
The synthesis of novel analogsof ML375 required a simple two-step synthesis involving condensation of ethylene diamine and an appropriately substituted 2-benzoylbenzoic acid 3 (or heteroaromatic/cyclo(hetero)alkyl congener) to provide 4, followed by a subsequent acylation reaction (Scheme 1) to deliver ML375 analogs 5–7.20,21 However, we quickly exhausted the commercial analogs of 3. Fortunately, we were able to employ three synthetic routes to access key intermediates 3 with either diverse substituents or encompassing heterocycles.
Scheme 1.

Reagents and conditions: (a) ethylene diamine, p-TSA, toluene (+1,4-dioxane), reflux, Dean-Stark trap, or microwave irradiation 130–150 °C 4–77%; (b) Ar(Het)COCl, CH2Cl2, DIPEA, 16–91%; (c) RMgX, THF, −65 °C to 0 °C or rt, 8–68%; (d) R-H, AlCl3, PhNO2, rt, 41–88%; (e) i. RCOCl, cat. Ni(acac)2, THF, rt, ii. Aq. NaOH, EtOH/THF, rt, 31–55%.
In the first round of library synthesis, we held the p-Cl 9b phenyl moiety of ML375 constant, and scanned alternate amides within a racemic core to provide analogs 5. Here, (Table 1) we found that heterocycles were generally not tolerated (5i-l) in the context of the p-Cl 9b phenyl core, but two amide congeners, the 4-isoproxyphenyl (5f) and the 3,4,5-trifluorophenyl (5g), displayed submicromolar human M5 activity (hM5 IC50s of 790 nM and 610 nM, respectively), yet were less potent than racemic ML375 (5a, hM5 IC50 = 480 nM). Within a conserved series of ethers, hM5 potency, e.g, M5 IC50s, was enhanced as steric bulk increased”- OMe (5d)<OEt (5e)<Oi-Pr (5f). Despite the lower hM5 potency, 5d displayed a moderate improvement in clogP relative to 5a (4.6 versus 5.2), so we elected to evaluate how diminished lipophilicity would impact plasma protein binding. While racemic 5a displayed high plasma protein binding (rat fu = 0.031, human fu = 0.015), binding of 5f was slightly decreased rat fu = 0.037, human fu = 0.027). These findings then led us to pursue second generation libraries where we aimed to incorporate polar, basic and sp3-hybridized ring systems into the 9b position, while holding the 3,4-difluorobenzoyl moietyconstant, to assess if we could improve both physiochemical properties as well as hM5 potency.
Table 1.
Structures and activities of analogs 5.
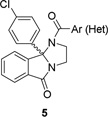 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Ar(Het) | hM5 IC50 (µM) |
hM5 pIC50a | Entry | Ar(Het) | hM5 IC50 (µM) |
hM5 pIC50a |
| 5a | 0.48 | 6.32±0.03 | 5g | 0.61 | 6.21±0.02 | ||
| 5b | >10 | >5 | 5h | >10 | >5 | ||
| 5c | 6.6 | 5.18±0.16 | 5i | >10 | >5 | ||
| 5d | 1.8 | 5.74±0.08 | 5j | 5.8 | 5.23±0.09 | ||
| 5e | 0.85 | 6.07±0.05 | 5k | >10 | >5 | ||
| 5f | 0.79 | 6.10±0.03 | 5l | >10 | >5 | ||
hM5 pIC50 reported as average ±SEM from our calcium mobilization assay; n = 3.
Following scheme 1, analogs 6 were rapidly prepared and screened against hM5 (Table 2). Once again, SAR was shallow, with all sp3-based systems, as well as heterocycles, devoid of hM5 activity. A similar pattern emerged for ethers between analog series 5 and 6, with 6c, the 4-OMe phenyl analog, superior potency (hM5 IC50 = 1.3 µM), but insufficient to advance as an in vivo probe. As before, 6c possessed a lower clogP (4.21), which translated into improved plasma free fraction (rat fu = 0.064, human fu = 0.037). Interestingly, the addition of more sp3-character, in the form of the cyclohexyl congener 6e, led to a higher clogP (5.2) and diminished free fraction (rat fu = 0.016, human fu = 0.008). In parallel, we replaced the phenyl ring at the 9b position with the three regioisomeric pyridines, and all were not tolerated (hM5 IC50 >5 µM), as were ring expansions and substitutions of the 1H-imidazo[2,1-a]isoindol-5(9bH)-one core.
Table 2.
Structures and activities of analogs 6.
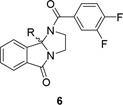 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R | hM5 IC50 (µM) |
hM5 pIC50a | Entry | R | hM5 IC50 (µM) |
hM5 pIC50a |
| 6a | 5.5 | 5.25±0.07 | 6g | >10 | >5 | ||
| 6b | 1.6 | 5.79±0.04 | 6h | >10 | >5 | ||
| 6c | 1.3 | 5.88±0.01 | 6i | >10 | >5 | ||
| 6d | >10 | >5 | 6j | >10 | >5 | ||
| 6e | 2.1 | 5.67±0.05 | 6k | >10 | >5 | ||
| 6f | 2.4 | 5.61±0.06 | 6l | >10 | >5 | ||
hM5 pIC50 reported as average ±SEM from our calcium mobilization assay; n = 3.
At a loss for a rational, singleton approach to build-in hM5 potency and improved physiochemical properties, we elected to pursue a 3 × 9 matrix library of analogs 7 to systematically evaluate all the possible combinations of monomers that showed either hM5 potency enhancement or improved physiochemical properties (Table 3).22,23 While we have generated, on numerous occasions, robust, tractable SAR within GPCR allosteric ligand chemotypes, we have also reported on numerous accounts of chemotypes that possess shallow or flat SAR,8–11,24 and this matrix library is an example of the latter. Here, the clear stand-out was racemic 7B-6 (also referred to as VU0652483, hM5 IC50 = 517 nM, pIC50 = 6.29±0.02), possessing a 3,4,5-trifluorobenzoyl amide and a 3-methyl-4-methoxy phenyl moiety in the 9b position. Due to the increased hM5 potency, we evaluated VU0652483 potency at rat M5 and found submicromolar activity (rat M5 IC50 = 963 nM, pIC50 = 6.02±0.04) as well. As all of the activity of ML375 resided in the (S)-enantiomer, we resolved the enantiomers of VU0652483 via chiral SFC to afford (S)-7B-6 (VU6000181) and (R)-7B-6 (VU6000180); here again, the (R)-enantiomer was inactive (Fig. 2) and the (S)-enantiomer, VU6000181, possessed all of the M5 M5 NAM reported to date and maintaining selectivity versus M1–M4 (IC50s > 30 µM).20 Moreover, the clogP for VU6000181 (4.6) was improved over ML375 (5.2), and this once again translated into a slight improvement over ML375 (rat fu = 0.031, human fu = 0.013, rat brain fu = 0.006). In addition, VU6000181 was highly centrally penetrant (brain:plasma Kp = 2.7 at 0.25 ht post-administration), yet a high clearance compound in vitro (rat hepatic microsome CLINT = 332 mL/min/kg, predicted CLHEP = 57.8 mL/min/kg and human hepatic microsome CLINT = 359 mL/min/kg, predicted CLHEP = 19.8 mL/min/kg) and in vivo (rat CLp = 80 mL/min/kg, t1/2 = 65 min, Vss = 4.9 L/kg). The PK profile of VU6000181 rendered it unsuitable as an in vivo probe.
Table 3.
Structures and activities of matrix library analogs 7.
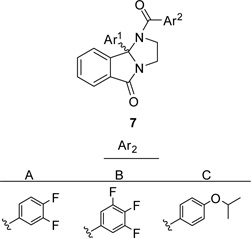 |
Ar1 | |||
|---|---|---|---|---|
| Entry | hM5IC50 (µM)a |
hM5IC50 (µM)a |
hM5IC50 (µM)a |
|
| 1 | 3.2 | >30 | 4.1 | |
| 2 | >30 | 2.4 | 1.9 | |
| 3 | 1.3 | 1.0 | 1.1 | |
| 4 | 3.3 | >30 | 3.2 | |
| 5 | 5.5 | 1.4 | 3.3 | |
| 6 | 1.2 | 0.51 | 1.3 | |
| 7 | 2.7 | 0.7 | 1.3 | |
| 8 | 2.0 | 1.6 | 6.4 | |
| 9 | 1.3 | 0.9 | 2.2 | |
hM5 IC50 data, n = 1; hM5 IC50 for 7B-6, n = 6, pIC50 = 6.29±0.02
Figure 2.

Structures and M5 functional assay concentration-response-curves (CRCs) obtained in the presence of a fixed acetylcholine EC80 in recombinant cells. A) rat M5 CRCs of racemic VU0652483, the active (S)-enantiomer, VU6000181 and the inactive (R)-enantiomer, VU6000180; B) human M5 CRCs of racemic VU0652483, the active (S)-enantiomer, VU6000181 and the inactive (R)-enantiomer, VU6000180; C) structures of VU0652483, VU6000181, VU6000180.
Previously, we productively utilized deuterium incorporation to overcome high clearance coupled with flat SAR in a series of mGlu3 NAMs,25 reducing rat in vitro and in vivo clearance by ~50% and affording an in vivo probe. Replacing the OCH3 moiety of VU6000181 with a OCD3 moiety (10, VU6001005 did in fact reduce human in vitro intrinsic clearance ~5-fold (human CL = 72.9 mL/min/kg) but provided negligible improvement to rat intrinsic clearance, and thus, was not a productive path towards an in vivo tool compound (Fig. 3).
Figure 3.

The impact of deuterium incorportion into VU6000181 on human, but not rat, intrinsic clearance determined in hepatic microsomes fortified with NADPH (values represent means from one independent determination performed in triplicate).
Finally, we performed an in vivo PK study by predosing rats with the pan-P450 inhibitor 1-aminobenzotriazole (ABT) in an attempt to potentially achieve therapeutically relevant drug levels and provide target validation for M5 NAMs.26 In this instance, we first pre-treated rats with an oral vehicle (10% tween 80 in 0.5% MC in water), followed 1.5 hours later by VU6000181 at 10 mg/kg, P.O. in 30% HPBCD in water (10 mg/mL). This control protocol revealed a VU6000181 mean residence time (MRT) of 6.5 hours, a Tmax of 2.8 hours, an AUC0-inf of 3,600 (hr*ng/mL) and low oral bioavailabiltiy (%F = 2.0). Pre-treatment of rats with a 56.6. mg/kg dose of ABT P.O. (10% tween 80 in 0.5% MC in water), followed 1.5 hours later by VU6000181 at 10 mg/kg, P.O. in 30% HPBCD in water (10 mg/mL), provided a ~5-fold increase in exposure (AUC0-inf 17,700 (hr*ng/mL), %F = 9.5) relative to that in the vehicle pre-treated animals(Fig. 4). In addition, ABT pre-treatment afforded a ~3-fold increase in Cmax (plasma = 2.0 µM, ~ 62 nM unbound) and, based on the a brain:plasma Kp (2.7), projected Cmax levels in brain in the presence of ABT would be ~ 5.4 µM (~ 32 nM unbound). Thus, ABT pretreatment may serve as a viable strategy to increase levels of VU6000181 for future fMRI and addiction studies to test selective M5 inhibition hypotheses.
Figure 4.

Oral plasma pharmacokinetics of VU6000181 in the absence (blue) or presence (red) of ABT pre-treatment in rats (male, Sprague-Dawley). ABT pre-treatment improved exposure ~ 5-fold and maximum concentrations ~ 3-fold.
In summary, we reported on the continued optimization of the MLPCN probe ML375, a highly selective M5 NAM, through a combination of matrix libraries and iterative parallel synthesis. While SAR can be highly tractable for certain allosteric ligands, this chemotype was a clear example of ‘flat’ or ‘shallow’ SAR, wherein an matrix library was critical in identifying a productive lead compound, VU6000181, with improved activity at rat M5 and disposition relative to ML375. Attempts to improve PK by deuterium incorporation substantially impacted only human in vitro CLINT, but ABT pre-treatment significantly increased exposure in rats, potentially affording a strategy to achieve target validation for selective M5 inhibition. Efforts continue, and work is in progress to pharmacologically probe M5 neurobiology and therapeutic potential with small molecules.
Acknowledgments
Vanderbilt is a member of the MLPCN and houses the Vanderbilt Specialized Chemistry Center for Accelerated Probe Development. This work was supported by the NIH/MLPCN grant U54 MH084659 (C.W.L.), the Vanderbilt Department of Pharmacology and William K. Warren, Jr. who funded the William K. Warren, Jr. Chair in Medicine (to C.W.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smythies J. Int. Rev. Neurobiol. 2005;64:1–122. doi: 10.1016/S0074-7742(05)64001-9. [DOI] [PubMed] [Google Scholar]
- 2.Wess J, Eglen RM, Gautam D. Nat. Rev. Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 3.Langmead CJ, Watson J, Reavill C. Pharmacol. Ther. 2008;117:232–243. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 4.Denker D, Thomsen M, Wortwein G, Weikop G, Cui Y, Jeon J, Wess J, Fink-Jensen A. ACS Chem. Neurosci. 2012;3:80–89. doi: 10.1021/cn200110q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caulfield MP, Birdsall NJM. Pharmacol. Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- 6.Weiner DM, Levey AI, Brann MR. Proc. Natl. Acad. Sci. U.S.A. 1990;87:7050–7054. doi: 10.1073/pnas.87.18.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yasuda RP, Ciesla W, Flores LR, Wall SJ, Li M, Satkus SA, Weisstein JS, Spagnola BV, Wolfe BB. Mol. Pharmacol. 1993;43:149–157. [PubMed] [Google Scholar]
- 8.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. J. Med. Chem. 2012;55:1445–1464. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conn PJ, Jones C, Lindsley CW. Trends in Pharm. Sci. 2009;30:148–156. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindsley CW. J. Med. Chem. 2014;57:7485–7498. doi: 10.1021/jm5011786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basile AS, Fedorova I, Zapata A, Liu X, Shippenberg T, Yamada M, Wess J. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11452–11457. doi: 10.1073/pnas.162371899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fink-Jensen A, Fedorova I, Wörtwein G, Woldbye DP, Thomsen M, Bolwig TG, Knitowski KM, McKinzie DL, Yamada M, Wess J, Basile AJ. Neurosci. Res. 2003;74:91–96. doi: 10.1002/jnr.10728. [DOI] [PubMed] [Google Scholar]
- 13.Thomsen M, Woldbye DP, Wortwein G, Fink-Jensen A, Wess J, Caine SB. Neurosci. 2005;25:8141–8149. doi: 10.1523/JNEUROSCI.2077-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anney RJL, Lotfi-Miri M, Olsson CA, Reid SC, Hemphill SA, Patton GC. BMC Genet. 2007;8:46. doi: 10.1186/1471-2156-8-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bridges TM, Marlo JE, Niswender CM, Jones JK, Jadhav SB, Gentry PR, Weaver CD, Conn PJ, Lindsley CW. J. Med. Chem. 2009;52:3445–3448. doi: 10.1021/jm900286j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridges TM, Kennedy JP, Hopkins CR, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2010;20:5617–5622. doi: 10.1016/j.bmcl.2010.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gentry PR, Bridges TM, Lamsal A, Vinson PN, Smith E, Chase P, Hodder PS, Engers JL, Niswender CM, Daniels JS, Conn PJ, Wood MR, Lindsley CW. Bioorg. Med. Chem. Lett. 2013;23:2996–3000. doi: 10.1016/j.bmcl.2013.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gentry PR, Kokubo M, Bridges TM, Byun N, Cho HP, Smith E, Hodder PS, Niswender CM, Daniels JS, Conn PJ, Lindsley CW, Wood MR. J. Med. Chem. 2014;57:7804–7810. doi: 10.1021/jm500995y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gentry PR, Kokubo M, Bridges TM, Cho HP, Smith E, Chase P, Hodder PS, Utley TJ, Rajapakse A, Byers F, Niswender CM, Morrison RD, Daniels JS, Wood MR, Conn PJ, Lindsley CW. Chem Med Chem. 2014;9:1677–1682. doi: 10.1002/cmdc.201402051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gentry PR, Kokubo M, Bridges TM, Kett NR, Harp JM, Cho HP, Smith E, Chase P, Hodder PS, Niswender CM, Daniels SJ, Conn PJ, Wood MR, Lindsley CW. J. Med. Chem. 2013;56:9351–9355. doi: 10.1021/jm4013246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Synthesis of VU6000181. To a suspension of Mg (936 mg, 38.5 mmol) and iodine (5 mg) in THF (2 mL) was added a part of a solution of 4-bromo-2-methylanisole (7.39 g, 36.8 mmol) in THF (9 mL) at ambient temperature. After initiating the reaction, a solution of 4-bromo-2-methylanisole diluted with THF (19 mL) was added dropwise to the mixture diluted with THF (10 mL). After the mixture was allowed to stir at ambient temperature for 2 hours, resulting Grignardreagent was added to a suspension of phthalic anhydride 8 (5.18 g, 35.0 mmol) in THF (50 mL) at −65 °C. The mixture was allowed to stir for 2.5 hours as temperature was elevated up to 0 °C. The reaction was quenched with cold water and the aqueous layer was separated. The organic layer was extracted with 1 N NaOH aqueous solution and the combined aqueous layer was acidified with 2 N HCl solution. The aqueous layer was extracted with ethyl acetate twice and the combined organic layer was washed with brine and dried over magnesium sulfate. The filtrate was evaporated under reduced pressure. The residue was triturated with ethyl acetate/diethyl ether to give compound 3 as a white powder (5.61 g, 59% yield). To a solution of compound 3 (1.0 g, 3.7 mmol) and para-toluenesulfonic acid monohydrate (10 mg) in PhMe (8 mL) and 1,4-dioxane (8 mL) was added ethylenediamine (0.50 mL, 7.4 mmol) at ambient temperature. The resulting white suspension was subjected to microwave irradiation at 150 °C for 30 minutes. After removing insoluble material, the filtrate was concentrated. The procedure above was repeated twice more and the three portions were combined and purified on silica gel using hexane/ethyl acetate as an eluent. Crude product was triturated with diethyl ether to give compound 4 as an off-white powder (1.69 g, 52% yield). To a solution of compound 4 (500 mg, 1.70 mmol) in dichloromethane (8 mL) was added DIPEA (0.74 mL, 4.25 mmol) and 3,4,5-trifluorobenzoyl chloride (0.33 mL, 2.55 mmol) at ambient temperature. After stirring for 30 minutes, cold NaHCO3-aq was added to the mixture which was extracted with dichloromethane twice. The combined organic layer was concentrated under reduced pressure and the residue was purified on silica gel using hexane/ethyl acetate as an eluent. Crude product was triturated with diethyl ether/hexane to yield compound 7B-6 as a white powder (491 mg, 64% yield). Chiral resolution by SFC (Agilent 1260, Column: LUX cellulose-3, Column dimensions: 10 × 250 mm, Co-solvent: MeOH, Modifier: none, Gradient Profile: 10% isocratic, Flow Rate: 15 mL/min, Backpressure: 100, Column temperature: 40 degrees, retention time: 2.814 min, 30 dated on July 30th) followed by concentration afforded (S)-7B-6 (VU6000181) as a white powder. 1H NMR (400.1 MHz, DMSO-d6): 7.89 (d, J = 7.4 Hz, 1H), 7.80-7.71 7.78 (m, 1H), 7.71-7.57 (m, 4H), 7.03 (dd, J = 8.5, 2.5 Hz, 1H), 6.91 (d, J = 8.5 Hz, 1H), 6.86 (d, J = 2.5 Hz, 1H), (dt, J = 252, 15.6 Hz), 133.87, 133.68 (q, J = 6.1 Hz), 132.84, 131.18, 130.35, 129.48, 128.74, 126.61, 125.75, 124.18, 113.29 (dd, J = 16.5, 6.0 Hz), 110.94, 87.66, 56.25, 52.45, 40.24, 17.11. HRMS calc’d for: C25H19F3N2O3 (M+H), 425.1348; found 452.1352. Specific rotation (c = 0.75, CHCl3).
- 22.Lavieri R, Scott SA, Selvy PE, Brown HA, Lindsley CW. J. Med. Chem. 2010;53:6706–6719. doi: 10.1021/jm100814g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. J. Comb. Chem. 2008;10:345–354. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]
- 24.Zhao Z, Wisnoski DD, O’Brien JA, Lemiare W, Williams DL, Jr, Jacobson MA, Wittman M, Ha S, Schaffhauser H, Sur C, Pettibone DJ, Duggan ME, Conn PJ, Hartman GD, Lindsley CW. Bioorg. Med. Chem. Lett. 2007;17:1386–1389. doi: 10.1016/j.bmcl.2006.11.081. [DOI] [PubMed] [Google Scholar]
- 25.Wenthur CJ, Niswender CM, Morrison R, Daniels JS, Conn PJ, Lindsley J. J. Med. Chem. 2013;56:5208–5212. doi: 10.1021/jm400439t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mico BA, Federowicz DA, Ripple MG, Kerns W. Biochem. Pharmacol. 1988;37:2515–2519. doi: 10.1016/0006-2952(88)90240-7. [DOI] [PubMed] [Google Scholar]