

Abstract
While a number of DNA sequence motifs have been functionally characterized, the full repertoire of motifs in an organism (the motifome) is yet to be characterized. The present study wishes to widen the scope of motif content analysis in different monocot and dicot species that include both rice species, Brachypodium, corn, wheat as monocots and Arabidopsis, Lotus japonica, Medicago truncatula, and Populus tremula as dicots. All possible existing motifs were analyzed in different regions of genomes such as were found in different sets of sequences in these species: the whole genome, core proximal and distal promoters, 5′ and 3′ UTRs, and the 1st introns. Due to the increased number of species involved in this study compared to previous works, species relationships were analyzed based on the similarity of common motif content. Certain secondary structure elements were inferred in the genomes of these species as well as new unknown motifs. The distribution of 20 motifs common to the studied species were found to have a significantly larger occurrence within the promoters and 3′ UTRs of genes, both being regulatory regions. Motifs common to the promoter regions of japonica rice, Brachypodium, and corn were also found in a number of orthologous and paralogous genes. Some of our motifs were found to be complementary to miRNA elements in Brachypodium distachyon and japonica rice.
Keywords: Monocot, Dicot, Genome, Motif, Promoter
Background
A motif is a conserved or frequently occurring sequence of defined length, usually 4–10 base pairs in the case of DNA sequences. Motifs can be found in DNA/RNA or protein sequences, where each motif is typically associated with certain biological function(s). While a number of DNA sequence motifs have been functionally characterized, the full repertoire of motifs in an organism (referred to as the motifome) is yet to be characterized. In this study, we focus on motifs in plants. The total motif content of differing lengths of Arabidopsis thaliana and Oryza sativa japonica has been determined [8], [9], which then allowed for cross-species comparison between a dicot and a monocot species. The present study wishes to widen the scope of motif content analysis in different monocot and dicot species that include Oryza sativa japonica, Oryza sativa indica, Brachypodium distachyon, Zea mays, Triticum aestivum as monocots and A. thaliana, Lotus japonica, Medicago truncatula, and Populus tremula as dicots. The reason that O. sativa indica and Brachypodium were chosen was that their promoter and UTR regions have been determined besides their genome sequences. O. sativa indica is also a close relative to O. sativa japonica, therefore we can expect that their motif content overlaps highly, compared to other species. The four dicot species were chosen because they are well-known model organisms and the whole genome sequences are available for all these species. The list of species, general characteristics of their genomes (ACGT%, number of genes, number of chromosomes, and size of genome) as well as a link or reference to their genomic information is provided in Table 1, Table 2.
Table 1.
Available data sets for the studied species.
| Species | genome | Core promoters | Proximal promoters | Distal promoters | 1st introns | 5′ UTRs | 3′ UTRs |
|---|---|---|---|---|---|---|---|
| Monocots | |||||||
| Oryza sativa japonica | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Oryza sativa japonica | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Brachypodium distachyon | 1 | 1 | 1 | X | X | X | 1 |
| Triticum aestivum | 1 | X | X | X | X | X | X |
| Zea mays | 1 | 1 | 1 | 1 | X | X | X |
| Dicots | |||||||
| Arabidopsis thaliana | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Lotus japonica | 1 | X | X | X | X | X | X |
| Medicago truncatula | 1 | X | X | X | X | X | X |
| Populus tremula | 1 | X | X | X | X | X | X |
Table 2.
General information on the genomes of the studied organisms.
| Species | A% | C% | G% | T% | Chrom. no. | Genome size (bp) | No. of genes | Reference |
|---|---|---|---|---|---|---|---|---|
| Monocots | ||||||||
| Brachypodium distachyon | 26.8 | 23.2 | 23.2 | 26.8 | 5 | 271,923,306 | 12,825 | [1] |
| Oryza sativa japonica | 28.2 | 21.8 | 21.8 | 28.2 | 12 | 382,150,945 | 30,294 | [8] |
| Oryza sativa indica | 28.6 | 21.4 | 21.4 | 28.6 | 12 | 427,026,737 | 49,710 | [2] |
| Zea mays | 26.5 | 23.5 | 23.5 | 26.5 | 10 | 2,065,722,704 | 54,814 | [3] |
| Triticum aestivum | 27.3 | 22.7 | 22.7 | 27.3 | 7 | 6,846,530,000 | ~ 94,000–96,000 | [4] |
| Dicots | ||||||||
| Arabidopsis thaliana | 32.0 | 18.0 | 18.0 | 32.0 | 5 | 147,812,252 | 33,323 | [9] |
| Lotus japonica | 33.4 | 16.6 | 16.6 | 33.4 | 6 | 119,146,348 | ~ 20,800 | [7] |
| Medicago truncatula | 33.4 | 16.6 | 16.6 | 33.4 | 9 | 307,511,856 | ~ 18,844 | [7] |
| Populus tremula | 33.2 | 16.8 | 16.8 | 33.4 | 19 | 417,640,243 | n.a. | [5] |
The present analysis endeavors to draw up a catalogue of all possible existing octamer motifs (48 = 65,536 in total) in the genomes of the above-named 9 species, as well as determining their statistical significance. In this study these motifs serve as regulatory signals or transcription factor binding sites in promoters, 1st introns, or UTR sequences. Octamers were studied in our previous study of O. sativa japonica [8] and in [9] of Arabidopsis. Motifs that are 8 bp long are long enough to be both diverse enough and statistically significant at the same time. Motifs can be found in different regions of genomes such as were found in 1–7 different sets of sequences, according to its availability in these species: the whole genome, core promoters (250 or 300 bp), proximal promoters (1000 bp), and distal promoters (3000 bp), 5′ and 3′ UTRs, and the 1st introns. The motifs that we found were matched with experimentally validated regulatory motifs in the Plant Cis-acting Regulatory Elements (PLACE) database [12]. The motifs in this database are characterized by a PLACE ID and a representative sequence, and are also cross-linked to papers describing these motifs in greater detail.
We published similar work on rice in a previous paper [8], and the methodology to find and determine the statistical significance of motifs used in this study is similar to that in previous works [9], [10], therefore we refer the reader to these specific references. Furthermore, what makes the present study significant is that it also makes it possible to compare motif content between more or less related species as well as two different groups of plants, monocots and dicots, making it possible to draw further insights from a wider variety of cross-species comparisons.
Results and discussion
Principle of investigation
According to the statistical measure, the total occurrence of all possible octamer motifs (65,536 in number) were enumerated in the genomes of the 5 named monocot and the 4 named dicot species. Octamers in the appropriate whole genomes, 5′ UTR, 3′ UTR, 1st introns, core, proximal, and distal promoters were analyzed in some of the monocot species and A. thaliana. Only motifs not containing ambiguous IUPAC symbols (M, R, W, S, Y, K, B, D, H, V, or N) were retained. According to the algorithm, the statistical significance value , where S is the number of sequences that the motif m occurs in, and ES is the expected number of sequences that the motif is expected to occur in according to the background base distribution of the different species. The genome sequences were masked using the RepeatMasker program prior to analysis to exclude repetitive sequences which might skew the results.
The detailed method of the algorithm is described in previous works [8], [9], [10]. However, a short description will be given in the Materials and methods section. All 7 sequence sets were analyzed in O. sativa japonica and indica, while only the whole genome, the 3′ UTRs, and the core promoters were analyzed in Brachypodium. The whole genome, the core, proximal, and distal promoters were studied in Z. mays. Only the genome motifs of T. aestivum were used to measure similarity between that species and the other grass species as only the genome sequence was available for this species [11]. The available data sets for each species can be seen in Table 2.
Analysis of motifs in the whole genome
The top 25 octamer motifs found in the monocot and dicot species are listed in Supplementary Tables 1 and 2, respectively; while the entire list of motifs with their significance scores can be found in the Supplementary Excel files for each species (Supplementary file 1 — O. sativa japonica, Suppl. file 2 — O. sativa indica, Suppl. file 3 — Brachypodium, Suppl. file 4 — Z. mays, Suppl. file 5 — Triticum, Suppl. file 6 — dicot genomes, Suppl. file 7 — Arabidopsis compared to monocots). The intersections of octamer motifs between different combinations of monocot and dicot species can be seen in the Venn diagrams in Fig. 1a and b. For all species, the top 100 motifs were further analyzed for their functional significance. The top 100 whole genome motifs were also checked against the PLACE database [12] to see whether they matched any experimentally verified regulatory motifs. The PLACE database [6] is a well-known plant motif database. Each motif is characterized by a PLACE id as well as a functional description as well as Pubmed ID's which link to papers describing the given motif. The functional definitions of each PLACE id can be found in the supplementary Excel file “PLACE_id_functional_dictionary.xlsx”. Z. mays had the least number of motifs in common with the other 4 species, with 65 of its top 100 high scoring motifs being unique only to itself. Triticum came in a close second with 44 motifs distinct to itself. This is no surprise as Z. mays belongs to a completely different clade (PACC clade) as do the two Oryza species and Brachypodium [13].
Fig. 1.
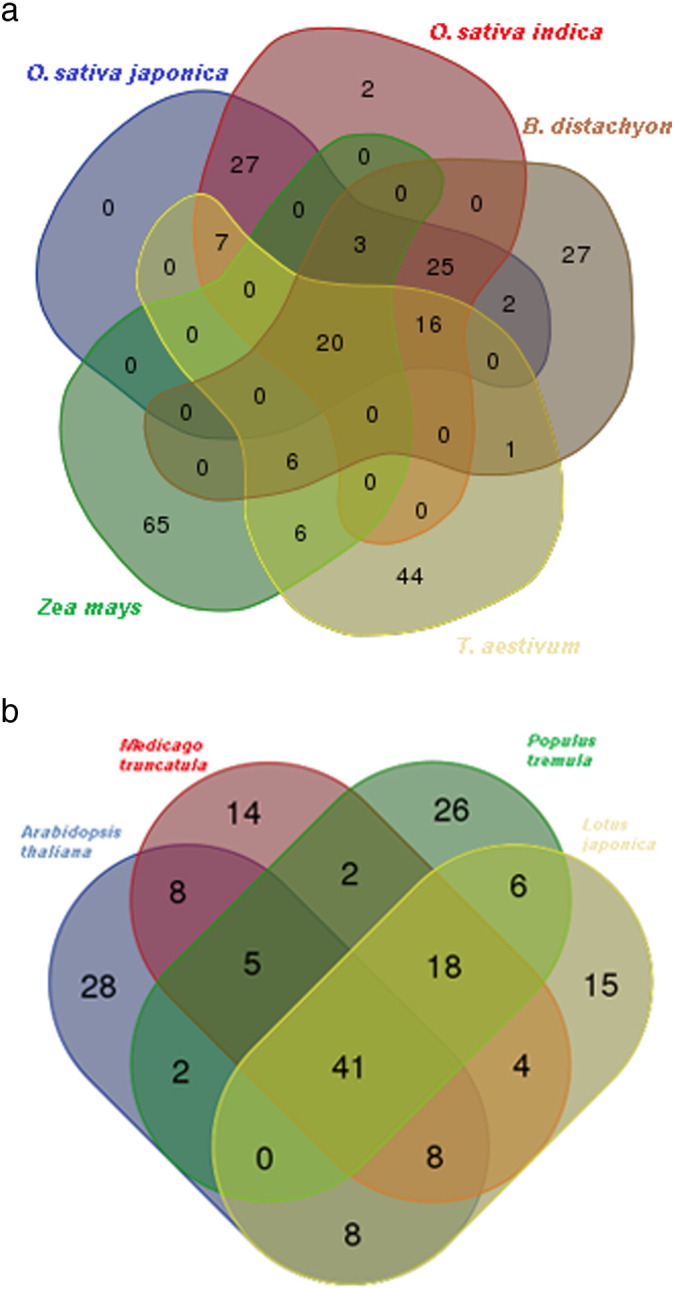
a. Number of putative top 100 genomic motifs common to different combinations of the five monocot species studied.
b. Number of putative top 100 genomics motifs common to different combinations of the four dicot species studied.
Overall, 20 motifs were common to all monocot species, while 41 motifs were common to the dicot species. Of these, 15 were common to all monocot and all dicot species, making them the general plant motif candidates. These motifs can be seen in Table 3. In Table 4 we can see the number of top 100 genomic motifs shared by each of the monocot and dicot species shared between 1, 2, 3, 4, and 5 other species. We can see again that Z. mays has a relatively low number of motifs shared by any number of other species, whereas O. sativa japonica and indica and Brachypodium have 71, 73, and 70 motifs shared by at least three species, 64 motifs common to all three of these species. 98 motifs are common to both rice species, which is also significant.
Table 3.
List of 15 motifs common to monocots and dicots and their annotation. Reverse complement motifs underlined.
| Motif | PLACE annotation |
|---|---|
| AAAAAAAA | ATRICHPSPETE CARGCW8GAT CARGNCAT MARTBOX |
| AAAAAGAA | |
| AAAAGAAA | |
| AAAATAAA | -314MOTIFZMSBE1 CARGCW8GAT CARGNCAT ELEMENT1GMLBC3 MARTBOX |
| AAAGAAAA | |
| AAATAAAA | -314MOTIFZMSBE1 3AF1BOXPSRBCS3 CARGCW8GAT CARGNCAT ELEMENT1GMLBC3 MARTBOX |
| AAGAAAAA | |
| TTCTTTTT | |
| TTTATTTT | -314MOTIFZMSBE1 CARGCW8GAT CARGNCAT ELEMENT1GMLBC3 MARTBOX |
| TTTCTTTT | |
| TTTGTTTT | |
| TTTTATTT | -314MOTIFZMSBE1 3AF1BOXPSRBCS3 CARGCW8GAT CARGNCAT ELEMENT1GMLBC3 MARTBOX |
| TTTTCTTT | |
| TTTTTCTT | |
| TTTTTTTT | ATRICHPSPETE CARGCW8GAT CARGNCAT MARTBOX |
Table 4.
Number of putative genomic top 100 motifs shared between different numbers of species for all monocot and dicot species.
| Species | Motifs shared with 1 species | Motifs shared with 2 species | Motifs shared with 3 species | Motifs shared with 4 species | Motifs shared with 5 species |
|---|---|---|---|---|---|
| Monocots | |||||
| Oryza sativa japonica | 0 | 29 | 32 | 19 | 20 |
| Oryza sativa indica | 2 | 27 | 34 | 19 | 20 |
| Brachypodium distachyon | 27 | 3 | 31 | 19 | 20 |
| Triticum aestivum | 44 | 7 | 13 | 16 | 20 |
| Zea mays | 65 | 6 | 6 | 3 | 20 |
| Dicots | |||||
| Arabidopsis thaliana | 28 | 18 | 13 | 41 | – |
| Medicago truncatula | 14 | 14 | 31 | 41 | – |
| Lotus japonica | 15 | 18 | 26 | 41 | – |
| Populus tremula | 26 | 10 | 23 | 41 | – |
What is interesting about these motifs is that a large number of them form reverse complementary pairs (AAAAAAAA|TTTTTTTT, AAAAAGAA|TTCTTTTT, AAAAGAAA|TTTCTTTT, AAAGAAAA|TTTTCTTT, AAATAAAA|TTTTATTT and AAGAAAAA|TTTTTCTT). These might possibly form parts of the secondary stem-loop structure in microRNAs. Indeed, the octamers, AAGAAAAA, AAAGAAAA, AAAAGAAA, AAAAAGAA all match part of the stem-loop structure of MIR169 in Arabidopsis [14]. 6 of these 12 motifs are not annotated yet, thus they could be associated with microRNA structures. Meng and coworkers [15] also found that miRNA mimic sites were found to be denser in the untranslated regions than in the other sequences.
This suggests that these elements are common regulatory elements that are generally present in plant species. Furthermore, this type of analysis can be useful in finding other secondary structures in the 3′ UTR regions of genes or possibly in other sequences. We searched for these top 15 motifs common to monocots and dicots in the genomes of those species which had sufficient gene annotation (A. thaliana, M. truncatula, O. sativa japonica and indica, B. distachyon, and Z. mays). As described in the Materials and methods section we downloaded the gene annotation for these species, and located the position of each occurrence of each of the 15 motifs. We counted the number of times they occurred in the promoter region (3 kbp upstream region from the ATG start), within the gene body, and within the 3′ UTR (defined as the 3 kbp region downstream of the gene). The number of occurrences of each motif in each of the 3 gene subregions can be seen in Tables 5a and b. We measured how significantly different the number of occurrences was between the promoter region and the gene body and between the 3′ UTR and the gene body. We found that the difference was significant for promoters and 3′ UTRs in all six species. This shows that these top 15 motifs occur more significantly within promoters and 3′ UTR regions, both being regulatory regions, in these species.
Table 5a.
Distribution of the 15 common motifs in three of the monocot species in promoters, within genes, and 3′ UTRs.
| Motif | O. sativa japonica promoter (p = 1.1e − 4) | O. sativa japonica gene | O. sativa japonica 3′ UTR (p = 1.3e − 4) | O. sativa indica promoter (p = 6.7e − 4) | O. sativa indica gene | O. sativa indica 3′ UTR (p = 0.001) | B. distachyon promoter (p = 0.0017) | B. distachyon gene | B. distachyon 3′ UTR (p = 0.0016) | Z. mays promoter (p = 2.8e − 9) | Z. mays gene | Z. mays 3′ UTR (p = 1.9e − 5) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AAAAAAAA | 9704 (35.77%) | 7490 (27.61%) | 9930 (36.6%) | 16932 (39.88%) | 8504 (20.03%) | 17018 (40.08%) | 43339 (43.53%) | 18516 (18.59%) | 37700 (37.86%) | 17139 (33.3%) | 15953 (31%) | 18365 (35.68%) |
| AAAAAGAA | 2203 (35.44%) | 1715 (27.59%) | 2297 (36.95%) | 3418 (38.53%) | 2069 (23.32%) | 3384 (38.14%) | 11752 (39.67%) | 6936 (23.41%) | 10936 (36.91%) | 11730 (33.76%) | 11156 (32.11%) | 11856 (34.12%) |
| AAAAGAAA | 2601 (35.28%) | 2068 (28.05%) | 2702 (36.65%) | 3989 (38.47%) | 2385 (23%) | 3994 (38.52%) | 13977 (39.81%) | 8380 (23.87%) | 12747 (36.31%) | 13282 (34%) | 12455 (31.88%) | 13320 (34.1%) |
| AAAATAAA | 2839 (35.63%) | 2186 (27.44%) | 2941 (36.91%) | 4780 (42.49%) | 2200 (19.55%) | 4269 (37.95%) | 11265 (38.79%) | 7550 (25.99%) | 10225 (35.21%) | 12529 (33.94%) | 11713 (31.73%) | 12667 (34.31%) |
| AAAGAAAA | 2549 (35.57%) | 2014 (28.1%) | 2602 (36.31%) | 3971 (38.76%) | 2366 (23.09%) | 3906 (38.13%) | 14279 (39.81%) | 8569 (23.89%) | 13012 (36.28%) | 13220 (33.98%) | 12425 (31.94%) | 13255 (34.07%) |
| AAATAAAA | 2562 (36.46%) | 1911 (27.19%) | 2553 (36.33%) | 4024 (41.28%) | 2010 (20.62%) | 3712 (38.08%) | 10116 (38.35%) | 6900 (26.15%) | 9362 (35.49%) | 11660 (33.62%) | 11041 (31.84%) | 11975 (34.53%) |
| AAGAAAAA | 2379 (35.15%) | 1922 (28.4%) | 2466 (36.44%) | 3719 (38.68%) | 2169 (22.56%) | 3726 (38.75%) | 13257 (39.94%) | 7871 (23.71%) | 12057 (36.33%) | 11076 (33.46%) | 10595 (32.01%) | 11423 (34.51%) |
| AGAGAGAG | 2551 (36.59%) | 1836 (26.33%) | 2584 (37.06%) | 4628 (48.11%) | 1646 (17.11%) | 3344 (34.76%) | 10559 (50.19%) | 3682 (17.5%) | 6797 (32.3%) | 10652 (33.9%) | 10039 (31.95%) | 10723 (34.13%) |
| TTCTTTTT | 2218 (36.02%) | 1758 (28.55%) | 2181 (35.42%) | 3418 (39.22%) | 1943 (22.3%) | 3352 (38.47%) | 11210 (38.79%) | 7047 (24.38%) | 10642 (36.82%) | 11767 (34.3%) | 11088 (32.32%) | 11450 (33.37%) |
| TTTATTTT | 2922 (37.1%) | 2150 (27.29%) | 2804 (35.6%) | 4647 (42.39%) | 2169 (19.78%) | 4145 (37.81%) | 11273 (38.73%) | 7516 (25.82%) | 10312 (35.43%) | 12345 (34.3%) | 11420 (31.73%) | 12223 (33.96%) |
| TTTCTTTT | 2657 (36.71%) | 2031 (28.06%) | 2548 (35.21%) | 4025 (39.63%) | 2296 (22.61%) | 3833 (37.74%) | 13510 (39.04%) | 8483 (24.51%) | 12608 (36.43%) | 13187 (34.42%) | 12412 (32.4%) | 12709 (33.17%) |
| TTTGTTTT | 1778 (36.12%) | 1396 (28.36%) | 1748 (35.51%) | 2784 (38.12%) | 1758 (24.07%) | 2761 (37.81%) | 10184 (37.63%) | 7230 (26.72%) | 9647 (35.65%) | 12370 (33.61%) | 11994 (32.59%) | 12441 (33.8%) |
| TTTTATTT | 2488 (36.49%) | 1947 (28.55%) | 2383 (34.95%) | 3953 (41.83%) | 1883 (19.92%) | 3614 (38.24%) | 10097 (38.3%) | 6885 (26.11%) | 9378 (35.57%) | 11546 (34.34%) | 10712 (31.86%) | 11356 (33.78%) |
| TTTTCTTT | 2631 (36.65%) | 1983 (27.62%) | 2564 (35.72%) | 3997 (39.41%) | 2292 (22.59%) | 3853 (37.99%) | 14112 (39.71%) | 8669 (24.39%) | 12753 (35.88%) | 13030 (34.1%) | 12358 (32.34%) | 12818 (33.54%) |
| TTTTTCTT | 2464 (36.23%) | 1899 (27.92%) | 2437 (35.83%) | 3807 (39.83%) | 2104 (22.01%) | 3645 (38.14%) | 12886 (39.24%) | 8168 (24.87%) | 11784 (35.88%) | 11114 (34.54%) | 10423 (32.39%) | 10639 (33.06%) |
| TTTTTTTT | 9638 (36.22%) | 7599 (28.56%) | 9366 (35.2%) | 16845 (40.59%) | 8570 (20.65%) | 16076 (38.74%) | 43035 (43.19%) | 18886 (18.95%) | 37706 (37.84%) | 17833 (34.62%) | 16465 (31.96%) | 17210 (33.41%) |
Motif content analysis of grass species
Genetic similarity and therefore species relationships may be measured based on the similarity in the motif content. Species with higher content of common motifs with similar ranking according to motif score would be closer relatives than those species that have fewer common motifs. This is because the longer two species have been diverged from each other; more mutations have been allowed to accumulate between them, allowing a larger motif turnover to have taken place. The advantage of this method over sequence similarities between single genes is that it takes a global genomic sequence composition into account.
To test such phylogenetic sequence changes, we studied the number of common top 1000 genome motifs between the five monocot species. Two rice species belong to the family Ehrhartoideae, while Triticum and Brachypodium belong to the family Pooideae, and Zea belongs to the more distantly related family, Panicoideae. As we can see in Table 6, O. sativa and indica, the two rice species had the highest number of common genome motifs (939). The Spearman coefficient computed for these common motifs is also relatively high (0.710). The number of common motifs between the two Oryza species and Brachypodium is also proportionate (O. sativa japonica: 704 vs. O. sativa indica: 716). Brachypodium is also more related to the two rice species than it is to corn and wheat (only 414 and 498 common motifs, respectively). Conversely, Triticum has less motifs in common with the 2 rice species than does Brachypodium (446 and 448 for japonica and indica rice, respectively). This means that although Triticum and Brachypodium are in the same family, there might be a tradeoff when comparing their motif content with another species regarding the total number of motifs versus the motifs' ranking. (See Table 5b.)
Table 6.
Common genome motifs and their Spearman coefficient from the top 1000 motifs from the monocot species and Arabidopsis as an outlier species.
| O. sativa japonica | O. sativa indica | B. distachyon | T. aestivum | Z. mays | |
|---|---|---|---|---|---|
| O. sativa indica | 939/0.710 | ||||
| B. distachyon | 704/0.417 | 716/0.449 | |||
| T. aestivum | 446/0.646 | 448/0.604 | 498/0.621 | ||
| Z. mays | 373/0.756 | 376/0.782 | 414/0.674 | 404/0.653 | |
| A. thaliana | 392/0.571 | 407/0.521 | 492/0.392 | 433/0.541 | 385/0.597 |
Table 5b.
Distribution of the 15 common motifs in two of the dicot species in promoters, within genes, and 3′ UTRs.
| Motif | Arabidopsis promoter (p = 5.1e − 4) | Arabidopsis gene | Arabidopsis 3′ UTR (p = 5.9e − 4) | Medicago promoter (1.9e − 3) | Medicago gene | Medicago 3′ UTR (2e − 3) |
|---|---|---|---|---|---|---|
| AAAAAAAA | 113367 (49.58%) | 21263 (9.3%) | 93997 (41.11%) | 176179 (37.06%) | 123956 (26.08%) | 175149 (36.85%) |
| AAAAAGAA | 20023 (44.96%) | 6323 (14.19%) | 18188 (40.84%) | 21275 (36.99%) | 15009 (26.09%) | 21224 (36.9%) |
| AAAAGAAA | 23235 (44.66%) | 7359 (14.14%) | 21421 (41.18%) | 25438 (36.84%) | 18090 (26.19%) | 25518 (36.95%) |
| AAAATAAA | 27470 (50.34%) | 5025 (9.2%) | 22073 (40.45%) | 46731 (37%) | 33212 (26.29%) | 46353 (36.7%) |
| AAAGAAAA | 24919 (44.73%) | 7887 (14.15%) | 22899 (41.1%) | 27648 (37.09%) | 19527 (26.19%) | 27365 (36.71%) |
| AAATAAAA | 26280 (49.71%) | 4922 (9.31%) | 21657 (40.97%) | 43908 (36.98%) | 31295 (26.35%) | 43526 (36.65%) |
| AAGAAAAA | 25016 (45.3%) | 7900 (14.3%) | 22302 (40.38%) | 28103 (36.92%) | 20056 (26.35%) | 27943 (36.71%) |
| TTCTTTTT | 19697 (44.9%) | 6015 (13.71%) | 18147 (41.37%) | 21078 (37.14%) | 14781 (26.04%) | 20885 (36.8%) |
| TTTATTTT | 26774 (49.75%) | 5057 (9.39%) | 21986 (40.85%) | 46196 (36.8%) | 33009 (26.29%) | 46310 (36.89%) |
| TTTCTTTT | 23091 (44.74%) | 7315 (14.17%) | 21204 (41.08%) | 25449 (36.95%) | 17949 (26.06%) | 25458 (36.97%) |
| TTTGTTTT | 26116 (45.24%) | 8194 (14.2%) | 23419 (40.57%) | 24684 (36.71%) | 17745 (26.39%) | 24814 (36.9%) |
| TTTTATTT | 25977 (49.31%) | 4984 (9.46%) | 21719 (41.22%) | 43667 (36.87%) | 30965 (26.14%) | 43790 (36.97%) |
| TTTTCTTT | 24914 (45.01%) | 7814 (14.11%) | 22619 (40.86%) | 27275 (36.84%) | 19404 (26.2%) | 27354 (36.94%) |
| TTTTTCTT | 25058 (45.5%) | 7743 (14.05%) | 22271 (40.43%) | 28005 (36.85%) | 19829 (26.09%) | 28143 (37.04%) |
| TTTTTTTT | 112239 (49.57%) | 20617 (9.1%) | 93565 (41.32%) | 173501 (36.76%) | 123670 (26.2%) | 174684 (37.02%) |
When comparing common motifs between the monocot species and an outlier species such as A. thaliana, we found that the number of common motifs was consistently low (Table 6). On average, there were 532 common motifs between any two monocot species, but only 422 common motifs between A. thaliana and any given monocot species. In the case of the core promoter motifs (Fig. 2), we found only 10, 1, and 3 common motifs with Arabidopsis out of the top 100 with O. sativa japonica, indica, and B. distachyon. For the proximal promoters, this number was 11, 1, and 0. When comparing the number of common motifs between O. sativa japonica and A. thaliana within the 7 sequence sets, we also found them to be quite low (Fig. 3).
Fig. 2.

Pairwise comparison of common putative motifs in the whole genome, core and proximal promoters between all monocot and dicot species.
Fig. 3.

Number of common motifs within the 7 sequence subsets between Oryza sativa japonica and Arabidopsis thaliana.
The scenario was somewhat different when the common top 1000 motif content of the core, proximal, and distal promoters were compared between the monocot species themselves (Tables 6a–c). The number of common motifs was greatest between the two rice species in the proximal and distal promoters (642 and 707, respectively). Surprisingly the number of common motifs in the core promoter between the two rice species was less than compared to either species versus Brachypodium. However, in the case of the core promoters and the 3′ UTRs (Table 7d), the Spearman correlation was greater when comparing the two rice species than when either rice species was compared to Brachypodium. About the same number of common motifs was found in all three species' proximal promoters, and the Spearman coefficients were also roughly the same. This could be because the longer the promoter sequence gets, the larger the noise, allowing many more dissimilar statistically significant motifs to accumulate. (See Table 7b, Table 7c.)
Table 7d.
3′ UTR motif content similarity and Spearman ranking between the studied monocot species.
| O. sativa japonica | O. sativa indica | B. distachyon | |
|---|---|---|---|
| O. sativa japonica | 167 (0.947) | 376 (0.589) | |
| O. sativa indica | 515 (0.498) | ||
| B. distachyon |
Table 7b.
Proximal promoter motif content similarity and Spearman ranking between the studied monocot species.
| O. sativa japonica | O. sativa indica | B. distachyon | Zea mays | |
|---|---|---|---|---|
| O. sativa japonica | 642 (0.344) | 645 (0.416) | 655 (0.409) | |
| O. sativa indica | 686 (0.399) | 439 (0.564) | ||
| B. distachyon | 518 (0.447) | |||
| Zea mays |
Table 7c.
Distal promoter motif content similarity and Spearman ranking between the studied monocot species.
| O. sativa japonica | O. sativa indica | Zea mays | |
|---|---|---|---|
| O. sativa japonica | 707 (0.341) | 619 (0.384) | |
| O. sativa indica | 460 (0.545) | ||
| Zea mays |
Analysis of promoter motifs in monocots
We compared the top 100 octamer motif content in the core, proximal, and distal promoters of the four monocot species that had promoter sequence sets available. The reason that this was not done for dicots was because only the genome sequence was available for those species. In case of Brachypodium, the distal promoter dataset is not available; hence it was not included in the distal promoter motif comparison. We found 18, 37, and 43 motifs in the core, proximal and distal promoter datasets, respectively, that occurred in all of the species compared. A list of these motifs and their PLACE annotations can be seen in Supplementary Tables 3a–c. The two most common motifs were CARGNCAT and CARGCW8GAT, which are binding sites for the AGL15 transcription factor [16]. MARTBOX corresponds to the T-box, which can be found in scaffold attachment regions [17]. ATRICHPSPETE serves as an A/T-rich quantitative enhancer [18]. GAGAGMGSA1 and GAGA8HVBKN3 both correspond to GA-rich elements which bind the BBR and GBR transcription factors [19]. CTRMCAMV35S serves as an inverted GAGA-element which enhances gene expression [20]. GT1MOTIFPSRBCS corresponds to the GT-1 motif [21], and AGL3ATCONSENSUS serves as a binding site for the transcription factor AGL3 [22].
We also looked at the distribution of motifs specific to the core, proximal and distal promoter regions. Our results showed that 61 distinct motifs from the top 100 are present only in the core promoter regions, 8 in the proximal promoter and 16 are found only in the distal promoter regions. A list of these motifs can be seen in Supplementary Tables 4a–c along with their PLACE annotation. The reason that the number of core promoter-specific motifs is so high is because this is the place where most of the regulation-specific molecular machinery assembles. A number of the motifs found by us lack annotation in the PLACE database; thus, some of them can be novel putative transcription factor binding sites that are specific to a promoter region. We found 42 such novel motifs in the core, 2 in the proximal, and 14 in the distal promoter regions.
We then took these 61, 8, and 16 common motifs from the monocot species which were common to the core, proximal, and distal promoters, and looked for them in the appropriate promoter sets in O. sativa japonica, B. distachyon, and Z. mays. The genomes of both O. sativa indica and T. aestivum had poor quality annotation; the wheat genome itself is very fragmentary and thus not usable for this analysis. We report a list of the top 50 genes from these 3 species in Supplementary Excel file 8 (SupplementaryFile8_MonocotPromoterMotifDistribution.xls) which have a high number of these motifs in their core, proximal, and distal promoters.
Here the Z. mays annotation proved to be fairly scant; however, we discovered a number of orthologous and paralogous gene sets in the core, proximal, and distal promoter gene sets. Orthologs are highlighted in bold in the Supplementary Excel file #8, whereas paralogs within species are underlined.
In the core promoter sets between O. sativa japonica And B. distachyon, we found a pair of transducin/WD-40 repeat family genes, 3 myb-like proteins, with 2 paralogs in Brachypodium. A RING/U-box superfamily gene pair was found in O. sativa japonica, and a pair of zinc-finger genes and a pentatricopeptide repeat gene pair was found in Brachypodium.
In the proximal promoter sets we found a WD-40 repeat gene in all 3 species, a pair of NB-ARC domain genes, RING/U-box genes, Class I peptide chain release factor genes, nodulin genes, purple acid phosphatase genes, and major facilitator superfamily genes in O. sativa japonica and B. distachyon. A pair of NAD(P) oxireductase paralogs was found in O. sativa japonica, and a pair of glycosyltransferase genes was found in Brachypodium.
In the distal promoter sets, we found two pairs of paralog genes in O. sativa japonica; a pair of NB-ARC domain-containing disease resistance genes and a pair of cellulose synthase genes.
Analysis of 3′ UTR regions
We compared the top 100 octamer motifs in the 3′ UTRs of three of the five monocot species (O. sativa japonica and indica and B. distachyon). We found 46 motifs to be present in all three species. These motifs can be seen in Supplementary Table 5 along with their annotation in the PLACE database. 24 of the 46 motifs had no annotation and therefore could be potentially novel 3′ UTR motifs. 16 of these 46 motifs were found to be complementary with one another (these ones are underlined in Supplementary Table 5).
We were interested in seeing what kinds of genes common to these three monocot species contain any of these reverse complementary motifs in their 3′ UTRs. We found that the 3′ UTRs of 154, 57, and 22 genes from O. sativa japonica, indica, and B. distachyon, respectively contained at least 50 occurrences of these 16 reverse complementary motifs. When checking their annotations (the annotation of O. sativa indica does not have good quality), we found 155 O. sativa japonica and B. distachyon genes with annotations. Out of these there were 7 pairs of genes from both species which either had similar or the same annotations, or take part in the same physiological process. Among them we can find a β-hydroxyisobutyryl-coA hydrolase/β-ketoacyl-reductase gene pair, a calcium-binding EF-hand family protein/calmodulin-binding protein — encoding gene pair, a pair of genes encoding cysteine proteinases, a pair of genes coding proteins with F-boxes, a pair of genes encoding RING/U-boxes, and a terpene synthase/terpenoid cyclase gene pair. A list of these genes can be seen in bold in Table 8.
Table 8.
List of Oryza sativa japonica and Brachypodium distachyon genes with more than 50 occurrences of reverse complementary motifs in their 3′ UTR regions.
| Gene ID | Number of reverse complementary 3′ UTR motifs | Gene annotation |
|---|---|---|
| Bradi1g32590 | 53 | 6-Phosphogluconate dehydrogenase family protein |
| Bradi1g56250 | 60 | A20/AN1-like zinc finger family protein |
| Bradi4g16400 | 73 | Agenet domain-containing protein |
| Os12g18729 | 75 | ARM repeat superfamily protein |
| Os08g43090 | 64 | Basic-leucine zipper (bZIP) transcription factor family protein |
| Bradi1g11310 | 61 | B-box type zinc finger protein with CCT domain |
| Os12g16350 | 57 | Beta-hydroxyisobutyryl-CoA hydrolase 1 |
| Bradi3g30120 | 61 | Beta-ketoacyl reductase 2 |
| Os01g72530 | 74 | Calcium-binding EF-hand family protein |
| Bradi5g24460 | 61 | Calmodulin-binding protein |
| Os08g03310 | 67 | CCCH-type zinc fingerfamily protein with RNA-binding domain |
| Os02g57410 | 75 | Cysteine proteinases superfamily protein |
| Bradi3g01980 | 81 | Cysteine proteinases superfamily protein |
| Os01g10040 | 55 | Cytochrome P450, family 90, subfamily D, polypeptide 1 |
| Os11g05970 | 72 | FAD/NAD(P)-binding oxidoreductase family protein |
| Os01g08830 | 54 | F-box family protein with a domain of unknown function (DUF295) |
| Bradi2g48880 | 61 | F-box/RNI-like superfamily protein |
| Bradi2g05226 | 64 | Gigantea protein (GI) |
| Os11g47870 | 50 | GRAS family transcription factor |
| Os08g33750 | 58 | Homeodomain-like superfamily protein |
| Os06g06080 | 59 | Hydrolase-like protein family |
| Os02g01150 | 61 | hydroxypyruvate reductase |
| Os04g56500 | 52 | ILI1 binding bHLH 1 |
| Os01g61720 | 54 | IQ-domain 2 |
| Os01g10504 | 87 | K-box region and MADS-box transcription factor family protein |
| Os07g01490 | 156 | Kinesin 5 |
| Os10g13970 | 68 | Leucine-rich repeat protein kinase family protein |
| Os06g19990 | 56 | LORELEI-LIKE-GPI ANCHORED PROTEIN 3 |
| Os06g49380 | 52 | LRR and NB-ARC domains-containing disease resistance protein |
| Os07g44090 | 60 | myb domain protein 61 |
| Os01g09550 | 65 | NAC domain containing protein 75 |
| Bradi3g08890 | 51 | PEBP (phosphatidylethanolamine-binding protein) family protein |
| Bradi1g04820 | 56 | Peptidase S24/S26A/S26B/S26C family protein |
| Os01g53880 | 60 | Phytochrome-associated protein 1 |
| Os12g37480 | 53 | Plant invertase/pectin methylesterase inhibitor superfamily protein |
| Os02g11000 | 59 | Plant Tudor-like RNA-binding protein |
| Os06g41930 | 50 | PLATZ transcription factor family protein |
| Os07g28260 | 72 | P-loop containing nucleoside triphosphate hydrolases superfamily protein |
| Os05g01380 | 53 | Polygalacturonase inhibiting protein 1 |
| Os03g57940 | 56 | Protein kinase family protein |
| Os04g21340 | 52 | Protein of unknown function (DUF1685) |
| Os08g45170 | 74 | Protein of Unknown Function (DUF239) |
| Os02g08364 | 76 | Protein phosphatase 2C family protein |
| Bradi2g03860 | 67 | Protein with RING/U-box and TRAF-like domains |
| Bradi3g52740 | 57 | Pyrophosphorylase 1 |
| Bradi2g40040 | 63 | Ribosomal L28 family |
| Os06g03580 | 52 | RING/U-box superfamily protein |
| Bradi4g44500 | 56 | Saposin B domain-containing protein |
| Os03g27590 | 56 | Serine carboxypeptidase-like 51 |
| Bradi2g39275 | 152 | Serine protease inhibitor, potato inhibitor I-type family protein |
| Bradi1g21510 | 80 | SPX domain gene 3 |
| Bradi5g01823 | 61 | Terpene synthase 21 |
| Os02g36210 | 57 | Terpenoid cyclases/protein prenyltransferases superfamily protein |
| Os12g08260 | 63 | Thiamin diphosphate-binding fold (THDP-binding) superfamily protein |
| Os04g20400 | 118 | UDP-Glycosyltransferase superfamily protein |
Furthermore, we downloaded known miRNA sequences from the PMRD (plant microRNA database) [23]. 3 of the 16 reverse complementary motifs matched with two of the O. sativa japonica miRNA sequences from the database (the underlined part shows where our motifs match with the database sequence): osa-miR1867: TTTTTTTTCTAGGACAGAGGGAGT and osa-miRf11270-akr: GTACTCCTTTCGTCCCAAAAAAAA. These 2 miRNAs in turn targeted 3 of the O. sativa japonica genes also found in our search using all 16 reverse complementary motifs. These genes are Os06g06080, a hydrolase-like protein family, Os06g41930, a PLATZ transcription factor family protein, and Os10g28570, which as of yet has no annotation. 1 of the 16 motifs matched an miRNA sequence from B. distachyon: bdi-miR319: TGAGGGAGCTTTCTTCTGTCC.
Motif pair analysis
The distribution of motif pairs within individual sequences belonging to the core, proximal, and distal promoters, 5′ UTRs, 3′ UTRs, and 1st introns was also examined in the monocot species for which these sequence sets were available. The top 5050 possible motif pairs were examined, coming from all possible pairing of the top 100 motifs from each sequence set from each species. The sequences for all of these motif pairs, their real and expected occurrences and their motif pair score can be found in the supplementary Excel files for each monocot species (Supplementary files 1–5). The reason dicot genomes were not examined is because motif pairs form the building blocks of transcription factor modules, and as such, require being in close proximity with each other, and this is not possible with the genome where motifs would be far away because of the sheer size of the genome.
The number of common motif pairs was examined for the core, proximal, and distal promoters, and 3′ UTRs, as can be seen in Supplementary Tables 7a–d. The Spearman correlation coefficient was also calculated for the common motif pair content. As with the Spearman correlation coefficient calculations performed with only single motifs, a trade-off between the number of common motif pairs and the Spearman correlation between them. It occurs many times that there are more motif pairs between either rice species and B. distachyon or Z. mays, compared to the number of common motifs between the two rice species; however, the Spearman coefficient is still larger between the two rice species. Conversely, the opposite is true, that there are less motif pairs between either rice species and B. distachyon and Z. mays; however, the Spearman coefficient is greater between these species and both rice species. This may be due to the fact that it is more likely to get a higher Spearman coefficient value with fewer common elements than with a larger number of common elements, which might easily introduce noise.
Table 7a.
Core promoter motif content similarity and Spearman ranking between the studied monocot species.
| O. sativa japonica | O. sativa indica | B. distachyon | Zea mays | |
|---|---|---|---|---|
| O. sativa japonica | 271 (0.688) | 404 (0.531) | 432 (0.544) | |
| O. sativa indica | 515 (0.498) | 569 (0.420) | ||
| B. distachyon | 580 (0.479) | |||
| Zea mays |
Otherwise, the similarity between the two rice species and Z. mays can be seen in that the common motif pair content drops off sharply to only 231 (4.6% of all 5,050 pairs) between O. sativa indica, and 351 (7% of all 5050 pairs) between B. distachyon and maize. The number is also relatively low between these two previously mentioned species when looking at the common motif pairs in proximal promoters (820, which is 16.2% of all 5050 pairs), and in distal promoters (990, which is 19.6% of all 5050 pairs).
Dicot consensus sequences
For the dicot species we defined consensus sequences for the top 100 genome. This we did by putting motifs with similar sequences into the same cluster. Afterwards, a multiple alignment was made for each cluster, and the consensus sequence was defined from the alignment. These consensus sequences can be seen in the tab “consensus sequences” in Supplementary File #6.
Conclusion
Comparing the motif content of different species at different taxonomical levels broadens our horizons and allows a deeper analysis, making it possible to uncover newer aspects of their genomes. For example, by looking at the motif content of whole genomes, we can capture global similarity between them in a holistic manner, instead of looking at similarities between genes on only a local level. This is especially evident in the number of common top-ranking motifs, for example within the studied monocot species, which reflect the degree of relationships between individual species. Using the top-ranking motifs from the whole genome is especially useful for this, compared to the motif content of different parts of the gene (such as the promoter or 3′ UTR), since this way we can draw general conclusions about the whole genome.
The similar content of top-ranking motifs between two given species can be quantified to measure how similar the two species are to one another. This can be done by comparing the number of common motifs, and/or by calculating the Spearman coefficient between sets of motifs between the two species. The number of common motifs between two sets shows how similar two species are, because we would expect that the closer two species are to one another, they would share more motifs in common which also occur more abundantly, and thus have a high score. Using the Spearman coefficient also gives a good measure of this similarity. This can be seen in the case of motif content similarity in the 3′ UTR regions between the two rice species compared to B. distachyon. Here even though there are a smaller number of common motifs between the two rice species compared to B. distachyon (167 compared to 376 and 515), their Spearman coefficient is larger (0.947 compared to 0.589 and 0.498). This also might be due to less selective pressure on the 3′ UTR region allowing for larger sequence divergence. Taking both measures into account shows how related two species are to each other. When comparing the common motif content with an outlier species such as A. thaliana, which consistently gave a low number of common motifs as well as a low Spearman coefficient.
For further study, it would be intriguing to analyze the motif content of a large number of species (30 or more), in order to perform motif content analysis on a larger scale. To this end we have written a perl script (genomotifome.pl) which analyzes the whole genome sequence of a given species for motifs of a given length, returning a set of the top-ranking motifs from that genome.
Furthermore, the analysis of top-ranking motifs also makes it possible to predict parts of secondary structures of different kinds of genetic elements, such as miRNA sequences, tandem repeats, or microsatellites. This is a novel property of the analysis of the motif content of different parts of the genome, and can complement other algorithms or models which do the same. We were also able to find sets of motifs which have a significantly higher occurrence in regulatory regions (promoters and 3′ UTRs). Furthermore, by analyzing the distribution of conserved motifs common to different species, we were able to find genes which could possibly take part in the same physiological processes under the same regulation.
In summary, motif cross-comparison between a number of different plant species provides new and exciting results which can be applied and broadened to other organisms as well to deepen our understanding of the regulation of their genomes.
Materials and methods
Sequence sets
Links and references to the data sets used in this analysis can be seen in Table 1. Here general information on the genomes of each species is listed, such as the number of chromosomes, number of genes, and total genome size. For the dicot species excluding Arabidopsis and also for the monocot, T. aestivum, only the whole genome sequence was available (for Triticum it was available only in contig sequences). B. distachyon had only the whole genome, the core promoter and proximal promoter sets and the 3′ UTRs available, while Z. mays had only the whole genome sequence, and the core, proximal and distal promoter sets available. While these sequence sets may be incomplete as of yet, we felt it worthwhile to analyze the available data and try to draw conclusions from them through multiple species comparisons.
The genome sequence of Arabidopsis has already been done by Lichtenberg at al. [9], however we performed our own analysis on the genome of this plant species, thus we were working with our own Arabidopsis data sets. Since the analysis had already been performed for O. sativa japonica by Cserhati [8], we simply used the data sets already available for that species. The genomes for eight of the nine species were masked using RepeatMasker [24] in order to purge them of repeat sequences. There were technical difficulties with the T. aestivum genome due to the fragmented nature of the genome as it had not yet been assembled into whole chromosome sequences, the program took too long to run.
Motif statistical measure
The statistical significance of a given motif m is sign(m) = S ⋅ ln(S/ES), where S is the number of sequences the motif m occurs in, and ES is the number of sequences the motif is expected to occur in, by calculating the probability of the motif's occurrence based on the background base distribution in a given species. The probability pm can be calculated with the following formula: , where n is the length of the motif, i is a running variable from 1 to n, and pX,i is the ith base in the motif, where X = {A,C,G,T}, and n is the length of the motif (6–10 for hexamers to decamers). Only motifs not containing ambiguous IUPAC letters (M, R, W, S, Y, K, B, D, H, V, or N) were retained. Thus, motifs which are overrepresented in the genome (compared to their expected occurrence) will receive a higher score. Not only are those motifs scored higher which occur relatively more than their expected value (S/E), but especially those with a high occurrence in general (that is why we multiple ln(S/ES) with S, the number of occurrences).
In the whole genome, the expected occurrence of w is ES(m) = Ngenome × pm, where Ngenome is the size of the species' genome, and pm is the occurrence probability of the motif. In the case of the other six sequence sets, ES is calculated somewhat differently. Here we assume that the occurrence of a given motif follows a Poisson distribution. Hence, the number of times the motif is expected to occur is , where Nsequences is the number of sequences within a given sequence set, Ns is the length of all sequences belonging to sequence set S, and pm is the occurrence probability of the motif. According to the Poisson distribution, the number of occurrences of a given motif (defined by the parameter λ) is NS ⋅ pm. The probability pn > 0 of finding at least 1 occurrence of the motif in any sequence is . Thus the expected occurrence of the motif is E = Nsequences⋅ pn > 0.
Motif content comparison
The motif content similarity between two species based on a given sequence set was measured by taking the top 1000 motifs found in the given sequence set and ranking them according to their sequence scores. Only motifs common between both species were retained for the calculation. Both the number of common motifs and the Spearman-coefficient were reported to measure the motif content similarity between the two species.
Motif clustering
For a given sequence set we matched all possible motif pairs from the top 100 highest scoring motifs with each other. Two motifs belonged to the same cluster if the Hamming distance was at most 1 bp. The two motifs were also allowed to slide 1 bp alongside each other. The consensus sequence for a given sequence set was determined with the Clustalw2, version 2.0.12 software [25]. The statistical significance for a consensus sequence was determined similar to the way it was determined for a single motif containing non-ambiguous letters.
The consensus sequences, their observed and expected occurrence, their score can be seen in the Supplementary Excel files for each individual sequence set in each individual species.
Motif pair statistical measure
For a motif pair m1;m2, the probability of finding such a pair is equal to the product of the individual motif probabilities: pm1;m2 = pm1 ⋅ pm2. The significance value for a motif pair can also be calculated similarly with pm1;m2.
Determination of consensus sequences in dicots
For the four dicot species, the top 100 octamers were all compared with one another to determine which sequence was similar to the other, and to what degree. Clusters of motifs from the top 100 motifs were defined where the members of the cluster were similar to at least one other cluster member by a sequence similarity of at least 87.5% (allowing for 1 mismatch). For each cluster the motif members were put into a multi-fasta file and the ClustalW2 program was run on it to determine a consensus sequence for that motif.
Other data and methods
Gene annotations for O. sativa japonica and B. distachyon were downloaded from http://www.plantgdb.org. A list of miRNA sequences and their targets can be found at http://bioinformatics.cau.edu.cn/PMRD/adjunct/osa_miR_target.txt. A multifasta sequence file containing known microRNA sequences was downloaded from http://bioinformatics.cau.edu.cn/PMRD/adjunct/osa_mature [23]. Gtf files for O. sativa japonica and indica, T. aestivum, Z. mays, B. distachyon, A. thaliana, and M. truncatula were all downloaded from http://plants.ensembl.org/info/website/ftp/index.html. The conversion between O. sativa japonica gene ids was performed with this file: http://rapdb.dna.affrc.go.jp/download/archive/RAP-MSU.txt.gz. p-Values for the differences in motif numbers between promoters and 3′ UTRs versus gene were calculated by the Wilcoxon-test in R.
Venn diagrams
The Venn diagrams were calculated using the software at the Bioinformatics and Evolutionary Genomics Workgroup at http://bioinformatics.psb.ugent.be/cgi-bin/liste/Venn/calculate_venn.htpl.
PLACE motif definitions
The supplementary dictionary for defining the functions of the PLACE database motif ids was adapted from http://ftp.dna.affrc.go.jp/pub/dna_place/place.fasta.
Perl script
The perl script can be downloaded from the author's webpage at http://unmc.edu/bsbc/docs/motifome_script.zip.
Footnotes
Supplementary data is available for all the calculations done on Oryza sativa indica, Brachypodium distachyon, Triticum aestivum, Zea mays, Arabidopsis thaliana, Lotus japonicus, Medicago truncatula, and Populus tremula. For supplementary information on Oryza sativa japonica, the reader is referred to [8]. Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.gdata.2014.12.006.
Appendix A. Supplementary data
Supplementary material 1.
Supplementary tables.
Supplementary material 2.
Supplementary material 3.
Supplementary material 4.
Supplementary material 5.
Supplementary material 6.
Supplementary material 7.
Supplementary material 8.
Supplementary material 9.
References
- 1.Web Reference 1. http://www.brachypodium.org/
- 2.Web reference 2. http://rice.genomics.org.cn/rice/index2.jsp
- 3.Web reference 3. ftp://ftp.plantgdb.org/download/Genomes/ZmGDB/
- 4.Web reference 4. http://www.cerealsdb.uk.net/CerealsDB/Documents/DOC_copyright.php
- 5.Web reference 5. ftp://ftp.plantgdb.org/download/Genomes/PtGDB/
- 6.Web reference 6. http://www.dna.affrc.go.jp/PLACE/
- 7.Cannon S.B., Sterck L., Rombauts S., Sato S., Cheung F. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc. Natl. Acad. Sci. U. S. A. Oct. 3 2006;103(40):14959–14964. doi: 10.1073/pnas.0603228103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cserháti M., Turóczy Z., Dudits D., Györgey J. The rice word landscape — a detailed catalog of the rice motif content in the noncoding regions. OMICS. Nov. 2011;15(11):819–828. doi: 10.1089/omi.2011.0132. [DOI] [PubMed] [Google Scholar]
- 9.Lichtenberg J., Yilmaz A., Welch J.D., Kurz K., Liang X., Drews F., Ecker K., Lee S.S., Geisler M., Grotewold E., Welch L.R. The word landscape of the non-coding segments of the Arabidopsis thaliana genome. BMC Genomics. Oct. 8 2009;10:463. doi: 10.1186/1471-2164-10-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geisler M., Kleczkowski L.A., Karpinski S. A universal algorithm for genome-wide in silicio identification of biologically significant gene promoter putative cis-regulatory-elements; identification of new elements for reactive oxygen species and sucrose signaling in Arabidopsis. Plant J. Feb. 2006;45(3):384–398. doi: 10.1111/j.1365-313X.2005.02634.x. [DOI] [PubMed] [Google Scholar]
- 11.Brenchley R., Spannagl M., Pfeifer M., Barker G.L., D'Amore R. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature. Nov. 29 2012;491(7426):705–710. doi: 10.1038/nature11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Higo K., Ugawa Y., Iwamoto M., Korenaga Y. Plant cis-acting regulatory DNA elements (PLACE) database. Nucleic Acids Res. 1999;27(1):297–300. doi: 10.1093/nar/27.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kellogg E.A. Evolutionary history of the grasses. Plant Physiol. Mar. 2001;125(3):1198–1205. doi: 10.1104/pp.125.3.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reinhart B.J., Weinstein E.G., Rhoades M.W., Bartel B., Bartel D.P. MicroRNAs in plants. Genes Dev. Jul. 1 2002;16(13):1616–1626. doi: 10.1101/gad.1004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meng Y., Shao C., Wang H., Jin Y. Target mimics: an embedded layer of microRNA-involved gene regulatory networks in plants. BMC Genomics. May 21 2012;13(1):197. doi: 10.1186/1471-2164-13-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H., Caruso L.V., Downie A.B., Perry S.E. The embryo MADS domain protein AGAMOUS-Like 15 directly regulates expression of a gene encoding an enzyme involved in gibberellin metabolism. Plant Cell. May 2004;16(5):1206–1219. doi: 10.1105/tpc.021261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gasser S.M., Amati B.B., Cardenas M.E., Hofmann J.F. Studies on scaffold attachment sites and their relation to genome function. Int. Rev. Cytol. 1989;119:57–96. doi: 10.1016/s0074-7696(08)60649-x. [DOI] [PubMed] [Google Scholar]
- 18.Sandhu J.S., Webster C.I., Gray J.C. A/T-rich sequences act as quantitative enhancers of gene expression in transgenic tobacco and potato plants. Plant Mol. Biol. Jul. 1998;37(5):885–896. doi: 10.1023/A:1006051832213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santi L., Wang Y., Stile M.R., Berendzen K., Wanke D., Roig C., Pozzi C., Müller K., Müller J., Rohde W., Salamini F. The GA octodinucleotide repeat binding factor BBR participates in the transcriptional regulation of the homeobox gene Bkn3. Plant J. Jun. 2003;34(6):813–826. doi: 10.1046/j.1365-313x.2003.01767.x. [DOI] [PubMed] [Google Scholar]
- 20.Pauli S., Rothnie H.M., Chen G., He X., Hohn T. The cauliflower mosaic virus 35S promoter extends into the transcribed region. J. Virol. Nov. 2004;78(22):12120–12128. doi: 10.1128/JVI.78.22.12120-12128.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou D.X. Regulatory mechanism of plant gene transcription by GT-elements and GT-factors. Trends Plant Sci. Jun. 1999;4(6):210–214. doi: 10.1016/s1360-1385(99)01418-1. [DOI] [PubMed] [Google Scholar]
- 22.Huang H., Tudor M., Weiss C.A., Hu Y., Ma H. The Arabidopsis MADS-box gene AGL3 is widely expressed and encodes a sequence-specific DNA-binding protein. Plant Mol. Biol. Jun. 1995;28(3):549–567. doi: 10.1007/BF00020401. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z., Yu J., Li D., Zhang Z., Liu F., Zhou X., Wang T., Ling Y., Su Z. PMRD: plant microRNA database. Nucleic Acids Res. Jan. 2010;38 doi: 10.1093/nar/gkp818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tempel S. Using and understanding RepeatMasker. Methods Mol. Biol. 2012;859:29–51. doi: 10.1007/978-1-61779-603-6_2. [DOI] [PubMed] [Google Scholar]
- 25.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A. Clustal W and Clustal X version 2.0. Bioinformatics. Nov. 1 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 1.
Supplementary tables.
Supplementary material 2.
Supplementary material 3.
Supplementary material 4.
Supplementary material 5.
Supplementary material 6.
Supplementary material 7.
Supplementary material 8.
Supplementary material 9.