

Abstract
A debilitating complication of breast cancer is the metastatic spread of tumor cells to the leptomeninges or cerebrospinal fluid (CSF). Patients diagnosed with this aggressive clinical syndrome, known as leptomeningeal carcinomatosis, have very poor prognosis. Despite improvements in detecting cerebrospinal fluid tumor cells (CSFTCs), information regarding their molecular biology is extremely limited. In our recent work, we utilized a protocol previously used for circulating tumor cell isolation to purify tumor cells from the CSF. We then performed genomic characterization of CSFTCs as well as archival tumors from the same patient. Here, we describe the microarray data and quality controls associated with our study published in the Cancer Research journal in 2013 [1]. We also provide an R script containing code for quality control of microarray data and assessment of copy number calls. The microarray data has been deposited into Gene Expression Omnibus under accession # GSE46068.
| Specifications | |
|---|---|
| Organism/cell line/tissue | Tumor cells isolated from the cerebrospinal fluid of breast cancer patients diagnosed with leptomeningeal carcinomatosis |
| Sex | Female |
| Sequencer or array type | 2.4 K Bacterial Artificial Chromosome (BAC) Array |
| Data format | Raw data: Sproc; normalized data: TXT |
| Experimental features | Array comparative genomic hybridization analysis was performed on amplified tumor DNA versus normal male genomic reference |
| Consent | All patients gave written informed consent under a protocol approved by the UCSF Institutional Review Board |
| Sample source location | University of California San Francisco |
Direct link to deposited data
Deposited data can be found here http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE46068.
Experimental design, materials and methods
Patient and clinical information
Fifteen (15) metastatic breast cancer patients who were diagnosed with leptomeningeal carcinomatosis by standard cytology or by imaging were enrolled in this study. Clinical information was obtained from the patients' medical records. A majority of the patients were estrogen receptor (ER) positive (73%). Almost half were HER2 positive (47%) while two cases were triple-negative (13%). Additionally, two-thirds had concurrent brain metastasis (67%).
Isolation of CSFTCs
Approximately 4 to 10 mL of CSF samples were obtained via lumbar puncture or via an Ommaya reservoir. Tumor cells were isolated from the CSF samples via a two-step process involving immunomagnetic enrichment followed by fluorescence-activated cell sorting or IE/FACS [2]. Briefly, samples were first enriched for tumor cells using a magnetic capture method involving iron particles coated with monoclonal antibodies to the epithelial cell adhesion marker or EPCAM. Tumor cells were further purified using FACS analysis. During cell sorting, events that were positive for nuclear and EPCAM stains but were negative for CD45 (leukocyte-specific marker) were considered CSFTCs.
Primary and metastatic tumor samples
In a subset of patients (6 of 15), archival formalin-fixed paraffin embedded (FFPE) primary tumors, and in some cases loco-regional or distant metastasis including circulating tumor cells from blood, were available and processed, as previously described [3]. Briefly, whole cell lysates were prepared from microdissected areas containing 70% tumors. DNA from lysates was processed in parallel with matched CSFTCs, as outlined in the next section.
Whole genome amplification and array comparative genomic hybridization
Array comparative genomic hybridization (ACGH) analysis usually requires DNA input that is roughly equivalent to genomic material from several thousands of cells. Since CSFTCs are rare, we can only isolate small pools of CSFTCs. The few hundred picograms of genomic material from these few cells require whole genome amplification (WGA) prior to downstream molecular analysis. To reduce the likelihood of detecting false positives (due to amplification bias) when comparing CSFTCs versus matched archival tumors, we subjected both sets of tumors to the same WGA method [4]. Samples from 2 of the 15 patients failed WGA product quality testing [4] and were excluded from further analysis.
Amplified tumor DNA samples were then subjected to ACGH analysis using a bacterial artificial chromosome (BAC) array containing 2464 clones printed in triplicate [5]. The BAC arrays were printed at the UCSF Helen Diller Family Comprehensive Cancer Center Array Core. The ACGH experimental protocol has been previously described in detail [4]. Briefly, the tumor (test sample) and reference DNA samples were differentially labeled with Cy3 and Cy5 dyes, respectively, and co-hybridized to a BAC array. A sex-mismatched (i.e., female vs. male) hybridization was used as an internal control to detect a copy number gain in X- and loss of Y-chromosomes in the female test sample. Post-hybridization imaging data and analysis of the BAC array were done as previously described [6]. The intensity values were used to calculate Cy3/Cy5 ratios using the UCSF Spot Program. An in-house R package ‘Spot Correction’ was also used to remove systematic variations of unknown origin across the array [7], including a correction that is based on the GC content of the BAC clones [4].
Microarray data processing
The aCGH data was processed using the custom program SPROC [8] in order to automatically filter out data points with low DAPI intensity, low correlation between Cy3 and Cy5 within each spot, and low reference/DAPI signal intensity. Clones whose ratios that were derived from only one of the triplicate spots or with a triplicate log2 SD > 0.2 were set as “missing”. The clones were mapped to the May 2004 freeze of the human DNA sequence.
Quality control of aCGH data
The median absolute deviation (MAD) estimates (see below) were used as a measure of the quality of the microarray data. Array data with a MAD estimate < 0.25 was considered “good quality”. A histogram of MAD estimates showed that all samples passed the MAD threshold (Fig. 1). Visual inspection of each aCGH profile was also performed to confirm results. The microarray and sample annotation data for 30 samples from 13 patients were deposited in Gene expression Omnibus under accession number GSE46068.
Fig. 1.
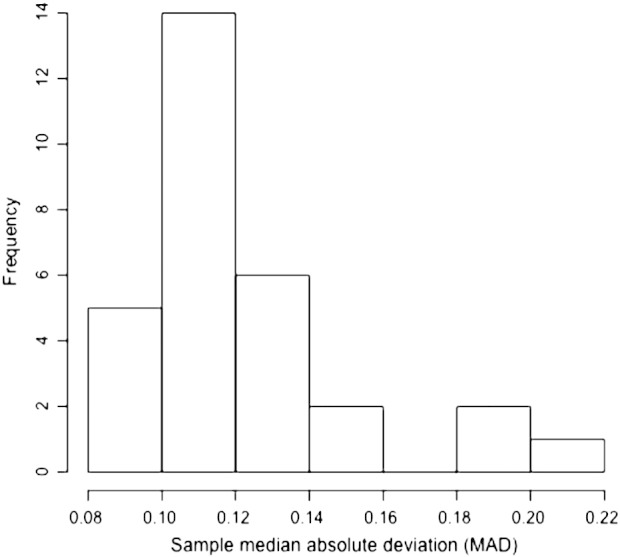
Quality control of microarray data. The median absolute deviation (MAD) estimates were used as a measure of array data quality. The plot shows the frequency distribution of MAD estimates for microarray data generated from 30 cerebrospinal fluid tumor cell (CSFTC)-samples collected from 13 metastatic breast cancer patients diagnosed with leptomeningeal carcinomatosis. All the samples were considered evaluable since the MAD estimates were < 0.25, which is the threshold chosen for good quality array data.
MAD and copy number calls
The microarray data was subjected to circular binary segmentation (CBS) [9] as implemented in the DNAcopy package from Bioconductor [10]. The algorithm translates intensity measurements into regions of equal copy number to make gain/loss/amplification calls (see Supplementary Information for R code). The median absolute deviation (MAD), scaled by the factor 1.4826, of the difference between the observed and segmented values of the autosomal clones was used to estimate sample-specific experimental variation. For copy number calls in each sample, a segment was declared to be gained or lost if the average log2 ratio was at least twice the sample MAD away from the median segmented value of the autosomal clones.
Basic analysis
Genomic alterations
To quantitate the extent of the genomic instability in each sample, we calculated the fraction of genome altered (FGA; i.e., the fraction of genome lost and gained), as previously described [11]. Briefly, the FGA was calculated by assigning each clone a distance equal to the sum of one half of the distance between its own center and that of its neighboring clones or to the end of the chromosome for the clones with only one neighbor.
Next, we compared the FGA between CSFTCs and matched primary tumors from patients 4011, 4015, 4037, 4038, 4039, and CSF6. Interestingly, the FGA in CSFTCs was significantly higher as compared to that of the matched primary tumors (lost: 11% vs. 4%; gained: 12% vs. 10% respectively) (p = 0.0277 sign test) [1] (Fig. 2).
Fig. 2.

Fraction of genome altered (FGA) in cerebrospinal fluid tumor cells (CSFTC) and corresponding primary tumors (PT). The plot shows the medians for the fraction of genome lost and fraction of genome gained for 6 pairs of matched CSFTCs and PT. The sum of the fraction of genome lost and gained is equal to the FGA.
Copy number analysis using Nexus
We also analyzed the CSFTC ACGH microarray data from 13 patients using Nexus 6.1 software (Biodiscovery) [3]. To determine chromosome gains and losses, we chose 0.20 and − 0.20, and 0.6 and − 0.6, as the thresholds of log2 ratio values for single copy number gains and losses, and high-level number gains and homozygous deletions, respectively. Using the rank segmentation algorithm, copy number was estimated in each sample, with the significance threshold set at p-value < 0.001. We considered regions of gains and losses that are present in ≥ 50% of the 13 samples as recurrent. We showed that CSFTCs exhibited a wide array of alterations, including frequent gains in 1q and 8q and loss in 8p and 16q [1] (Fig. 3). These aberrations are also frequently found in primary breast cancers [12], [13].
Fig. 3.

Copy number alterations in cerebrospinal fluid tumor cells (CSFTCs). An ideogram showing cumulative genomic aberrations in CSFTCs from 13 metastatic breast cancer patients diagnosed with leptomeningeal carcinomatosis. Chromosome regions with gains and losses are depicted in blue and red, respectively. Analysis and visualization was performed in Nexus 6.1. The observed gain and loss of X- and Y-chromosomes, respectively, are a result of internal controls performed in each sample using sex-mismatch co-hybridization (i.e., female test versus male reference).
Discussion
We describe, to our knowledge, the first genome-wide ACGH data on tumor cells isolated from the CSF (i.e., CSFTCs). We also provide, in some patients, the ACGH data on corresponding primary and/or metastatic tumors. We showed that the data are of high quality as indicated by low MAD estimates. In addition, the genomic aberrations we found in CSFTCs were similar to those frequently observed in primary breast cancers. Furthermore, in the original paper [1], we demonstrated the clonal relationship of CSFTCs and their corresponding primary tumors. Interestingly, we found more copy number alterations in CSFTCs as compared to the latter, suggesting the acquisition of additional aberrations in CSFTCs or that less normal DNA contamination was present in our CSFTC samples. Finally, our genome-wide copy number analysis was performed using BAC arrays with a 1.2 Mb resolution. Therefore, further molecular studies on CSFTCs using oligoarrays and next generation sequencing are needed to facilitate further interrogation of the CSFTC genome.
Conflict of interest
J.W. Park has received commercial research grant support from Veridex LLC. No potential conflicts of interest were disclosed by the other authors.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.gdata.2014.04.003.
Appendix A. Supplementary data
List of all the samples and file names.
List of start and end base pair positions of centromeres for chromosome 1-22, X (23) and Y (24).
List of all BAC clones and their chromosomal location and start and end base pair positions.
R script for preprocessing of microarray data, circular binary segmentation, sample MAD calculations.
R script for analysis of the CSFTC data.
References
- 1.Magbanua M.J., Melisko M., Roy R., Sosa E.V., Hauranieh L., Kablanian A., Eisenbud L.E., Ryazantsev A., Au A., Scott J.H. Molecular profiling of tumor cells in cerebrospinal fluid and matched primary tumors from metastatic breast cancer patients with leptomeningeal carcinomatosis. Cancer Res. 2013;73:7134–7143. doi: 10.1158/0008-5472.CAN-13-2051. [DOI] [PubMed] [Google Scholar]
- 2.Magbanua M.J., Park J.W. Isolation of circulating tumor cells by immunomagnetic enrichment and fluorescence-activated cell sorting (IE/FACS) for molecular profiling. Methods. 2013;64:114–118. doi: 10.1016/j.ymeth.2013.07.029. [DOI] [PubMed] [Google Scholar]
- 3.Magbanua M.J., Sosa E.V., Scott J.H., Simko J., Collins C., Pinkel D., Ryan C.J., Park J.W. Isolation and genomic analysis of circulating tumor cells from castration resistant metastatic prostate cancer. BMC Cancer. 2012;12:78. doi: 10.1186/1471-2407-12-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magbanua M.J., Sosa E.V., Roy R., Eisenbud L.E., Scott J.H., Olshen A., Pinkel D., Rugo H.S., Park J.W. Genomic profiling of isolated circulating tumor cells from metastatic breast cancer patients. Cancer Res. 2013;73:30–40. doi: 10.1158/0008-5472.CAN-11-3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snijders A.M., Nowak N., Segraves R., Blackwood S., Brown N., Conroy J., Hamilton G., Hindle A.K., Huey B., Kimura K. Assembly of microarrays for genome-wide measurement of DNA copy number. Nat. Genet. 2001;29:263–264. doi: 10.1038/ng754. [DOI] [PubMed] [Google Scholar]
- 6.Snijders A.M., Fridlyand J., Mans D.A., Segraves R., Jain A.N., Pinkel D., Albertson D.G. Shaping of tumor and drug-resistant genomes by instability and selection. Oncogene. 2003;22:4370–4379. doi: 10.1038/sj.onc.1206482. [DOI] [PubMed] [Google Scholar]
- 7.Neuvial P., Hupe P., Brito I., Liva S., Manie E., Brennetot C., Radvanyi F., Aurias A., Barillot E. Spatial normalization of array-CGH data. BMC Bioinforma. 2006;7:264. doi: 10.1186/1471-2105-7-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jain A.N., Tokuyasu T.A., Snijders A.M., Segraves R., Albertson D.G., Pinkel D. Fully automatic quantification of microarray image data. Genome Res. 2002;12:325–332. doi: 10.1101/gr.210902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olshen A.B., Venkatraman E.S., Lucito R., Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 10.Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blaveri E., Brewer J.L., Roydasgupta R., Fridlyand J., DeVries S., Koppie T., Pejavar S., Mehta K., Carroll P., Simko J.P. Bladder cancer stage and outcome by array-based comparative genomic hybridization. Clin. Cancer Res. 2005;11:7012–7022. doi: 10.1158/1078-0432.CCR-05-0177. [DOI] [PubMed] [Google Scholar]
- 12.Chin K., DeVries S., Fridlyand J., Spellman P.T., Roydasgupta R., Kuo W.L., Lapuk A., Neve R.M., Qian Z., Ryder T. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006;10:529–541. doi: 10.1016/j.ccr.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 13.Fridlyand J., Snijders A.M., Ylstra B., Li H., Olshen A., Segraves R., Dairkee S., Tokuyasu T., Ljung B.M., Jain A.N. Breast tumor copy number aberration phenotypes and genomic instability. BMC Cancer. 2006;6:96. doi: 10.1186/1471-2407-6-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of all the samples and file names.
List of start and end base pair positions of centromeres for chromosome 1-22, X (23) and Y (24).
List of all BAC clones and their chromosomal location and start and end base pair positions.
R script for preprocessing of microarray data, circular binary segmentation, sample MAD calculations.
R script for analysis of the CSFTC data.