

Abstract
Background
In Eucalyptus genus, studies on genome composition and transposable elements (TEs) are particularly scarce. Nearly half of the recently released Eucalyptus grandis genome is composed by retrotransposons and this data provides an important opportunity to understand TE dynamics in Eucalyptus genome and transcriptome.
Results
We characterized nine families of transcriptionally active LTR retrotransposons from Copia and Gypsy superfamilies in Eucalyptus grandis genome and we depicted genomic distribution and copy number in two Eucalyptus species. We also evaluated genomic polymorphism and transcriptional profile in three organs of five Eucalyptus species. We observed contrasting genomic and transcriptional behavior in the same family among different species. RLC_egMax_1 was the most prevalent family and RLC_egAngela_1 was the family with the lowest copy number. Most families of both superfamilies have their insertions occurring <3 million years, except one Copia family, RLC_egBianca_1. Protein theoretical models suggest different properties between Copia and Gypsy domains. IRAP and REMAP markers suggested genomic polymorphisms among Eucalyptus species. Using EST analysis and qRT-PCRs, we observed transcriptional activity in several tissues and in all evaluated species. In some families, osmotic stress increases transcript values.
Conclusion
Our strategy was successful in isolating transcriptionally active retrotransposons in Eucalyptus, and each family has a particular genomic and transcriptional pattern. Overall, our results show that retrotransposon activity have differentially affected genome and transcriptome among Eucalyptus species.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-015-0550-1) contains supplementary material, which is available to authorized users.
Keywords: LTR retrotransposons, Dynamics, Genomic distribution, Comparative analysis, Eucalyptus genomes
Background
Retrotransposons correspond to class I transposable elements, inserting into a host genome through an RNA intermediate [1]. Based on structural features and phylogenetic relationships, five orders of retrotransposons were defined [1]. LTR retrotransposons (LTR-RTEs), an order related to retroviruses, usually encode two open reading frames (ORFs): one called gag, which encodes a structural protein for virus-like particles, and another called pol, which encodes enzymatic domains involved in the transposition cycle, such as an aspartic protease, a reverse transcriptase, an RNaseH and an integrase [1]. The two major superfamilies of plant LTR retrotransposons are Copia and Gypsy, in which pol genes differ in their domain order [1].
In most angiosperm genomes, the LTR-RTEs are the most significant contributor to genome size, contributing over 70 % of the nuclear DNA in grasses like maize [2]. Most LTR retrotransposons families exist in low copy numbers, but the amplification of few individual families contribute with large differences in genome size among closely related species [3, 4].
Despite their stringent regulation, LTR-RTEs are transcriptionally active in plants [5–7]. Although epigenetic regulation is an important feature of most plant transposable elements [reviewed in [8], cis-regulation has a crucial role regulating LTR-RTE transcription, since LTRs represent promoter sequences. The modulation of LTR-RTEs transcriptional levels has been observed in different tissues, organs and development stages i.e. [5, 9].
The eucalypts are the most widely planted hardwoods in the world due to their ability to adapt, grow and provide quality wood for multiple applications [10]. Species of subgenus Symphyomyrtus account for > 95 % of the world's planted eucalypts [11]. These include three members of section Latoangulatae: E. grandis, E. urophylla and E. saligna, broadly planted in tropical areas due to their fast growth and disease resistance; and E. tereticornis (section Exsertaria), known for their drought tolerance and rapid growth [10]. Other species are better known for their potential for introgressing new traits in breeding, i.e., E. brassiana [12].
The Eucalyptus grandis genome assembly into 11 pseudochromosomes (605 megabases (Mb)), is composed by 44.5 % of retrotransposons, and LTR-RTEs are the most representative, constituting 21.9 % of the E. grandis genome [13]. Up to now, TE dynamics were scarcely studied in the Eucalyptus genus and most analysis were based on private EST data [i.e. [14]. In the present study, we identify and comprehensively characterize a selected group of Eucalyptus LTR-RTEs, emphasizing the characterization of elements with putative transcriptional activity. We analyzed the phylogenetic pattern of nine E. grandis LTR-RTEs families and we extended this analysis, understanding DNA interaction properties of selected encoding domains. Comparative classifications of LTR-RTEs from closely related species were performed on monocots in which transposable elements (TEs) had previously been well characterized [15, 16], however this is the first time this approach was employed in forest trees. Our study is the first to exhaustively sample and classify transcriptionally active TEs in Eucalyptus species, identifying their structure, genomic distribution, insertion time estimation, genomic polymorphism and transcriptional activity in five Eucalyptus species and one intrageneric hybrid.
Methods
Eucalyptus grandis transcriptionally active LTR-RTEs: selection and annotation
LTR-RTEs of Copia and Gypsy superfamilies described in Wicker and Keller [17] and Lloréns et al. [18] were used as queries in a BLASTX against Eucalyptus spp ESTs from the dbEST database at the National Center for Biotechnology Information (NCBI), website on 10/10/2011. We selected ESTs that aligned over 200 bp or more (e-value <1e − 50) for further analyses, similar to Rossi et al. [19]. In order to confirm whether the EST codes for a LTR-RTE, we analyzed sequences using CENSOR implemented in RepBase [20] and the ones where the LTR-RTE sequence matched more than 80 % of an EST were selected for other analyses.
These selected ESTs were used as queries in a BLASTN search to identify full-length LTR-RTEs in E. grandis genome v. 0.6 (http://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Egrandis). The first 50 regions (hits) with over 85 % identity in a region over 250 bp were selected for full-length LTR-RTE screening. Regions 20 kb up- and downstream from these hits were analyzed using the LTR-Finder [21] and LTR_STRUC [22]. Only full-length LTR-RTEs that aligned with ESTs in BLASTN were retrieved for further analyses, and redundant sequences were discarded. These reference sequences were deposited at GenBank under accessions KM196471 to KM196479. Target site duplications (TSDs) were identified by submitting the full-length sequences as a query and subject to a blast2seq on NCBI website. Putative ORFs were retrieved using FGENESH + tool [23] on Softberry platform (http://linux1.softberry.com/berry.phtml) and manually inspected. Conserved domains were annotated using Pfam (http://pfam.xfam.org/). In the case of the two RLC_egAngela families, 5’ non coding leader sequences were compared using LALIGN (http://fasta.bioch.virginia.edu/fasta_www2/fasta_www.cgi?rm=lalign). RNA minimum free energy secondary structure of aligned regions (376 and 204 nt) was performed using RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi).
Phylogenetic analysis and RTE family name assignment
All sequences from two previous large-scale analyses of plant LTR-RTEs [17, 24] were used to develop phylogenetic trees based on a reverse transcriptase fragment. We used a total of 95 Copia [17, 24] and 37 Gypsy [24] families to classify the nine Eucalyptus LTR-RTE families. DNA sequences were aligned using MUSCLE [25] with default parameters and the phylogenetic trees were made using MEGA 6.0 [26], applying the Maximum Likelihood method, with 1,000 bootstrap replicates. We used the Kimura-2-parameter substitution model and gap positions were excluded when present in more than 5 % of the sequences.
Eucalyptus LTR-RTEs were assigned to families within lineages on the basis of 80 % sequence identity in at least 80 % of their LTRs, based on the universal classification of TEs [1]. We standardized the name of Eucalyptus LTR-RTE sequences similar to Domingues et al. [6] rationale: they were named 'RLC' (Copia) or 'RLG' (Gypsy), 'eg' for 'Eucalyptus grandis', the lineage name (e.g. 'Ale') and the family number (e.g. '1').
Theoretical protein modeling and molecular dynamics simulations
We selected amino acid sequence of RLG_egTekay_1 chromodomain, integrase and reverse transcriptase, as well as the integrase of RLC_egAle_1 and reverse transcriptase of RLC_egBianca_1 by manual selected translation of nucleotide sequences. The sequences were submitted to HHpred server [27], and further to MUSCLE [25] and MUSTER (MUlti-Sources ThreadER) [28]. The best alignments were used to generate models with Modeller v.9.12 [29]. The models were calculated with RAMPAGE [30] and ProSA-web [31] based on structural comparison with template. DNA/RNA molecules were added to the Integrases and Reverse Transcriptases theoretical models by superimposition of DNA binding region.
The best 13 models were submitted to molecular dynamics (MD) simulations using Groningen Machine for Chemical Simulation (GROMACS) v.4.5.3. [32]. The Charmm force field [33] was chosen with explicit solvent [34] and a minimum of 50 ns unrestrained simulation. Theoretical models stability was evaluated by average root mean square deviation (rmsd)/time graph and by overall stereochemical and energy quality. Figure and surface illustrations were generated in CHIMERA [35], with the electrostatic surface generated by APBS [36], PDB2PQR server (http://nbcr-222.ucsd.edu/pdb2pqr_2.0.0/) and PROPKA [37].
Copy number determination in E. grandis genome
We used MEGABLAST to extract the full-length copies of the nine families from the E. grandis genome v 1.0 [13]. All matches that were at least 80 % of the length of the reference full-length sequences and had a similarity level higher than 80 % were considered for copy number analysis. Copies that did not harbor the canonical 5'TG..CA3' were manually removed. Complete copies were plotted in E. grandis genome using Circos [38].
Estimation LTR-RTE insertion time and average LTR divergence sequence
The insertion time of intact LTR-RTE families with two complete LTR sequences and TSDs was calculated based on the assumption that they are identical at the time of integration [39]. For each element, we aligned 5’ and 3’ LTRs using the MUSCLE program implemented in MEGA 6.0 [26], with default parameters. Divergence between LTRs (K) was calculated using MEGA 6.0, using Kimura-2-parameter distance [40]. The insertion time (T) for each intact element was calculated with the formula: T = K/2r. The value of 1.5 x 10-8 substitution per site per year (r) [41] was recently used for the calculation of LTR-RTEs age in grape [42].
Average divergence (Pi) of LTRs for each LTR-RTE family was also calculated, using DnaSp program [43].
In silico transcriptional analysis: Eucalyptus spp EST screening
LTR-RTEs full-length sequences were used as BLASTN queries against Eucalyptus ESTs from EUCANEXT database [44, 45], http://bioinfo03.ibi.unicamp.br/eucalyptusdb/). ESTs similar to LTR-RTEs were assigned to a family according to the criteria adapted from Wicker et al. [1]: 80 % coverage with 80 % nucleotide identity, in a region over 200 bp.
Plant Material and nucleic acid extraction
For IRAP (Inter-Retrotransposon Amplified Polymorphism) and REMAP (Retrotransposon-Microsatellite Ampliflied Polymorphism) analyses, leaves were collected from 10 unrelated individuals of five Eucalyptus species: Eucalyptus brassiana S.T. Blake, Eucalyptus grandis W. Hill ex Maid., Eucalyptus saligna Sm., Eucalyptus tereticornis Sm. and Eucalyptus urophylla S.T. Blake. These individuals were maintained in field by Suzano Papel and Cellulose breeding program. Total DNA was extracted from fresh young leaves using the protocol described in Ferreira and Grattapaglia [46].
For all other analyses, Eucalyptus seedlings were grown under naturally fluctuating conditions of temperature and air relative humidity, and were fertilized and irrigated as necessary in a greenhouse from the Suzano Papel and Cellulose breeding program, in Itapetininga, São Paulo, Brazil. Throughout the experiment, the plants were randomized periodically to minimize any variation within light environment. All plants were harvested 90 days after seed planting.
In the relative quantification of LTR-RTE families, we used the total DNA of E. grandis (clone GD 33) and E. urophylla (clone URO11). In this case, genomic DNA was obtained from young leaves using the DNeasy plant kit (QIAGEN), as recommended by the manufacturer.
For RNA extraction we used freshly harvested leaves, stalk and secondary roots from the five Eucalyptus species mentioned above and one hybrid E. grandis x E. urophylla (termed “E. urograndis”). For each tissue, total RNA was extracted from two groups composed by five plants each according to modificated CTAB protocol proposed by Korimbocus et al. [47]. RNA integrity was checked by electrophoresis, in denaturing agarose gel. RNA quality and quantification was analyzed by spectrophotometry at 260 nm and 280 nm (NanoDrop ND-1000, Thermo Scientific) and stored at -80 °C until used.
IRAP and REMAP profile and data analysis
Sixteen single IRAP primers were designed based on nine LTR-RTE families of E. grandis genome v. 1. (Additional file 1: Table S1). The procedures of PCR amplification were adapted from the protocol of Smýkal [48]. Reactions were done in a total volume of 10 μl, containing 25 ng of genomic DNA, 0.7 X PCR buffer (750 mM Tris-HCl, 200 mM (NH4)2SO4), 25 mM MgCl2, 40 ng of primer, 0.4 mM of each dNTP and 0.3 U of Taq DNA polymerase (Fermentas). The amplification profile consisted of an initial denaturation at 94 °C for 4 min, followed by 35 cycles at 94 °C for 40 s, 50 °C for 2 min, 72 °C for 3 min and 50 s and a final extension of 5 min at 72 °C.
For REMAP, we used the combination of sixteen IRAP primers with 10 SSR primers described in Kalendar et al. [49] (Additional file 1: Table S1). PCR reactions contained 40 ng of genomic DNA, 1.0 X PCR buffer (750 mM Tris-HCl, 200 mM (NH4)2SO4), 25 mM MgCl2, 10 ng of each primer, 0.6 mM of each dNTP, and 0.3 U of Taq DNA polymerase (Fermentas). The amplification profile composed of an initial denaturation at 94 °C for 4 min, followed by 35 cycles at 94 °C for 1 min, 56 °C for 1 min, 72 °C for 1 min and 50 s, and a final extension of 5 min at 72 °C. All IRAP and REMAP reactions were carried out in a PTC-100 thermocycler (MJ Research, Inc.) and PCR products were resolved in 1.8 % agarose gels stained with ethidium bromide.
Each IRAP and REMAP band was treated as a single locus. The presence or absence of a given fragment length in each sample was recorded manually in binary code. DNA marker data was processed by NTSYS-pc version 2.10 [50] and using the SIMQUAL module with the Jaccard genetic similarity coefficient (GSj), and the similarity data was used to perform an unweighted pair group method with arithmetic mean (UPGMA) cluster analysis using the SHAN module, following Fan et al. [51].
LTR-RTE quantification in Eucalyptus urophylla: quantitative real time PCR
Eight LTR-RTE families from E. grandis were quantified in E. urophylla using quantitative real time PCR (qPCR) using the method of Kraitshtein et al. [52] and Baruch and Kashkush [53], which is based on a comparative 2−ΔΔCt method, using a single-copy gene as a reference. Our reference gene was DUR3, a urea transporter, which is a single-copy gene in several eukaryotes [54, 55], and in E. grandis genome we found only a single copy in chromosome 5 (data not shown).
Primers were designed using PerlPrimer v1.1.17 software (http://perlprimer.sourceforge.net) in LTR and internal regions (Additional file 1: Table S1). In order to confirm target specificity we cloned and sequenced amplified regions (ABI 3130xl, Applied Biosystems). Quantitative PCR melt curves also revealed single and unique peaks for each primer pair, confirming high specificity to the target sequence fragments.
PCR efficiencies of the target and reference genes were determined by generating standard curves, based on serial dilutions prepared from cloned DNA templates. We made serial dilutions of the control DNA from 5x10-1 to 5x10-6 ng/μl, with 0.15 ng of each primer.
Fold amplification in each cycle was calculated according to PCR efficiency, which was deduced by the software from the slope of the regression line (y) according to the equation E = [(10 − 1/y) − 1] × 100. For primers with 100 % efficiency, the fold equals 2.
qPCR reactions were conducted in a Step One Plus Real Time PCR System (Applied Biosystems) and analyzed in Step One 2.1 software (Applied Biosystems).
Each qPCR reaction was performed in 5 μl of GoTaq® qPCR Master Mix (Promega), with 1.0 ng of each primer and 3.7 μl of ultra-pure water. The cycling conditions were as follows: 5 min at 95 °C, followed by 45 cycles each of 15 s at 95 °C, 60 s at 60 °C. In order to confirm the reproducibility of our results, reactions were done in technical triplicates in three independent experiments using 0.125, 0.25 and 0.5 ng of genomic DNA.
The relative quantities of LTRs and internal regions of Copia and Gypsy-like LTR-RTEs families were calculated according to Kraitshtein et al. [52] and Baruch and Kashkush [53].
LTR-RTE Transcriptional analysis by RT-qPCR
In addition to plant material mentioned above, in RT-qPCR analysis, we also evaluated the transcriptional impact of osmotic shock in secondary roots of E. grandis submerged in hydroponic solution with PEG. Plants were harvested 0, 6 and 24 h after osmotic stress. Additional details of this experiment are detailed in Rodrigues et al. [56].
All RNA samples were treated with DNase I (Fermentas) and reverse transcribed using GoScript™ Reverse Transcription System kit (Promega). RT-qPCR reactions were performed in technical triplicates from at least two biological replicates in a StepOnePlus Real Time PCR System (Applied Biosystems). Primers used for RT-qPCR are described in Additional file 1: Table S1, and fragments were cloned and sequenced confirming target amplification. Each biological replicate was represented by bulks of leaves, stalk and secondary roots from five plants.
Reactions contained 5 μl of 2 x mix GoTaq® Master Mix (Promega), 1.0 ng of each primer, 150 to 280 ng of cDNA and ultra-pure water up to a final volume of 10 μl. The cycling conditions were as follows: 5 min at 95 °C, followed by 45 cycles each of 15 s at 95 °C, 60 s at 60 °C. Relative expression levels were normalized using the GAPDH gene [57]. The GAPDH stability was evaluated using geNorm and NormFinder. Expression stability was <0.15 in geNorm and <0.03 in NormFinder for all species and tissues (data not shown). These values were compatible with the most stable normalizers from a previous publication in Eucalyptus [57]. In geNorm, stability values below 0.15 do not require an additional gene as a reference [58, 59].
The tissue/organ with lowest expression (highest Ct) was used as calibrator (expression value = 1).
RT-qPCR efficiency was calculated using Linreg v. 2013.0 [60], and reactions with efficiency >90 % were used for analysis. Relative expression was calculated using ΔΔCt method [61] with the formula (1 + E)ΔΔCt, where E represents the efficiency. The statistical analysis was performed using Assistat 7.7 beta [62]. We used one-way analysis of variance (ANOVA) and in cases where significant differences were found, the Least Square Deviation (LSD) method for multiple comparisons was performed. Results were considered significant at P < 0.05.
Results
LTR-retrotransposons in Eucalyptus: overall view and phylogenetic structure
By using a homology-based approach, we mined a total of nine transcriptionally active LTR-RTE families in the E. grandis genome. Seven families belong to the Copia superfamily and two correspond to LTR-RTE elements from the Gypsy superfamily. Copia superfamily LTR-RTEs were classified into five major plant evolutionary lineages [17] (Fig. 1a) and two Gypsy lineages [24] (Fig. 1b). We did not found any Copia element from GMR and Tar lineages. Three Gypsy lineages - CRM, Reina and Athila - did not harboured any transcriptionally active LTR-RTE in our analysis.
Fig. 1.
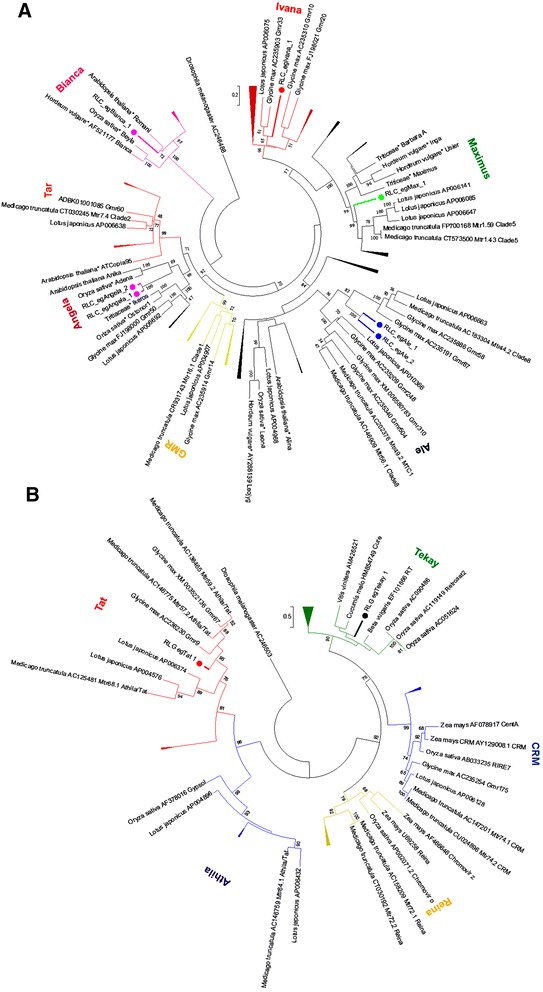
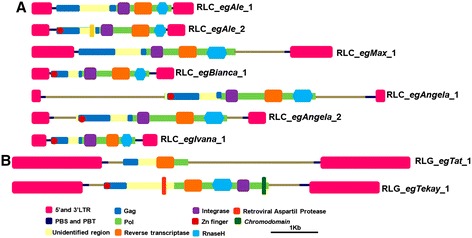
Domains in Eucalyptus LTR-RTEs. a Copia superfamily; b Gypsy superfamily. Abbreviations and domain color coding: LTR = long terminal repeat (pink); PBS = protein binding site and PPT = poly-purine tract (dark blue); Gag (blue); RNAseH = Ribonuclease H (light blue); integrase = Integrase (purple)
The reference full-length sequences of these families (Table 1) have between 93 and 99 % identity among 5’and 3’LTRs, and for all reference sequences we annotated at least two internal coding domains among gag, reverse transcriptase, integrase and RNAseH (Fig. 2). RLC_egAngela_1 and RLC_egAngela_2 families have > 95 % identity between internal coding domains; however, LTRs have < 80 % similarity; consequently, we preferred to classify these elements as separate families. We also compared 5’ non coding region in order to confirm if they correspond to distinct families, similar to Tanskanen et al. [63]. The folding pattern of 5’ leader region was completely distinct between the two families (Additional file 2: Figure S1), confirming that they represent two different families.
Table 1.
Overall features of LTR-RTEs identified in E. grandis genome
| Superfamilies | Families | Lineages | LTR length (bp) | LTRs Ida | LTR-RTE size (bp) | Copy number |
|---|---|---|---|---|---|---|
| Copia | RLC_egAle_1 | Ale | 458/457 | 99 % | 5509 | 12 |
| RLC_egAle_2 | Ale | 434/434 | 96 % | 5395 | 4 | |
| RLC_egMax_1 | Maximus | 904/903 | 98 % | 9670 | 623 | |
| RLC_egBianca_1 | Bianca | 424/423 | 99 % | 5008 | 77 | |
| RLC_egAngela_1 | Angela | 162/161 | 93 % | 11280 | 1 | |
| RLC_egAngela_2 | Angela | 392/392 | 92 % | 7473 | 67 | |
| RLC_egIvana_1 | Ivana | 247/239 | 93 % | 4440 | 49 | |
| Gypsy | RLG_egTat_1 | Tat | 2199/2257 | 93 % | 18300 | 15 |
| RLG_egTekay_1 | Tekay | 555/552 | 96 % | 12159 | 7 |
aId: identity among 5’ and 3’LTRs
Fig. 2.
Classification of Eucalyptus grandis LTR-RTEs Copia and Gypsy superfamily sequences into nine new families. Phylogenetic analyses used 95 Copia sequences from Wicker et al. [1] and 37 Gypsy sequences from Du et al. [24] a Evolutionary lineages of seven elements from Copia superfamily in Eucalyptus b Evolutionary lineages of two Eucalyptus Gypsy LTR-RTEs
Overall size followed previous large-scale reports for LTR-RTEs, as well for PBS and PPT sequences (Additional file 1: Table S2). Both Gypsy families have > 12 kbp and the average size of Copia is around 7 kpb (Table 1). LTRs from Gypsy elements are also larger: only RLC_egMax_1 has LTRs longer than 500 bp (Table 1). Five families contained a spacer region between internal coding domain and LTRs (Fig. 1).
Structural characterization and molecular dynamics simulations of LTR retrotransposon domains
By threading modeling, we generated the first theoretical models of LTR-RTE proteins in plants. We used three domains of RLG_egTekay_1: chromodomain (CD1) (Fig. 3a), integrase (INT1) (Fig. 3b) and reverse transcriptase (RT1) (Fig. 3c). We also generated domains theoretical models on the domains integrase of RLC_egAle_1 (INT2) (Fig. 4a) and reverse transcriptase of RLC_egBianca_1 (RT2) (Fig. 4b). The template used to generate CD1 was the NMR structure of a chromodomain (PDB id/monomer: 2RSO/A) from Schizosaccharomyces pombe [64]. The template used to generate INT1 was the crystallographic structure of an inhibited retroviral integrase (3OYM/A) from human Spumaretrovirus [65] and M-phase phosphoprotein 8 (PDB id: 3LWE/A) from Homo sapiens [66]. The template used to generate RT1 was the crystallographic structure of a reverse transcriptase/ribonuclease H p80 complexed with RNA/DNA hybrid (4HKQ/A) from Schizosaccharomyces pombe [67]. The template used to generate INT2 was the crystallographic structure of PFV integrase (3OYM/A) from Human spumaretrovirus [65]. The template used to generate RT2 was the crystallographic structure of Ty3 reverse transcriptase complexed to RNA/DNA hybrid (4OL8/A) from Saccharomyces cerevisiae [68]. The CD1, INT1, RT1, INT2 and RT2 had original models with 93.6 %, 98.3 %, 98.5 %, 94.2 % and 96.8 % of residues in favored and allowed region which were improved to 100 %, 99.7 %, 99.3 % 98.2 % and 98.7 %, respectively, after MD dynamics. These models started with a Z-score of -0.87, -5.49, -9.7, -4.41 and -3.82 which was either improved or maintained to -1.03, -5.41, -8.91, -5.04 and -4.02. All of the models were submitted to 50 ns and had its stabilization within the first 30 ns with a rmsd of 0.35 to 1.25 nm. Thus, the generated models were stabilized by long MD simulations and validated by different approaches.
Fig. 3.
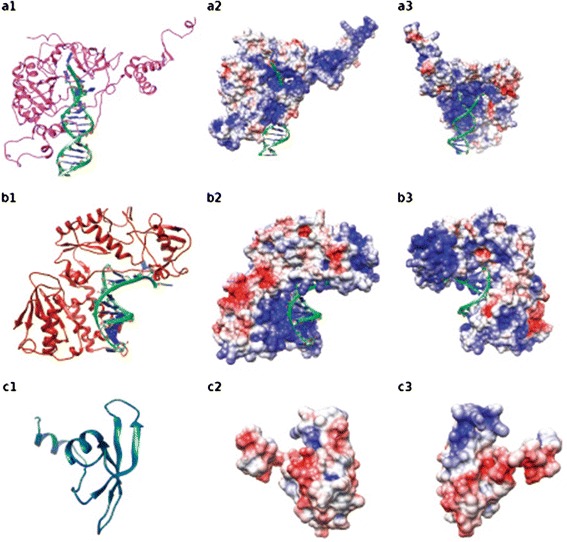
Three dimension theoretical models of RLG_egTekay_1: Integrase (INT1) (a), Reverse Transcriptase (RT1) (b) domains complexed with DNA and Chromodomain (c). In 1, protein cartoon representation. In 2 and 3, electrostatic surface in red (-4) acid regions and in blue (4) basic regions rotated in 180°
Fig. 4.
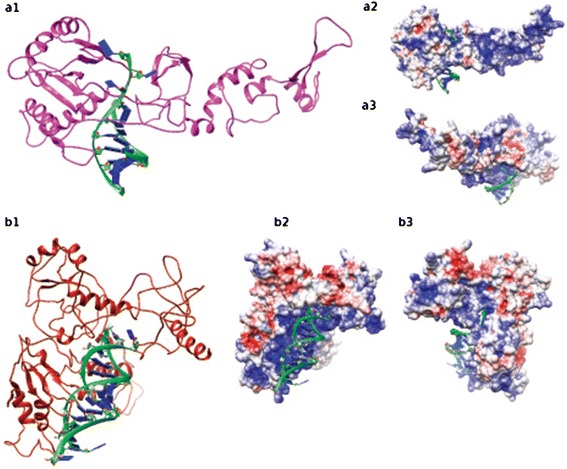
Three dimension theoretical models of Integrase (INT2) of RLC_egAle_1 (a) and Reverse Transcriptase (RT2) (b) of RLC_egBianca_1 proteins complexed with DNA. In 1, protein cartoon representation. In 2 and 3, electrostatic surface in red (-4) acid regions and in blue (4) blue regions rotated in 180°
Quantification of LTR-RTEs
A total of 843 full-length elements from Copia superfamily and 22 from Gypsy superfamily were identified, and copy number per family ranged from 1 to 623 (Table 1). The distribution of each family is distinct among E. grandis chromosomes when compared to gene and repeats density (Additional file 2: Figure S2). RLC_egMax_1 is the most ubiquitous, in accordance with the prevalence of Maximus LTR-RTE families in plants. This family is dispersed along the chromosome arms in both gene-rich and repetitive-rich regions (Additional file 2: Figure S2c) without any specific preference. Full-length RLC_egMax_1 copies correspond to ~1 % of E. grandis genome. RLC_egBianca_1 and RLC_egAngela_2 families presented a similar distribution, preferentially inserted in repeat-rich regions (Additional file 2: Figure S2d,f). RLC_egAngela_1 have only one copy in chromosome 3, also located in a repeat-rich region (Additional file 2: Figure S3e). Both LTR-RTEs from Gypsy superfamily also have an insertional preference over repeat-rich regions (Additional file 2: Figure S2j,k). On the other hand, RLC_egIvana_1 was preferentially inserted in gene-rich regions of chromosomes 2, 3, 7 and 9 (Additional file 2: Figure S2g).
Additionally, we also quantified LTRs and internal regions of families by qPCR in E. urophylla using E. grandis as a calibrator. E. urophylla is among the most commonly used species in the paper industry in Brazil and belongs to the same subgenus of E. grandis [69, 45].
In silico quantification and qPCR had similar results, except for RLC_egAle_2 which qPCR analyses showed more copies than in silico analyses. The proportion of LTRs for four retrotransposons families, RLC_egAle_1, RLC_egMax_1, RLC_egIvana_1 and RLG_Tat_1, is higher in E. urophylla than E. grandis (Fig. 6a). This pattern is different in internal regions, where only two, RLC_egAngela_1 and RLC_egTekay_1, have a higher copy number in E. urophylla, suggesting a diversification in LTR regions. RLC_egAngela_1 and RLC_egAngela_2 families display the most prominent difference in coding regions, increasing significantly in E. urophylla compared to E. grandis (Fig. 6b).
Fig. 6.
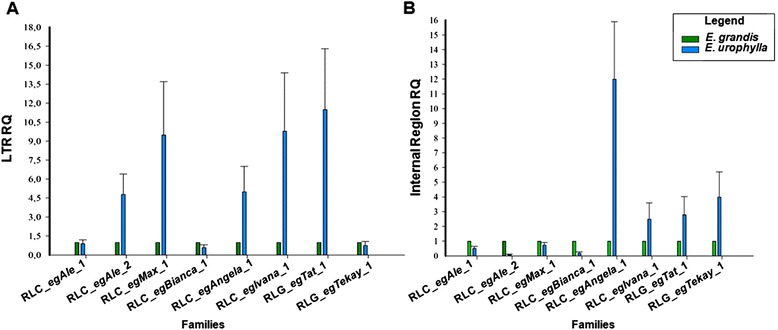
Copy number of Copia and Gypsy LTR-RTE families E. grandis and E. urophylla using qPCR. a RQ of LTRs; b RQ of internal regions
Insertion time of intact elements
We estimated the insertion time of all LTR-RTE families that harbor at least five intact copies. The 12 RLC_egAle_1 copies were inserted into the genome <1.5 Mya, whereas most RLC_egMax_1 copies (98.38 %) were inserted into the genome between 0 – 2.5 Mya (Fig. 5). RLC_egBianca_1 and RLC_egAngela_2 copies have a similar pattern of insertion time where most of the copies were inserted into the genome 0.5 – 4.5 Mya. Approximately one third of the copies from RLC_egBianca_1 (24) and RLC_egAngela_2 (23) insertion has >3.0 Mya, including two copies dated to >7.5 Mya in both families (Fig. 5). The majority of RLC_egIvana_1 copies were inserted in the genome between 0.25 - 2.0 Mya (Fig. 5). The two LTR-RTEs from Gypsy superfamily have distinct patterns: while RLG_egTat_1 have most of these copies inserted 0.5 – 3.5 Mya (Fig. 5), RLG_egTekay_1 has a uniform distribution. We found one copy with two identical LTRs, in RLC_egAngela_2, five identical LTRs in RLC_egMax_1. The diversity (Pi) of LTRs ranged from 0.25 (±0.00379) (RLC_egBianca_1) to 0.35 (±0.17742) (RLC_egAngela_1) (Additional file 1: Table S3) and both Gypsy families have an average diversity of 0.31 (±0.023 from RLG_egTat_1 and ± 0.045 from RLG_egTekay_1).
Fig. 5.
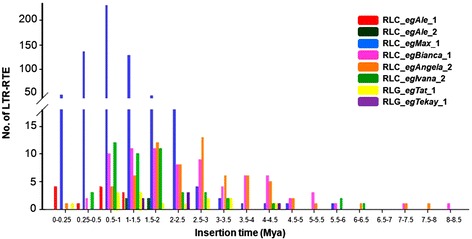
Estimative insertion time of LTR-RTE families in Eucalyptus grandis genome
IRAP and REMAP polymorphisms within Eucalyptus genus
Fifty Eucalyptus spp unrelated individuals were scored by IRAP and REMAP, yielding in total 3096 fragments, among which 700 were polymorphic. IRAP bands ranged from 250 bp to 2.25 kb (Additional file 2: Figure S3) and REMAP fragments ranged from 50 bp to 1 kb, except for primers from RLC_egAle_2 family (Additional file 2: Figure S4). Fifteen single IRAP primers and 23 REMAP primer combinations yielded reliable results. The number of scorable bands per primer in IRAP ranged from 18 (E. urophylla; 2.4 % polymorphic) to 20.8 (E. saligna; 4.6 % polymorphic). For REMAP bands ranged from 11.9 (E. brassiana; 4.3 % polymorphic) to 15.6 (E. urophylla; 2.1 % polymorphic) (Additional file 1: Table S3 and S4).
The genetic relationships of these genotypes were unrevealed using the UPGMA method based on Jaccard (ranging from 0 to 0.92) computed with IRAP and REMAP markers (Fig. 7, Additional file 2: Figure S5). E. brassiana remained an outgroup, and the most related species were E. tereticornis and E. urophylla. E. grandis was the second most external species, after E. brassiana.
Fig. 7.
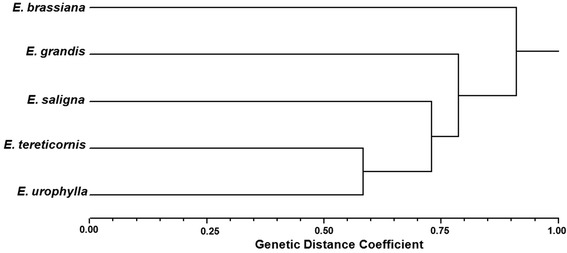
UPGMA dendrogram of five Eucalyptus species using IRAP and REMAP data based on Jaccard distance
Transcriptional activity of LTR retrotransposons families
In order to further characterize the transcriptional profile of LTR-RTE families, we performed BLAST searches against EST of six Eucalyptus species available in the EUCANEXT database [44, 45]. More than 150 ESTs matched the selected LTR-RTE families, and results are shown in Additional file 1: Table S3. RLC_egBianca_1 and RLG_egTat_1 were the most represented families (Additional file 1: Table S3). RLC_egAle_1, RLC_egAle_2, and RLG_egTekay_1 showed similarity with ESTs from three of six analyzed Eucalyptus species (Additional file 1: Table S3), and one EST of E. globulus displayed high similarity RLC_egIvana_1 (Additional file 1: Table S3). RLC_egMax_1 only had similarity with expressed sequences from E. globulus, (Additional file 1: Table S3).
A second approach, RT-qPCR of coding regions, was employed to detect transcriptional levels of LTR-RTE families in three tissues (leaves, stalk and secondary roots) from five Eucalyptus species (E. brassiana, E. grandis, E. saligna, E. tereticornis and E. urophylla) and one hybrid (E. grandis x E. urophylla – termed “E. urograndis” to facilitate discussion). E. grandis secondary roots were also evaluated in osmotic stress imposed by PEG treatment [61] (Fig. 8).
Fig. 8.
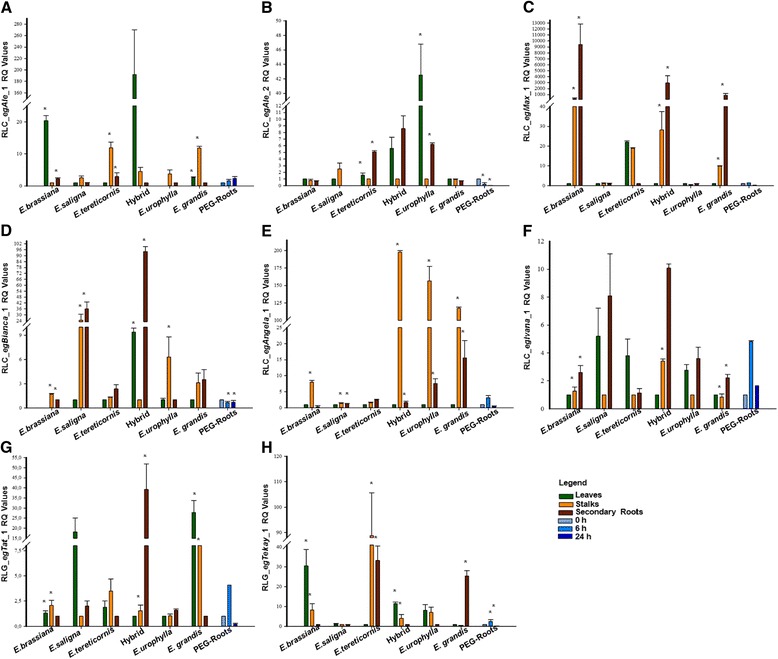
Transcriptional profile of Eucalyptus LTR-RTEs in three organs (detailed in panel "Legend") from five Eucalyptus species and one interspecific hybrid using RT-qPCR. Asterisk indicating standard error (n = 9) in bars (*p ≤ 0.05, ANOVA followed by LSD test)
This analysis expanded and detailed EST analysis, confirming that families have differential transcriptional activity Eucalyptus spp tissues. RLC_egAle_1 highest transcriptional levels were in leaves from “E. urograndis” and E. brassiana, with a remarkable level in stalks of E. tereticornis and E. grandis (Fig. 8a). Regarding the RLC_egAle_2 family, leaves from E. urophylla presented the highest transcriptional activity (Fig. 8b). RLC_egMax_1 family showed a high level of transcriptional activity in secondary roots from E. brassiana and E. grandis, and in “E. urograndis” we observed a notable difference compared to parentals E. grandis and E. urophylla (Fig. 8c). RLC_egBianca_1 family also displayed a higher transcriptional level in secondary roots of “E. urograndis”, and it was the most expressed family in stalks and roots of E. brassiana (Fig. 8d). LTR-RTEs of the Angela lineage (primers could not distinguish families) have a remarkable level in stalks in three Eucalyptus species and “E. urograndis”, and a low transcriptional level in leaves of all Eucalyptus species studied (Fig. 8e). The RLC_egIvana_1 family had a ubiquitous expression level (Fig. 8f).
Gypsy elements showed a distinct transcriptional level according to the family, tissue and Eucalyptus species evaluated. RLG_egTat_1 showed a remarkable expression in leaves of two Eucalyptus species (see Fig. 8g), and in stalks from E. saligna and secondary roots from “E. urograndis” (Fig. 8g). On the other hand, RLG_egTekay _1 family showed the highest transcriptional activity in stalks from E. tereticornis (Fig. 8h).
The transcriptional level of LTR-RTEs in roots submitted to osmotic stress by PEG treatment increased after 6 h in five families (RLC_egAle_1, RLC_egMax_1, RLC_egBianca_1, RLC_egIvana_1, RLG_egTat_1 and RLG_egTekay_1) and decreased after 24 h (Fig. 8). Only in the RLC_egAle_1 family did we observe a higher expression after 24 h (Fig. 8). RLC_egAle_2 and RLC_egBianca_1 families showed a reduction in transcription level following PEG application (Fig. 8).
Discussion
Novel LTR-RTE families in Eucalyptus genus have individual molecular patterns
This study represents a fine-scale analysis of transcriptionally active LTR-RTEs in Eucalyptus species, taking advantage of the recently released E. grandis genome and expanding in silico analyses to a comparative study in terms of copy number, polymorphism insertion and tissue-specific transcriptional pattern.
The majority of the identified LTR-RTE families were from the Copia superfamily, consistent with previous EST analysis of LTR-RTEs in sugarcane [6] and coffee trees [7] but in contrast to wheat [70] and maize [5]. These findings confirm that the superfamily transcriptional preference is variable among plant genomes.
Most families from the same evolutionary lineage have similar size, LTR length and high similarity between PPT and PBS regions with previous multi-scale reports [17, 24]. Three Copia families displayed a spacer region between ORF and LTRs: RLC_egMax_1, with a 1 kb spacer region at 3-UTR. The RLC_egAngela_1 and RLC_egAngela_2 has a spacer region of 5580 pb and 3205 pb at 5-UTR, respectively. Despite the high identity between internal coding domains between Angela families, their LTRs display differences in size and similarity; moreover, folding analyses of the 5’UTR also support that they represent distinct families (Additional file 2: Figure S1). RLC_egAngela_1 has a larger spacer region between LTRs and this region has 99 % identity with an Eucalyptus spp EST (GenBank accession HS066626, data not shown), suggesting that spacer regions are also transcriptionally active. RLC_egAngela_1 and RLC_egAngela_2 have strikingly high similarity in coding regions, but they habour dissimilar LTR and untranslated regions. This feature is particularly common in Copia superfamily, (i.e, BARE1 and 2) [63]. We also discovered that one transcriptionally active LTR-RTE family initially classified as a member of the Gypsy superfamily are in fact caulimoviruses, with 58 complete copies in the E. grandis genome with EST support (data not shown). Previous studies in plants provided similar results [15], and the identification of ESTs matching plant caulimoviruses suggest that these elements may have a significant impact in transcriptome of angiosperms.
Gypsy families can be roughly divided according to the presence of a Chromodomain in the C-terminal of the integrase domain. RLG_egTekay_1 family is a typical Chromoviridae member, closely related to the sugarbeet element Bongo3 [71]. But, differing from Bongo3, a high-copy element spread throughout all sugarbeet chromosomes, only 5 E. grandis chromosomes have full-length RLG_egTekay_1 copies.
Eucalyptus LTR-RTE proteins interact with DNA and RNA
Up to now, few studies address TEs molecular structure [72–74] and here we deliver the first study to theoretically model and stabilize with long MD simulations domains of LTR-RTE proteins in plants. The generated models posses a wide distribution of unordered regions, above all the Copia models (Fig. 4) [72]. Comparison of integrase models (Figs. 3a1 and 4a1 representation colored in pink) clearly show variability between members (rmsd of 7.2 Å), although superimposition of DNA interacting region yields lower value (3.17 Å). The same is observed in reverse transcriptases (Figs. 3b1 and 4b1 representation colored in dark red) with lower values, which shows higher structural conservancy. The high rmsd values are due to the flexibility between cores given by connecting loops and unordered regions, although the cores are similar explaining the conservancy of function. The variance between models corroborates the differences observed in previous sequence analysis, since RLC_egAle_1 and RLC_egBianca_1 are from different families and the RLG_egTekay_1 from a different superfamily. The electrostatic surface of Integrase and Reverse Transcriptase show the complementarities region with RNA/DNA (Figs. 3a3–b2, 4a3–b2). The Chromodomain was not modeled with RNA since its interaction region is not fully understood. This model consists in its general structure, an N-terminal three-stranded anti-parallel β-sheet (β-sandwich) packed against C-terminal α-helix (Fig. 3c1) and it has mostly acid regions. Further studies should be performed to aid transposons domains structural comprehension, such as modeling the whole ORFs from the LTR-RTE.
Most LTR-RTE families are recently inserted in Eucalyptusgrandis genome
Our analysis highlights the fact that even in transcriptionally active LTR-RTEs we could track contrasting insertion peaks. We dated the most part of full-length LTR-RTEs insertions dated <3 Mya (Fig. 5). This characteristic is similar to other plants, such as rice [75], Vitis [76] and tomato [77]. The identification of insertions >3Mya old is difficult in plant genomes [78, 77], because old LTR-RTEs insertions could be removed by recombination processes, as well as mutational events on nucleotide composition.
Six out of 8 analyzed families showed recently inserted copies (<1.0 Mya), with RLC_egAngela_2 and RLC_egMax_1 families having copies with estimated insertion of <0.1 Mya. RLC_egMax_1, which represents ~1 % of E. grandis genome, has a peak of amplification between 0.5 and 1.5 Mya. Most insertions over 4.5 Mya correspond to elements from 2 families: RLC_egBianca_1 and RLC_egAngela_2, suggesting that forces driving RLC_egAngela_2 amplification are evolutionarily old and still in action. This is consistent with analyses in other plants genomes that insertion >6 Mya, such as Populus tricocarpa [79], onion and asparagus [80].
LTR-RTEs in plants can be either localized in gene-rich regions [81, 77] or in repeat-rich regions [82], which also represents the scenario of Eucalyptus retrotransposons here characterized. Although chromosome 3, 5 and 8 concentrated most of annotated full-length copies, Eucalyptus LTR-RTE families had distinct copy distributions, both in genic and repetitive regions. The most part of younger (<3Mya) LTR-RTEs inserted in genic regions, similar to the gene-rich euchromatin distribution from SALIRE family in Beta vulgaris [83] and LTR-RTEs in tomato genome [74]. Few full length copies of RLC_egIvana_1 (5), RLC_egBianca_1 (2), RLC_egAngela_2 (1) and RLC_egMax_1 (9) are inserted near to telomeric regions, suggesting that these families are not directly related to telomeric repeats.
The quantification of LTR-RTE families by qPCR has been performed in several species [84, 53, 16] and we successfully used this strategy to compare amplification of Copia and Gypsy elements in E. grandis and E. urophylla. qPCR quantification did not follow in silico quantification just for RLC_egAle_2 which qPCR analyses showed more copies than in silico analyses. This fact may be due to the E. grandis genotype used in qPCR analyses (Clone GD 33) that is not the same from genome sequencing (BRASUZ1), and defective LTR-RTEs may be overrepresented in E. grandis genotype here sampled.
This approach also helped us to track out the proportion of non-autonomous LTRs and internal regions. E. grandis (1C = 630 Mb) and E. urophylla (1C = 640 Mb) genomes have similar size and diverged <20 Mya [13] enabling the comparison of LTR-RTE families shared by both genomes.
RLC_egAle_1 have similar LTR and internal domain proportions for both genomes, suggesting that this family have not gone through an expansion burst since E. grandis and E. urophylla divergence. RLC_egAle_2, RLC_egMax_1, RLC_egIvana_1 and RLG_egTat_1 families had an increase of LTRs compared to internal domains in E. urophylla, which may indicate the propagation of LTRs, including non-autonomous elements, in this genome and may also reflect a fast substitution processes among internal domains of these families. RLC_egAngela_1 and RLG_egTekay_1 had the opposite profile, with a higher proportion of internal domains. This observation may indicate a higher divergence of LTRs that were not recognized by the primer combination used in our approach and conservation in coding domains, similar to the pattern of the Angela family within the E. grandis genome.
Three LTR-RTEs families, RLC_egAle_1, RLC_egMax_1 and RLG_egTat_1, showed an approximate proportion of two LTRs to each element. These families have most of their copies have young insertions, indicating that they probably did not harboured recombination processes. The qPCR analyses from RLC_egAle_2, RLC_egAngela_1 and RLC_egBianca_1 families showed more copies of internal regions than LTRs, which suggest a loss of LTR and internal region conservation in E. grandis genomes.
IRAP and REMAP markers suggest distinct activities of LTR-RTEs families in Eucalyptus species
IRAP and REMAP markers may contribute to understanding the insertion activity of LTR-RTE families in Eucalyptus species. Most primer combinations were successfully applied in Eucalyptus species, showing the ubiquity character of families among the Eucalyptus genus. E. grandis showed more fragments and polymorphic bands, indicating that LTR-RTEs families had distinct activity after speciation events in this specie. Copia LTR-RTEs studied in four species from Vitis genus had a similar pattern, with polymorphic bands suggesting an amplification burst after speciation events [76].
The average size of REMAP fragments was probably the result of proximity between LTR and SSR regions than LTR-RTEs in tandem insertions. The pattern of REMAP fragments per Eucalyptus species reflect the preferential insertion events of LTR-RTE families in SSR regions with repetition motifs [(CT)10G] and [(AG)10 T], probably sampling LTR-RTEs located in pericentromeric regions, which are gene-poor and enriched in repetitive sequences, especially retrotransposons [85]. The high level of polymorphisms suggests that LTR-RTEs are extensively heterogeneous among Eucalyptus species, as observed in Diospyros sp. and Medicago sativa [24, 86].
This is the first report using IRAP and REMAP markers for genetic diversity in Eucalyptus, and genomic polymorphism suggests differential activity among RTEs within subgenus Symphyomyrtus. Those species occupy the same clade within subgenus Symphyomyrtus [87] but it is important to notice that the separation between those Eucalyptus species was not completely supported by bootstrap analyses. Distribution of Eucalyptus species in dendrogram has some differences comparing to molecular analyses based on DArT markers [87]. E. tereticornis and E. urophylla were the most close in RTE-based tree differently from the close relation usually present between E. grandis and E. urophylla, also observed for E. brassiana and E. tereticornis using other nuclear markers. On the other hand, our analysis shows a small distance between E. saligna and E. grandis, in agreement with a previous report using a nuclear gene [88].
Transcriptional activity of Eucalyptus LTR-RTE families is variable among organs and species
The annotation of ESTs related to LTR-RTEs was an initial assessment of transcriptional activity of these elements in Eucalyptus genomes. RLC_egAle_1 was the family with the largest number of ESTs. RLC_egBianca_1 was the most ubiquitous element, with EST in the six mined Eucalyptus species. Detailed information of ESTs matching LTR-RTEs is available in Supporting Information Additional file 1: Table S6.
RT-qPCR clearly demonstrates that families have differences of transcriptional activity among Eucalyptus spp. tissues and species (Fig. 8). LTR-RTEs copy number have been suggested a cause to transcriptional increase, because more copies inserted in gene-rich euchromatin region [77] and probably near genes. Nevertheless, our data showed that no relation between LTR-RTEs copy number and expression level increase.
The transcriptional activity of families with the highest copy number, RLC_egMax_1, RLC_egBianca_1 and RLC_egIvana_1, was higher in secondary roots when compared to leaves. On the other hand, families with lower genomic copies are highly expressed in stalks and lower expressed in roots, i.e, RLC_egAle_1, RLC_egAle_2 and RLC_egAngela_1.
RLC_egAle_1 and RLC_egAle_2 families, despite their similar structure, have remarkable differences in their transcriptional patterns. The most striking differences can be observed in E. brassiana, E. urophylla and “E. urograndis” leaves, where each element displayed a species-specific transcriptional pattern. LTR-RTEs activation in hybrids was also described in other eudicots, like sunflower [84, 89] and tobacco [90].The activation of LTR-RTEs in hybrids may reflect two issues: an organ-specific deregulation of transcription factors that target LTRs in “urograndis” and/or a specific deregulation of silencing mechanisms regulating TE transcription.
The expression levels from RLC_egAle_1 in roots were lower to other tissues in all Eucalyptus species and “E. urograndis”, RLC_egAle_2 has similar expression characteristic in stalks from Eucalyptus species and “E. urograndis”. Families from the same lineage in sugarcane had distinct transcriptional pattern in leaves and buds [6].
High transcriptional levels in roots were described for several Copia LTR-RTEs families in different plant species, such as citrus [91] and Quercus suber [9].
This is the first work that LTR-RTEs were evaluated in roots submitted to PEG osmotic stress. E. grandis roots submitted to osmotic stress showed variable transcriptional activity. Three families with young insertion (<3 Mya) and with more genomic copies, RLC_egIvana_1, RLC_egAle_1 and RLC_egAle_2, had transcriptional activity modification, except RLC_egMax_1.
The transcriptional activity from RLC_egAle_1, RLC_egAngela_1, RLC_egIvana_1 e RLG_egTekay_1 families had a peak after 6 h of osmotic stress by PEG followed by a decrease in expression level in RLC_egIvana_1 and RLG_egTat_1 families. This observation suggests that both families have their transcription triggered by similar stress conditions, a common feature among TEs [92]. Future functional studies validating the promoter specificity of these LTRs may shed a light on the stress activation of TEs in Eucalyptus.
Conclusions
This study demonstrated that each Copia and Gypsy families in Eucalyptus have a different amplification pattern. Particularly in E. grandis and E. urophylla, that have diverged from a common ancestor ~ 20 Mya ago, we observed lower copy number in most LTR-RTEs at E. urophylla compared to E. grandis. These differences warrant further investigation to determine if recombination, nucleotide divergence or a specific burst of amplification can explain this pattern. Despite conservation to LTR-RTEs between species, IRAP and REMAP markers analyses based on transcriptionally active LTR-RTEs suggest different level of transpositional activity within Eucalyptus genus. This hypothesis is reinforced taking account that transcriptional activity is not the same among Eucalyptus species. Future studies can address if LTR-RTEs are specifically modulated by other stresses beside osmotic shock. Another important issue is to address if Eucalyptus LTR-RTEs families characterized here are in expansion in Eucalyptus genus, or even if they are conserved across other families rather than Myrtaceae, which may indicate horizontal transfer and/or purifying selection.
Availability of supporting data
The data sets supporting the results of this article are available in the Dryad repository, doi:10.5061/dryad.h2t57.
Acknowledgments
Authors thank Drs. Ivan de Godoy Maia and Juliana Pereira Bravo by their assistance in transcriptional analyses and cession of plant material; to Alan P. Lorenzetti, by his help in LTR-RTE distribution analysis, and to Izabel Gava and Shinitiro Oda, who kindly provided access to plant material at Suzano Papel e Celulose. This work was supported by a CNPq grant (474123/2010-3). HSM was also supported by fellowships from CAPES and CNPq. CLM is a research fellow from CNPq.
Abbreviations
- TEs
Transposable elements
- LTR-RTEs
LTR retrotransposons
- ORFs
Open reading frames
- Mb
Megabases
- NCBI
National Center for Biotechnology Information
- IRAP
Inter-Retrotransposon Amplified Polymorphism
- REMAP
Retrotransposon-Microsatellite Ampliflied Polymorphism
- TSDs
Target site duplications
- MUSTER
MUlti-Sources ThreadER
- MD
Molecular dynamics
- GROMACS
Groningen Machine for Chemical Simulation
- Rmsd
Root mean square deviation
- UPGMA
Unweighted pair group method with arithmetic mean
- qPCR
Quantitative real time PCR
- INT1
RLG_egTekay_1 integrase
- CD1
RLG_egTekay_1 chromodomain
- RT1
RLG_egTekay_1 reverse transcriptase
- INT2
RLC_egAle_1 integrase
- RT2
reverse transcriptase of RLC_egBianca_1
Additional files
Table S1. Primers Sequences using different techniques from LTR-RTEs families. Table S2. PBS and PPT sequences from the LTR-RTE families characterized in E. grandis genome. Table S3. Diversity (Pi) between LTRs of retrotransposon families in Eucalyptus grandis genome. We used data from intact complete copies. Table S4. Total fragments and polymorphic bands in IRAP primers of five Eucalyptus species. Table S5. Total fragments and polymorphic bands in REMAP combinations of five Eucalyptus species. Table S6. Expressed sequence tags (EST) matched to complete LTR-RTEs elements.
Figure S1. RNA folding of RLC_egAngela_1 and RLC_egAngela_2 5’spacer region using RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) with minimum free energy (MFE). The (a) RLC_egAngela_1 region 185-561; (b) RLC_egAngela_2 region 404-648. Figure S2. Distribution of full-length Copia and Gypsy elements across 11 chromosomes from E. grandis. Data was plotted using Circus. Tracks: (a) gene density (number per Mb, range 6 - 131) (Myburg et al. 2014); (b) Repetitive coverage (22 – 88% per Mb) (Myburg et al. 2014); (c) RLC_egMax_1 density (number per Mb, range 0 – 8); (d) RLC_egBianca_1 density (number per Mb, range 0 – 3); (e) RLC_egAngela_1 density (number per Mb, range 0 – 1); (f) RLC_egAngela_2 density (number per Mb, range 0 – 3); (g) RLC_egIvana_1 density (number per Mb, range 0 – 2); (h) RLC_egAle_1 density (number per Mb, range 0 – 2 p); (i) RLC_egAle_2 density (number per Mb, range 0 – 1); (j) RLG_egTat_1 density (number per Mb, range 0 – 2); (k) RLG_egTekay_1 density (number per Mb, range 0 – 2). Figure S3. IRAP analysis for a set of four Eucalyptus species using primers EgBiancaLTR-3 (a) and EgTatLTR-5 (b). L: 1 kb molecular weight marker: Gene Ruler DNA Ladder Mix (Fermentas). Figure S4. REMAP analysis for a set of three Eucalyptus species with primer Micro1 and EgAle2LTR-3 (a) or EgTatLTR-5 (b). L: 1kb molecular weight marker: Gene Ruler DNA Ladder Mix (Fermentas). Figure S5. UPGMA dendrogram of individuals from five Eucalyptus species using IRAP and REMAP data based on Jaccard distance.
Footnotes
Helena Sanches Marcon and Douglas Silva Domingues contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
The study was conceived by HSM and DSD, and co-ordinated by CLM, who participated in its design and helped to draft the manuscript. HSM and DSD carried out the molecular genetic and phylogenetic studies. JCS, helped by HSM and DSD, did the bioinformatic analyses. RJB, FFM and MRMF carried out modelling studies. HSM, DSD and RJB wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Helena Sanches Marcon, Email: helenasmarcon@gmail.com.
Douglas Silva Domingues, Email: doug@rc.unesp.br.
Juliana Costa Silva, Email: julianacostasilvati@gmail.com.
Rafael Junqueira Borges, Email: rafaeljborges@hotmail.com.
Fábio Filippi Matioli, Email: fabiofima@ibb.unesp.br.
Marcos Roberto de Mattos Fontes, Email: fontes@ibb.unesp.br.
Celso Luis Marino, Phone: +55 14 38800365, Email: clmarino@ibb.unesp.br.
References
- 1.Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 2.Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–5. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 3.Hawkins JS, Kim HR, Nason JD, Wing RA, Wendel JF. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006;16:1252–1261. doi: 10.1101/gr.5282906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piegu B, Guyot R, Picault N, Roulin A, Saniyal A, Kim H, et al. Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006;16:1262–9. doi: 10.1101/gr.5290206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vicient CM. Transcriptional activity of transposable elements in maize. BMC Genomics. 2010;11:601. doi: 10.1186/1471-2164-11-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Domingues DS, Cruz GMK, Metcalfe CJ, Nogueira FTS, Vicentini R, Alves CS, et al. Analysis of plant LTR-retrotransposons at the fine scale family level reveals individual molecular patterns. BMC Genomics. 2012;13:137. doi: 10.1186/1471-2164-13-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopes FR, Jjingo D, Da Silva CRM, Andrade AC, Marraccini P, Teixeira JB, et al. Transcriptional activity, chromosomal distribution and expression effects of transposable elements in Coffea genomes. PLoS One. 2013;8:175–191. doi: 10.1371/journal.pone.0078931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lisch D. Epigenetic regulation of transposable elements in plants. Annu Rev Plant Physiol Plant Mol Biol. 2009;60:43–66. doi: 10.1146/annurev.arplant.59.032607.092744. [DOI] [PubMed] [Google Scholar]
- 9.Rocheta M, Carvalho L, Viegas W, Morais-Cecílio L. Corky, a Gypsy-like retrotransposon is differentially transcribed in Quercus suber tissues. BMC Res. 2012;5:432. doi: 10.1186/1756-0500-5-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myburg AA, Potts BM, Marques CM, Kirst M, Gion JM, Grattapaglia D, et al. Eucalyptus. In: Chittaranjan K, et al., editors. Genome mapping and molecular breeding in plants. New York, NY, USA: Springer; 2007. pp. 115–160. [Google Scholar]
- 11.Harwood C. New introductions – doing it right. In: Walker J, editor. Developing a eucalypt resource: learning from Australia and elsewhere: University of Canterbury. Christchurch, New Zealand: Wood Technology Research Centre; 2011. pp. 43–54. [Google Scholar]
- 12.Assis, TF. Melhoramenro genético de Eucalyptus: desafios e perspectivas. 3rd Brazilian Forestry Meeting, 2014.
- 13.Myburg AA, Grattapaglia D, Tuskan GA, Hellsten U, Hayes RD, Grimwood J, et al. The genome of Eucalyptus grandis. Nature. 2014;5410:356–362. doi: 10.1038/nature13308. [DOI] [PubMed] [Google Scholar]
- 14.Bacci Júnior M, Soares RBS, Tajara E, Ambar G, Fischer CN, Guilherme IR, et al. Identification and frequency of transposable elements in Eucalyptus. Gen Mol Biol. 2005;28:634–9. doi: 10.1590/S1415-47572005000400019. [DOI] [Google Scholar]
- 15.Piednoël M, Carrete-Veja G, Renners S. Characterization of the LTR retrotransposon repertoire of a plant clade of six diploid and one tetraploid species. Plant J. 2013;75:699–709. doi: 10.1111/tpj.12233. [DOI] [PubMed] [Google Scholar]
- 16.Yaakov B, Ben-David S, Kashkush K. Genome-wide analysis of stowaway-like MITEs in wheat reveals high sequence conservation, gene association, and genomic diversification. Plant Physiol. 2013;161:486–496. doi: 10.1104/pp.112.204404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wicker T, Keller B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 2007;17:1072–1081. doi: 10.1101/gr.6214107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lloréns C, Muñoz-Pomer A, Bernad L, Botella H, Moya A. Network dynamics of eukaryotic LTR retroelements beyond phylogenetic trees. Biol Direct. 2009;4:41. doi: 10.1186/1745-6150-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rossi M, Araujo PG, Van Sluys MA. Survey of transposable elements in sugarcane expressed sequence tags (ESTs) Gen Mol Biol. 2001;24:147–154. doi: 10.1590/S1415-47572001000100020. [DOI] [Google Scholar]
- 20.Kohany O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics. 2006;7:474. doi: 10.1186/1471-2105-7-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:265–8. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mccarthy EM, Mcdonald JF. LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics. 2003;19:362–7. doi: 10.1093/bioinformatics/btf878. [DOI] [PubMed] [Google Scholar]
- 23.Solovyev VV. Statistical approaches in eukaryotic gene prediction. In: Balding D, Cannings C, Bishop M, editors. Handbook of statistical genetics. London.: Wiley-Interscience; 2007. [Google Scholar]
- 24.Du J, Tian Z, Hans CS, Laten HM, Cannon SB, Jackson SA, et al. Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: insights from genome-wide analysis and multi-specific comparison. Plant J. 2010;63:584–598. doi: 10.1111/j.1365-313X.2010.04263.x. [DOI] [PubMed] [Google Scholar]
- 25.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol. 2013;30:2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Söding J. Protein homology detection by HMM-HMM comparison. Bioinformatics (Oxford, England) 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 28.Wu S, Zhang Y. MUSTER: Improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins. 2008;72:547–556. doi: 10.1002/prot.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- 30.Lovell SC, Davis IW, Arendall WB, de Bakker PIW, Word JM, Prisant MG, et al. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 31.Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:407–410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theor Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 33.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 34.Berendsen HJC, Postma JPM, Gunsteren WF, Hermans J. Interaction Models for Water in Relation to Protein Hydration. In: Pullman B. Editor. The Jerusalem Symposia on Quantum Chemistry and Biochemistry. Intermolecular Forces. Springer Netherlands. 1981; p. 331-342.
- 35.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 36.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J Chem Theor Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 38.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: An information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.San Miguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL. The paleontology of intergene retrotransposons of maize. Nat Genet. 1998;20:43–5. doi: 10.1038/1695. [DOI] [PubMed] [Google Scholar]
- 40.Kimura M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 41.Koch MA, Haubold B, Mitchell-Olds T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae) Mol Biol Evol. 2000;17:1483–1498. doi: 10.1093/oxfordjournals.molbev.a026248. [DOI] [PubMed] [Google Scholar]
- 42.Moisy C, Schulman AH, Kalendar R, Buchmann JP, Pelsy F. The Tvv1 retrotransposon family is conserved between plant genomes separated by over 100 million years. Theor Appl Genet. 2014;127:1223–1235. doi: 10.1007/s00122-014-2293-z. [DOI] [PubMed] [Google Scholar]
- 43.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 44.Nascimento LC, Lepikson JN, Salazar MM, Camargo ELO, Marques WL, Gonçalves DC, et al. An integrated database of Eucalyptus spp. genome Project. BMC Proceedings. 2011;5:170. doi: 10.1186/1753-6561-5-S7-P170. [DOI] [Google Scholar]
- 45.Salazar MM, Nascimento LC, Camargo ELO, Gonçalves DC, Lepikson Neto J, Marques WL, et al. Xylem transcription profiles indicate potential metabolic responses for economically relevant characteristics of Eucalyptus species. BMC Genomics. 2013;14:201. doi: 10.1186/1471-2164-14-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferreira ME, Grattapaglia D. Introdução ao uso de marcadores moleculares em análises genéticas. 3. Embrapa-Cenargen: Brasília; 1998. [Google Scholar]
- 47.Korimbocus J, Danks C, Boonham N, Barker I, Coates D. Improved detection of sugarcane leaf virus using a real-time fluorescent reverse-transcription polymerase chain reaction assay. Phytopathology. 2002;90:448–453. [Google Scholar]
- 48.Smýkal P. Development of an efficient retrotransposon-based fingerprinting method for rapid pea variety identification. J Appl Genet. 2006;47:221–230. doi: 10.1007/BF03194627. [DOI] [PubMed] [Google Scholar]
- 49.Kalendar R, Grob T, Regina M, Suoniemi A, Schulman AH. IRAP and REMAP: two new retrotransposon-based DNA fingerprinting techniques. Theor Appl Gen. 1999;98:704–711. doi: 10.1007/s001220051124. [DOI] [Google Scholar]
- 50.Rohlf FJ. NTSYS-pc: numerical taxonomy and multivariate analysis system. Setauket, NY: Exeter Software; 2000. [Google Scholar]
- 51.Fan F, Cui B, Zhang T, Ding G, Wen X. LTR-retrotransposon activation, IRAP marker development and its potential in genetic diversity assessment of masson pine (Pinus massoniana) Tree Gen Genom. 2013;10:213–222. doi: 10.1007/s11295-013-0677-x. [DOI] [Google Scholar]
- 52.Kraitshtein Z, Yaakov B, Khasdan V, Kashkush K. Genetic and epigenetic dynamics of a retrotransposon after allopolyploidization of wheat. Genetics. 2010;186:801–812. doi: 10.1534/genetics.110.120790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baruch O, Kashkush K. Analysis of copy-number variation, insertional polymorphism, and methylation status of the tiniest class I (TRIM) and class II (MITE) transposable element families in various rice strains. Plant Cell Rep. 2012;31:885–893. doi: 10.1007/s00299-011-1209-5. [DOI] [PubMed] [Google Scholar]
- 54.Navarathna DHMLP, Das A, Morschhäuser J, Nickerson KW, Roberts DD. Dur3 is the major urea transporter in Candida albicans and is co-regulated with the urea amidolyase Dur1,2. Microbiology. 2011;157:270–279. doi: 10.1099/mic.0.045005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Michele R, Loque D, Lalonde S, Frommer WB. Ammonium and urea transporter inventory of the Selaginella and Physcomitrella genomes. Frontiers in Plant Science. 2012;3:62. doi: 10.3389/fpls.2012.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rodrigues MR, Bravo JP, Sassaki FT, Severino FE, Maia IG. The tonoplast intrinsic aquaporin (TIP) subfamily of Eucalyptus grandis: Characterization of EgTIP2, a root-specific and osmotic stress-responsive gene. Plant Sci. 2013;213:106–113. doi: 10.1016/j.plantsci.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 57.Cassan-Wang H, Soler M, Yu H, Camargo ELO, Carocha V, Ladouce N, et al. Reference Genes for High-Throughput Quantitative RT-PCR Analysis of Gene Expression in Organs and Tissues of Eucalyptus Grown in Various Environmental Conditions. Plant and Cell Physiol. 2012;53:2101–2116. doi: 10.1093/pcp/pcs152. [DOI] [PubMed] [Google Scholar]
- 58.Vandesompele J, Preter K, Pattyn F, Poppe B, Roy NV, Paepe AD, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:34.1–34.11. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sinha DK, Smith CM. Selection of Reference Genes for Expression Analysis in Diuraphis noxia (Hemiptera: Aphididae) Fed on Resistant and Susceptible Wheat Plants. Sci Rep. 2014;4:5059. doi: 10.1038/srep05059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruijter JM, Ramakers C, Hoogaars WMH, Karlen Y, Bakker O, Van Den Hoff MJB, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37:1–12. doi: 10.1093/nar/gkp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 62.Silva FAS, Azevedo CAV. Principal components analysis in the software assistat-statistical attendance. In: World Congress on Computers in Agriculture, 7. American Society of Agricultural and Biological Engineers, Reno-NV-USA. 2009.
- 63.Tanskanen JA, Sabot F, Vicient C, Schulman AH. Life without GAG: The BARE-2 retrotransposon as a parasite's parasite. Gene. 2007;390:166–174. doi: 10.1016/j.gene.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 64.Ishida M, Shimojo H, Hayashi A, Kawaguchi R, Ohtani Y, Uegaki K, et al. Intrinsic nucleic acid-binding activity of Chp1 chromodomain is required for heterochromatic gene silencing. Mol Cell. 2012;47:228–241. doi: 10.1016/j.molcel.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 65.Hare S, Vos AM, Clayton RF, Thuring JW, Cummings MD, Cherepanov P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc Natl Acad Sci U S A. 2010;107:20057–20062. doi: 10.1073/pnas.1010246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li J, Li Z, Ruan J, Xu C, Tong Y, Pan PW, et al. Structural basis for specific binding of human MPP8 chromodomain to histone H3 methylated at lysine 9. PLoS One. 2011;6 doi: 10.1371/journal.pone.0025104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nowak E, Potrzebowski W, Konarev PV, Rausch JW, Bona MK, Svergun DI, et al. Structural analysis of monomeric retroviral reverse transcriptase in complex with an RNA/DNA hybrid. Nucleic Acids Res. 2013;41:3874–3887. doi: 10.1093/nar/gkt053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nowak E, Miller JT, Bona MK, Studnicka J, Szczepanowski RH, Jurkowski J, et al. Ty3 reverse transcriptase complexed with an RNA-DNA hybrid shows structural and functional asymmetry. Nature Structural Mol Biol. 2014;21:389–396. doi: 10.1038/nsmb.2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kullan ARK, van Dyk MM, Hefer CA, Jones N, Kanzler A, Myburg AA. Genetic dissection of growth, wood basic density and gene expression in interspecific backcrosses of Eucalyptus grandis and E. urophylla. BMC Genet. 2012;13:60–72. doi: 10.1186/1471-2156-13-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Echenique V, Stamova B, Wolters P, Lazo G, Carollo V, Dubcovsky J. Frequencies of Ty1-copia and Ty3-gypsy retroelements within the Triticeae EST databases. Theor Appl Genet. 2002;104:840–4. doi: 10.1007/s00122-001-0849-1. [DOI] [PubMed] [Google Scholar]
- 71.Weber B, Heitkam T, Holtgräwe D, Weisshaar B, Minoche AE, Dohm JC, et al. Highly diverse chromoviruses of Beta vulgaris are classified by chromodomains and chromosomal integration. Mobile DNA. 2013;4:8. doi: 10.1186/1759-8753-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Novikova O. Chromodomains and LTR retrotransposons in plants. Commun Integ Biol. 2009;2:158–162. doi: 10.4161/cib.7702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gao D, Jimenez-Lopez JC, Iwata A, Gill N, Jackson SA. Functional and Structural Divergence of an Unusual LTR Retrotransposon Family in Plants. PLoS One. 2012;7 doi: 10.1371/journal.pone.0048595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abrusán G, Zhang Y, Szilágyi A. Structure prediction and analysis of DNA transposon and LINE retrotransposon proteins. J Biol Chem. 2013;288:16127–16138. doi: 10.1074/jbc.M113.451500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ma J, Bennetzen JL. Recombination, rearrangement, reshuffling, and divergence in a centromeric region of rice. Proc Natl Acad Sci. 2006;103:383–8. doi: 10.1073/pnas.0509810102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moisy C, Garrison KE, Meredith CP, Pelsy F. Characterization of ten novel Ty1/Copia-like retrotransposon families of the grapevine genome. BMC Genomics. 2008;9:469. doi: 10.1186/1471-2164-9-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu Y, Du J. Young but not relatively old retrotransposons are preferentially located in gene-rich euchromatic regions in tomato (Solanum lycopersicum) plants. Plant J. 2014;80(4):582–591. doi: 10.1111/tpj.12656. [DOI] [PubMed] [Google Scholar]
- 78.Bennetzen JL, Ma J, Devos KM. Mechanisms of recent genome size variation in flowering plants. Ann Bot. 2005;95(1):127–132. [DOI] [PMC free article] [PubMed]
- 79.Cossu RM, Buti M, Giordani T, Natali L, Cavallini A. A computational study of the dynamics of LTR retrotransposons in the Populus trichocarpa genome. Tree Gen Genomes. 2011;8:61–75. doi: 10.1007/s11295-011-0421-3. [DOI] [Google Scholar]
- 80.Vitte C, Estep MC, Leebens-Mack J, Bennetzen JL. Young, intact and nested retrotransposons are abundant in the onion and asparagus genomes. Ann Bot. 2013 doi: 10.1093/aob/mct155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gao D, Chen J, Chen M, Meyers BC, Jackson S. A Highly Conserved, Small LTR Retrotransposon that Preferentially Targets Genes in Grass Genomes. PLoS One. 2012;7 doi: 10.1371/journal.pone.0032010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tsukahara S, Kawabe A, Kobayashi A, Ito T, Aizu T, Shin T, et al. Centromere-targeted de novo integrations of an LTR retrotransposon of Arabidopsis lyrata. Genes Dev. 2012;26:705–713. doi: 10.1101/gad.183871.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weber B, Wenke T, Frömmel U, Schmidt T, Heitkam T. The Ty1-copia families SALIRE and Cotzilla populating the Beta vulgaris genome show remarkable differences in abundance, chromosomal distribution, and age. Chromosome Res. 2010;18:247–263. doi: 10.1007/s10577-009-9104-4. [DOI] [PubMed] [Google Scholar]
- 84.Kawakami T, Strakosh SC, Zhen Y, Ungerer MC. Different scales of Ty1/Copia-like retrotransposon proliferation in the genomes of three diploid hybrid sunflower species. Heredity. 2010;104:341–350. doi: 10.1038/hdy.2009.182. [DOI] [PubMed] [Google Scholar]
- 85.Zhao M, Ma J. Co-evolution of plant LTR-retrotransposons and their host genomes. Protein Cell. 2013;4:493–501. doi: 10.1007/s13238-013-3037-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mandoulakani BA, Piri Y, Darvishzadeh R, Bernoosi I, Jafari M. Retroelement Insertional Polymorphism and Genetic Diversity in Medicago sativa Populations Revealed by IRAP and REMAP Markers. Plant Mol Biol Reporter. 2012;30:286–296. doi: 10.1007/s11105-011-0338-x. [DOI] [Google Scholar]
- 87.Steane DA, Nicolle D, Sansaloni CP, Petroli CD, Carling J, Kilian A, et al. Population genetic analysis and phylogeny reconstruction in Eucalyptus (Myrtaceae) using high-throughput, genome-wide genotyping. Mol Phylogenet Evol. 2011;59:206–224. doi: 10.1016/j.ympev.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 88.Poke FS, Martin DP, Steane DA, Vaillancourt RE, Reid JB. The impact of intragenic recombination on phylogenetic reconstruction at the sectional level in Eucalyptus when using a single copy nuclear gene (cinnamoyl CoA reductase) Mol Phylogenet Evol. 2006;39:160–170. doi: 10.1016/j.ympev.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 89.Ungerer MC, Kawakami T. Transcriptional Dynamics of LTR Retrotransposons in Early Generation and Ancient Sunflower Hybrids. Genome Biol Evol. 2013;5(2):329–337. doi: 10.1093/gbe/evt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Petit M, Guidat C, Daniel J, Denis E, Montoriol E, Bui QT, et al. Mobilization of retrotransposons in synthetic allotetraploid tobacco. New Phytologist. 2010;186:135–147. doi: 10.1111/j.1469-8137.2009.03140.x. [DOI] [PubMed] [Google Scholar]
- 91.Rico-Cabanas L, Martínez-Izquierdo JA. CIRE1, a novel ranscriptionally active Ty1-copia retrotransposon from Citrus sinensis. Mol Genet Genomics. 2007;227:365–377. doi: 10.1007/s00438-006-0200-2. [DOI] [PubMed] [Google Scholar]
- 92.Grandbastien MA, Audeon C, Bonnivard E, Casacuberta JM, Chalhoub B, Costa APP, et al. Stress activation and genomic impact of Tnt1 retrotransposons in Solanaceae. Cytogen Genome Res. 2005;110:1–4. doi: 10.1159/000084957. [DOI] [PubMed] [Google Scholar]