

Abstract
DNA methylation is the current strategy in the field of biomarker discovery due to its prognostic efficiency. Its role in prognosis and early diagnosis has been recognized in various types of cancer. Sepsis still remains one of the major causes of neonatal mortality. Delay in diagnosis of sepsis leads to treatment difficulties and poor outcome. In this study, we have done an epigenome wide search to identify potential markers for prognosis of neonatal sepsis which may improve the treatment strategies. We analyzed the CpG methylation status in the epigenome of three septic and non-septic babies using Illumina Infinium HumanMethylation450K methylation microarray. The microarray data was analyzed with Illumina GenomeStudio v2011.1. After screening for biological and clinical significance, we found 81 differentially methylated CpGs located in 64 genes. Bioinformatic analysis using DAVID and GeneMania revealed a panel of differentially methylated protocadherin beta (PCDHB) genes that play vital role in leukocyte cell adhesion and Wnt signaling pathway. Apart, genes like CCS, DNAJA3, and DEGS2 were potentially hyper/hypo methylated which can be utilized in the development of novel biomarkers. This study will be helpful in exploring the role of DNA methylation in the pathophysiology of neonatal sepsis. The complete microarray data can be accessed from the public domain, Gene Expression Omnibus of NCBI (http://www.ncbi.nlm.nih.gov/geo/). The accession number is GSE58651.
Keywords: DNA methylation, Epigenetics, Neonatal sepsis, Microarray, CpG sites
| Specifications | |
|---|---|
| Organism/cell line/tissue | Homo sapiens |
| Sex | Both |
| Sequencer or array type | Illumina 450K Human Methylation Microarray |
| Data format | Raw and processed |
| Experimental factors | Sepsis vs non-sepsis |
| Experimental features | Microarray based comparison study of genomic DNA methylation pattern among newborns with sepsis and without sepsis |
| Consent | Informed consent from parent/guardian |
| Sample source location | Dravidian population from Tamilnadu, India |
Direct link to deposited data
Experimental design, material and methods
Study population
We conducted this study in the division of Neonatology, Department of Pediatrics of our tertiary care referral hospital during the period of March, 2014 to April, 2014. Our study was approved by Institute Scientific Advisory and Human Ethics Committees. We enrolled six preterm newborns belonging to Dravidian population of Tamilnadu, India, with and without sepsis (two male and one female in each group) in this cohort study following inclusion and exclusion criteria for sepsis. The newborns were matched for basic demographic characteristics like age, sex, birth weight, Apgar, etc. Babies less than 28 days of age with blood culture positive sepsis were enrolled as cases. Babies who had no sepsis and underwent blood sampling for minor ailments were enrolled as controls. Newborns with surgical conditions, major congenital malformations, maternal history of infections/inflammations/antibiotic therapy and Apgar score < 6/10 at 5 min were excluded.
Genomic DNA isolation and bisulfite treatment
After getting written informed consent from the parents, we collected 200 μL of peripheral venous blood in an EDTA vacutainer from the newborns. Genomic DNA was extracted from the whole blood using QIAmp DNA Blood Mini kit (Qiagen, Hilden, Germany) following manufacturer's protocol. The concentration of DNA was measured using Nanodrop 2000 spectrophotometer (Thermo scientific, USA) and quality checked in 1% TopVision LE GQ Agarose (Thermo scientific, USA) gel electrophoresis. Intact genomic DNA was diluted to 50 ng/μL concentration based on Quant-iT Picogreen (Invitrogen) quantitation and 550 ng of DNA was subjected to heat based bisulfite treatment using EZ DNA methylation kit (Zymoresearch, USA).
Illumina 450 K methylation microarray
After bisulfite treatment, the whole genome was amplified, enzymatically fragmented and hybridized to the Illumina Infinium HumanMethylation450 BeadChip kits (Illumina, Inc., San Diego, USA). Following hybridization, allele specific single-base extension and staining were performed and then the BeadChips were imaged on Illumina BeadArray Reader. The image intensities were extracted using Illumina's BeadScan software (Illumina iScan scanner). The fluorescent signals from methylated and unmethylated alleles represented methylation data point from which the background intensity was then subtracted. The microarray service was provided by Macrogen Inc., South Korea.
Microarray data analysis
Array data export processing and analysis were performed using Illumina GenomeStudio v2011.1 (Methylatioin Module v1.9.0) and the statistical computing package R 3.0.2 (http://www.r-project.org). Background correction & Dye bias equalization were done using library (methylumi) in R and is represented in Fig. 1, Fig. 2, respectively.
Fig. 1.

Dye intensities before and after background correction for each sample.
Fig. 2.
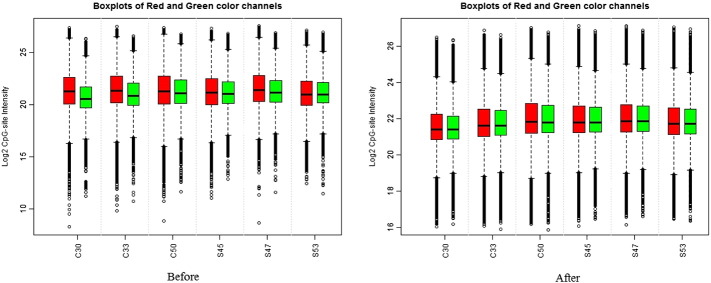
Intensity of red and green dyes before and after dye bias equalization.
The CpGs were filtered out using a detection P-value of < 0.05. On an average, about 485,337 and 485,405 CpGs were detected by detection P-value < 0.01 and detection P-value < 0.05, respectively (Fig. 3). Beta mixture quantile (BMIQ) normalization method was used to reduce the assay bias [1].
Fig. 3.
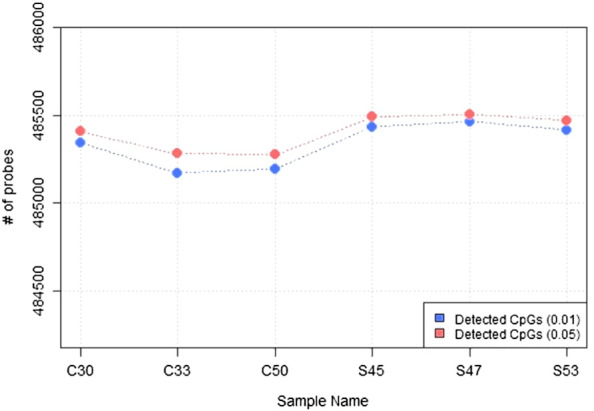
Detected CpGs satisfying detection P-value of < 0.05 and < 0.01.
β-Value reflects the methylation intensity at each CpG site. β-Value of 0 to 1 represented signifying percent methylation, from 0% to 100%, respectively, for each CpG site. Delta mean (Δm) is the average of β-value of cases and controls. The Average β-value (Δm) represented the methylation level but it has heteroscedasticity which is not suitable for the statistical analysis. Beta-value has more sensitive biological interpretation, but the M-value is more statistically applicable for differential analysis of methylation levels and also satisfies homoscedasticity. β-Value was transformed to M-value as the log2 ratio of the intensities of methylated probe versus unmethylated probe [2]. The box plot and density plot to compare the distribution of methylation data before/after BMIQ normalization and data transformation are given in Fig. 4.
Fig. 4.

Distribution of methylation data after BMIQ normalization and transformation. (a) Box plot; (b) density plot.
The degree of reproducibility between samples was assessed using Pearson's correlation scatter and level plots with M-values in the range of − 1 ≤ r ≤ 1 (Fig. 5). Out of 485,350 CpGs analyzed, only 201 CpGs were found differentially methylated with delta mean (Δm) P-value < 0.02 and detection P-value < 0.05. The heat map of hierarchical clustering on distance similarity for samples and significant 201 CpGs was generated by Euclidean distance and complete linkage method with Z-score which is a normalized form of M-value (Fig. 6). The odds ratio and fold change in methylation among cases and controls were calculated. The formulae used for all calculations are given in the supplementary data S1.
Fig. 5.
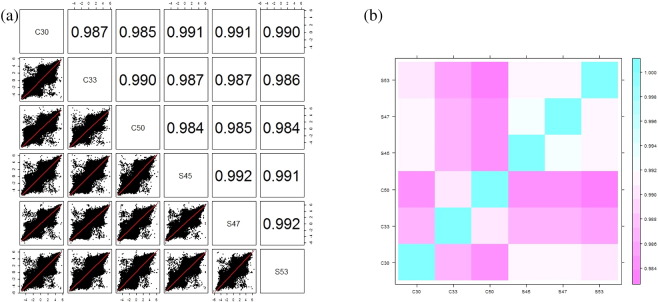
Degree of reproducibility between samples. (a) Scatter plot; (b) level plot.
Fig. 6.
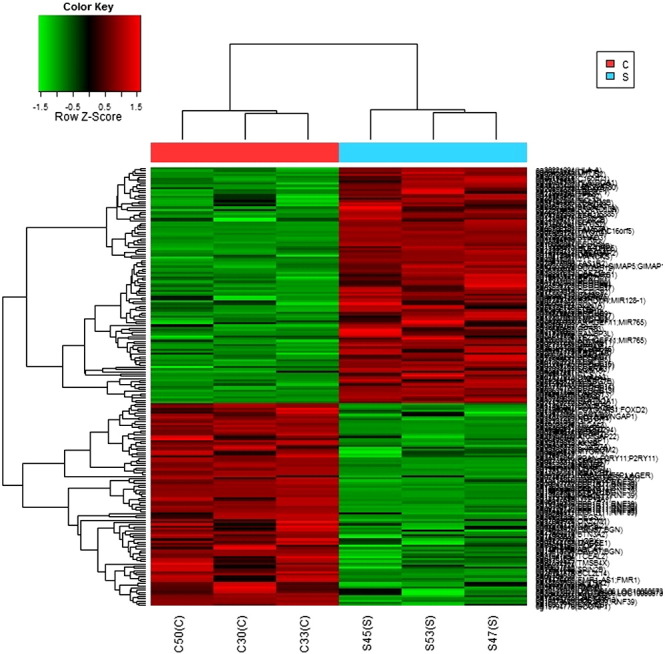
Heat map of hierarchical clustering of 201 significant CpGs.
Functional annotation
Screening of the 201 significant CpGs for gender-specific probes, SNP probes, repeats and unidentified locations, resulted in 81 CpGs (54 hypermethylated and 27 hypomethylated) located in 64 genes. The biological and clinical significance of these genes were identified by annotation clusters using DAVID v6.7 and GeneMania database which is based on Cytoscape plugin.
Statistical analysis
All statistical analyses were carried out at 95% confidence interval. Student t-test was used to compare the methylation levels of septic newborns and non-septic newborns. The false discovery rate was calculated using Benjamini–Hochberg procedure. The complete microarray data can be accessed from the public domain, Gene Expression Omnibus of NCBI (http://www.ncbi.nlm.nih.gov/geo/). The accession number is GSE58651.
Discussion
Functional annotation revealed that a group of PCDHB genes (PCDHB11, PCDHB12, PCDHB16, PCDHB5, PCDHB6, PCDHB7, and PCDHB9) was hypermethylated in newborns with sepsis. This panel of genes is located on chromosome 5q31. To our surprise, the genes in this panel have single exon with CpG island containing not less than 80 CpGs. It was also found that most of other PCDHB genes which were not covered in this study also have similar feature. An epigenome wide association study by Dallosso et al. (2009), showed similar results of protocadherin gene hypermethylation in Wilms' tumor in pediatric patients, especially protocadherin gamma genes [3]. They also found that the expression of protocadherin genes was suppressed due to hypermethylation regulating the WNT/β-catenin signaling pathway in animal models. In recent years, Toll-like-receptor mediated WNT/β-catenin signaling was found to be significant in inflammation during sepsis [4]. Also, a recent study showed that PCDHB10 gene was epigenetically regulated in multiple haematologic malignancies [5]. In our study, bioinformatic analysis showed that PCDHB genes play vital role in calcium dependent cell to cell adhesion and other immunological processes like antigen processing and presentation. In sepsis, suppression of leukocyte cell adhesion and migration may exaggerate disease severity and poor outcome due to multiple organ dysfunctions [6], [7].
Apart from PCDHB genes, other genes like CCS (Copper Chaperone for Superoxide Dismutase) and DEGS2 (Delta(4)-Desaturase, Sphingolipid 2) had the highest fold change in methylation status among hypermethylated and hypomethylated genes, respectively. The biological significance of these genes was not well studied except that they were known to be involved in anti-oxidation and sphingolipid metabolism respectively, during inflammation [8], [9]. This study provides some novel insights into the role of DNA methylation in neonatal sepsis.
Conflicts of interest
NIL.
Acknowledgments
This study was supported by Jawaharlal Institute of Postgraduate Medical Education and Research Intramural Research grant. We thank Indian Council of Medical Research for providing fellowship to the first author.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.gdata.2014.11.004.
Contributor Information
D. Benet Bosco Dhas, Email: benetbiotech@gmail.com.
A. Hiasindh Ashmi, Email: hiasindh.ashmi@gmail.com.
B. Vishnu Bhat, Email: drvishnubhat@yahoo.com.
S. Kalaivani, Email: kalaivani.sekar@ymail.com.
Subash Chandra Parija, Email: subhashparija@yahoo.co.in.
Appendix A. Supplementary data
Statistical Component 1.
References
- 1.Teschendorff A.E., Marabita F., Lechner M., Bartlett T., Tegner J., Gomez-Cabrero D. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29(2):189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Du P., Zhang X., Huang C.C., Jafari N., Kibbe W.A., Hou L., Lin S.M. Comparison of beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:1–9. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dallosso A.R., Hancock A.L., Szemes M., Moorwood K., Chilukamarri L., Tsai H.H. Frequent long-range epigenetic silencing of protocadherin gene clusters on chromosome 5q31 in Wilms' tumor. PLoS Genet. 2009;5(11):e1000745. doi: 10.1371/journal.pgen.1000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villar J., Cabrera N.E., Casula M., Valladares F., Flores C., López-Aguilar J. WNT/β-catenin signaling is modulated by mechanical ventilation in an experimental model of acute lung injury. Intensive Care Med. 2011;37(7):1201–1209. doi: 10.1007/s00134-011-2234-0. [DOI] [PubMed] [Google Scholar]
- 5.Ying J., Gao Z., Li H., Srivastava G., Murray P.G., Goh H.K. Frequent epigenetic silencing of protocadherin 10 by methylation in multiple haematologic malignancies. Br. J. Haematol. 2007;136:829–832. doi: 10.1111/j.1365-2141.2007.06512.x. [DOI] [PubMed] [Google Scholar]
- 6.Liu L., Kubes P. Molecular mechanisms of leukocyte recruitment: organ-specific mechanisms of action. Thromb. Haemost. 2003;89:213–220. [PubMed] [Google Scholar]
- 7.Watanabe S., Mukaida N., Ikeda N., Akiyama M., Harada A., Nakanishi I. Prevention of endotoxin shock by an antibody against leukocyte integrin beta 2 through inhibiting production and action of TNF. Int. Immunol. 1995;7:1037–1046. doi: 10.1093/intimm/7.7.1037. [DOI] [PubMed] [Google Scholar]
- 8.Gongora M.C., Lob H.E., Landmesser U., Guzik T.J., Martin W.D., Ozumi K. Loss of extracellular superoxide dismutase leads to acute lung damage in the presence of ambient air: a potential mechanism underlying adult respiratory distress syndrome. Am. J. Pathol. 2008;173:915–926. doi: 10.2353/ajpath.2008.080119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang S.C., Kim B.R., Lee S.Y., Park T.S. Sphingolipid metabolism and obesity-induced inflammation. Front Endocrinol (Lausanne) 2013;4:1–11. doi: 10.3389/fendo.2013.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Statistical Component 1.