

Summary
Purpose
Benzodiazepines such as diazepam may fail to effectively treat status epilepticus because benzodiazepine-sensitive GABAA receptors are progressively internalized with continued seizure activity. Ionotropic glutamate receptors, including AMPA receptors, are externalized, so that AMPA receptor antagonists, which are broadspectrum anticonvulsants, could be more effective treatments for status epilepticus. We assessed the ability of the noncompetitive AMPA receptor antagonist GYKI 52466 to protect against kainic acid–induced status epilepticus in mice.
Methods
Groups of animals treated with kainic acid received GYKI 52466 (50 mg/kg followed in 15 min by 50 mg/kg) or diazepam (25 mg/kg followed in 20 min by 12.5 mg/kg) beginning at 5 min of continuous seizure activity or 25 min later. The duration of seizure activity was determined by EEG recording from epidural cortical electrodes.
Results
Both GYKI 52466 and diazepam rapidly terminated electrographic and behavioral seizures when administered early. However, diazepam-treated animals exhibited more seizure recurrences. With late administration, GYKI 52466 also rapidly terminated seizures and they seldom recurred, whereas diazepam was slow to produce seizure control and recurrences were common. Although both treatments caused sedation, GYKI 52466-treated animals retained neurological responsiveness whereas diazepam-treated animals did not. GYKI 52466 did not affect blood pressure whereas diazepam caused a sustained drop in mean arterial pressure.
Discussion
Noncompetitive AMPA receptor antagonists represent a promising approach for early treatment of status epilepticus; they may also be effective at later times when there is refractoriness to benzodiazepines.
Keywords: Status epilepticus, Kainic acid, AMPA receptor antagonist, GYKI 52466, Diazepam, Blood pressure
Randomized controlled trials have demonstrated that benzodiazepines are effective in the initial treatment of patients with status epilepticus and they are the accepted standard first line therapy (Prasad et al., 2005). However, these agents successfully terminate status epilepticus in only 43–89% of patients (Leppik et al., 1983; Treiman et al., 1998; Alldredge et al., 2001). Refractoriness of status epilepticus to benzodiazepines likely results from progressive, seizure-induced alterations in GABAA receptors. In the transition from single selflimiting seizures to repeated prolonged seizures, benzodiazepine-sensitive GABAA receptors are internalized and become functionally inactive (Naylor et al., 2005; Goodkin et al., 2005, 2008). Extrasynaptic, benzodiazepine-insensitive GABAA receptors are not internalized, so that their relative abundance increases (Kapur & Macdonald, 1997). In contrast, as synaptic GABAA receptors are functionally inactivated, internal ionotropic glutamate receptors move to synaptic sites and become functionally active, which is believed to increase excitability and promote continued seizure activity (Chen et al., 2007). Both NMDA and AMPA receptors undergo such externalization.
Agents that block these glutamate receptor types could potentially be useful in the treatment of status epilepticus, including refractory status epilepticus resistant to benzodiazepines. In fact, NMDA receptor antagonists, such as ketamine, alone or in combination with benzodiazepine have been found to protect against status epilepticus in animal models (Borris et al., 2000; Martin & Kapur, 2008) and there are anecdotal reports of effectiveness in human status epilepticus (Sheth & Gidal, 1998; Ubogu et al., 2003; Abend & Dlugos, 2008). However, NMDA receptor antagonists may cause neurobehavioral side effects, irreversible neurotoxicity, and may not be effective in some types of human epilepsy (Meldrum & Rogawski, 2007; Abend & Dlugos, 2008). AMPA receptors mediate the bulk of excitatory synaptic neurotransmission in the central nervous system. Antagonists of this class of ionotropic glutamate receptor protect against seizures in diverse animal epilepsy models (Yamaguchi et al., 1993; Rogawski et al., 2001) and may be active even in situations where NMDA receptor antagonists are not (Rutecki et al., 2002). Recently, continuous infusion of a competitive AMPA receptor antagonist was reported to be efficacious in the treatment of experimental status epilepticus in rats (Pitkänen et al., 2007).
GYKI 52466, a 2,3-benzodiazepine, is a highly selective, noncompetitive AMPA receptor antagonist (Donevan & Rogawski, 1993) that has good bioavailability and rapidly penetrates the blood brain barrier due to its lipophilicity (Vizzi et al., 1996). Noncompetitive AMPA receptor antagonists like GYKI 52466 may be more effective than competitive antagonists in some seizure models, possibly because their blocking action cannot be overcome by high levels of glutamate that may be associated with intense seizure activity (Yamaguchi et al., 1993). In the present study, we determined whether GYKI 52466 can effectively treat kainic acid–induced status epilepticus in mice when systemically administered early (5 min) after the onset of continuous seizure activity or at a later time point (25 min after the time of dosing in the early group). For comparison, separate groups were treated with a high dose of diazepam. Drugs used in the treatment of acute status epilepticus, including diazepam, cause adverse cardiovascular effects in animals and humans (Bolme & Fuxe, 1977; Sunzel et al., 1988) that may lead to poor outcome even if seizures are stopped. Therefore, we also compared the effects of GYKI 52466 and diazepam on blood pressure at the doses used to treat status epilepticus.
Materials and Methods
Animals
One hundred twenty-nine SvEv male mice (6–8 weeks old at the beginning of the experiments) were used for this study. Animals were single housed with free access to food and water with controlled temperature (22–24°C), humidity (40–50%) and lighting (artificial 12 h light/dark cycle). The experiments were performed during the light cycle after at least 30 min of acclimation to the experimental room. The animal facilities are fully accredited by the American Association for Accreditation of Laboratory Animal Care and the experiments were performed under a protocol approved by the National Institute of Neurological Disorders and Stroke Animal Care and Use Committee, in full compliance with the Guide for Care and Use of Laboratory Animals of the National Research Council.
Electrode implantation
Mice were stereotaxically implanted with a tripolar electrode unit (MS333/1; Plastics One, Roanoke, VA, U.S.A.) under general anesthesia with ketamine (100 mg/kg, i.p.) and medetomidine (1 mg/kg, i.p.). The electrode tips were positioned through burr holes above the right and left frontal cortices (AP, 0 mm and ML, ±2.5 mm from bregma); the third electrode tip was positioned over the cerebellum to serve as reference (midline, 1 mm caudal to lambda). The electrode tips were seated to contact but did not penetrate the dura mater. The electrode assembly was fixed to the skull with two fixation screws and glued in place with a layer of Geristore resin (generous gift from Den-Mat Holding, Santa Maria, CA, U.S.A.) followed by standard dental acrylic cement. The anesthesia was reversed with atipamezole (0.5 mg/kg, i.p.). Mice were allowed 1 week of recovery before the start of the experiment.
Induction of status epilepticus and treatment protocols
Mice were injected intraperitoneally (i.p.) with kainic acid solution at either a dose of 45 mg/kg (5 min group) or 40 mg/kg (30 min group) during continuous video EEG recording as described below. In preliminary experiments, the 45-mg/kg dose was found to reproducibly induce status epilepticus in all animals. However, high mortality was experienced in the 5 min group without treatment (see Fig. 3). To insure that animals survived to receive the treatment doses, we reduced the kainic acid dose slightly in the 30 min group.
Figure 3.
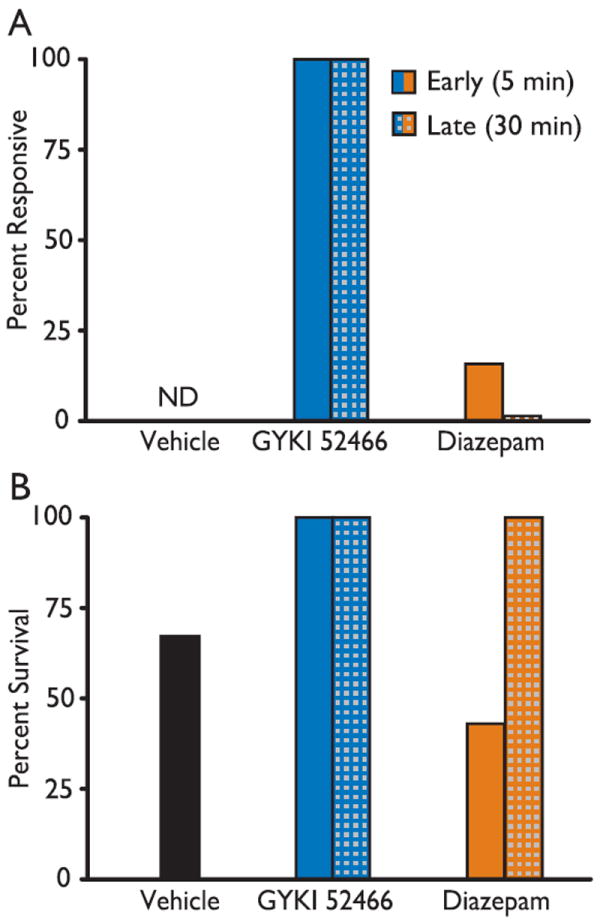
Outcome of the treatment experiment of Fig. 2. (A) Fifteen min after the second treatment dose, mice were mechanically stimulated on the vibrissae and snout to evaluate responsiveness. Animals exhibiting movement of all four limbs were designated as “responsive.” Responsiveness could not be assessed in the vehicle control group as all animals experienced ongoing behavioral seizures (ND). Animals in the 30 min diazepam group also experienced subtle ongoing seizure activity that may have affected the assessment of responsiveness. (B) Comparison of percent survival in the different treatment groups. All animals in the GYKI 52466 group survived whereas some animals died in the diazepam-treated and control groups (the difference did not reach statistical significance by the Fisher’s exact test). Note that animals in the 30 min groups received a lower dose of kainic acid (40 mg/kg), which was not associated with lethality.
Epilepsia © ILAE
In pilot experiments with rats undergoing kainic acid status epilepticus, we determined that a 50 mg/kg dose of GYKI 52466 reproducibly terminated seizure activity whereas lower doses were not effective. Therefore, in the present series of experiments with mice, we routinely used a 50 mg/kg dose administered as a bolus i.p. After i.p. administration, plasma levels of GYKI 52466 peak at 15 min and then fall to 21% of the peak value at 60 min (De Sarro et al., 1998). To maintain sufficient plasma levels to protect against prolonged status epilepticus, we administered a second 50 mg/kg dose 15 min after the first. For comparison, some animals received diazepam as an initial bolus injection of 25 mg/kg followed 20 min later by a 12.5 mg/kg dose, to maintain sufficient brain levels since diazepam reaches its peak brain levels between 5 and 15 min after i.p. administration and decays with a half-life of about 50 min (Friedman et al., 1986).
To investigate the efficacy of the treatments at different times during the status epilepticus, the first GYKI 52466 and diazepam injections were administered at two time points designated “early” and “late.” Following kainic acid treatment, animals exhibited a stuttering course of behavioral and electrographic seizure activity progressing to continuous electrographic discharges or periodic rhythmic discharges. The early group received treatment after the occurrence of 5 min of such continuous electrographic seizure activity with breaks of no longer than 10 s. The late group received treatment 25 min after a 5 min continuous period of seizure discharge (30 min after the onset of the 5 min episode).
Video-EEG recording and analysis
Seizure activity was monitored with a video-EEG monitoring system (Stellate Systems, Montreal, QC, Canada) modified for the use with rodents (Respitech Medical, Lancaster, PA, U.S.A.). Mice were acclimated to the recording cage for 30 min, and 20 min of baseline recording was obtained prior to induction of status epilepticus. The recording continued during the kainic acid treatment and was allowed to run for 5 h post treatment. EEG analysis was carried out offline with Stellate Reviewer software. Status epilepticus was considered terminated when the EEG returned to normal baseline or showed irregular spikes without recurrence of seizures in the subsequent observation period.
Assessment of drug effects on blood pressure and pulse rate
Due to intense body movements, it was not possible to obtain pulse rate or blood pressure measurements in animals undergoing status epilepticus. Therefore, we assessed the cardiovascular effects of GYKI 52466 and diazepam in groups of non–kainic acid treated mice. Arterial blood pressure and pulse rate were monitored from the tail artery with a commercially available blood pressure analysis system that allowed measurements to be obtained from two mice simultaneously (Model SC1000; Hatteras Instruments, Cary, NC, U.S.A.). Mice were restrained on a warming platform maintained at ~38°C. The base of the tail was placed in a computer-controlled pneumatic cuff, and the distal tail was fixed in a sensor assembly that consisted of a light-emitting diode and a photodiode detector. Mice were acclimated to the tail cuff procedure in two daily sessions prior to recording. On the third day, baseline blood pressure values and pulse rate measurements were taken approximately every minute for 10 consecutive readings. Two mice were studied simultaneously to assure comparability. One was treated with GYKI 52466 (50 mg/kg, i.p.) and the other with diazepam (25 mg/kg, i.p.). The first posttreatment measurements were obtained 45–60 s after the injection and 20 consecutive measurements were recorded over the subsequent 20- min period.
Drugs and solutions
Kainic acid [(2S,3S,4R)-carboxy-4-(1-methylethenyl)-3-pyrrolidineacetic acid] and GYKI 52466 were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.). Immediately before use, kainic acid was dissolved in phosphate buffered saline (40 or 45 mg/10 ml). GYKI 52466 was dissolved in 10% β-cyclodextrin (Cyclodextrin Technologies Development, High Springs, FL, U.S.A.) in saline (50 mg/10 ml). 10% β-cyclodextrin in sterile water was used for control (vehicle) treatments. Diazepam was purchased as a solution (50 mg/10 ml) from Hospira (Lake Forest, IL, U.S.A.).
Statistical analysis
Group comparisons of latency to termination of status epilepticus and cumulative seizure duration (as assessed by EEG) were performed by two-tailed Student’s t-test for independent samples after confirmation of normal distribution by the Kolmogorov–Smirnov test. Group comparisons in mortality rate and the rate of response were made with the Fisher exact probability test. Nonparametric tests were used to assess drug effects on pulse rate and blood pressure. A p-value of <0.05 was considered significant.
Results
Kainic acid–induced status epilepticus
Mice treated with kainic acid exhibited the following behaviors typically associated with kainic acid–induced status epilepticus: behavioral arrest with staring, facial twitching, chewing, hind limb scratching, head bobbing, unilateral and bilateral forelimb clonus, rearing with and without loss of postural control, and running and bouncing (Kokate et al., 1996). The behavioral seizure activity in all animals was rated class 5 according to the Racine scale (bilateral forelimb clonus with rearing and loss of postural control). In addition, kainic acid–treated mice exhibited continuous epileptiform EEG activity (Fig. 1). We used EEG activity to determine the time for initiation of the treatment and for outcome assessment rather than the clinical expression of seizures, since the clinical features varied between animals, were difficult to quantify, and were therefore considered to be unreliable as a means of assessing treatment success. Behavioral and EEG seizures began in a stuttering fashion, reaching continuity on average within 27.2 ± 4.3 min after kainic acid injection. When untreated, the seizures continued without interruption for up to 5 h (or until the animals expired, which occurred in a proportion of untreated and diazepam treated animals receiving 45 mg/kg kainic acid). Surviving animals no longer exhibited EEG seizures 24 h after kainic acid treatment.
Figure 1.
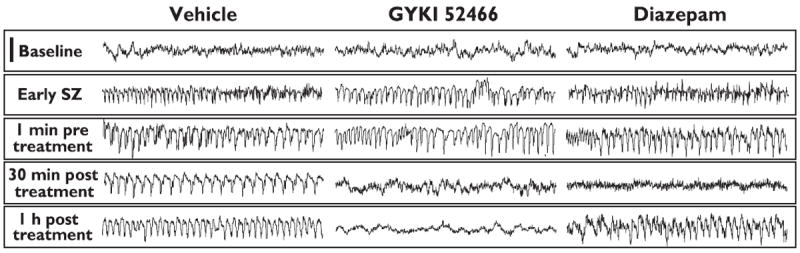
Representative EEG recordings from epidural cortical electrodes in vehicle-, GYKI 52466- and diazepam-treated mice. Status epilepticus was induced by injection of kainic acid (45 mg/kg, i.p.). After 5 min of status epilepticus, the mice were treated either with 10% cyclodextrin in saline (vehicle), two successive 50 mg/kg GYKI 52466 doses separated by 15 min, or 25 mg/kg and 12.5 mg/kg doses of diazepam separated by 15 min. Both active treatments initially eliminated the epileptiform EEG activity as illustrated in the 30 min posttreatment traces; behavioral seizure activity was also terminated. The status epilepticus EEG pattern recurred in the diazepam-treated animal at 1 h post treatment, which was accompanied by subtle behavioral seizures. Each trace represents a 10 s epoch. Scale bar (upper left), 500 μV.
Epilepsia © ILAE
Effects of GYKI 52466 and diazepam on electrographic status epilepticus
Figure 1 illustrates the typical EEG response to GYKI 52466 and diazepam treatment of mice in which status epilepticus had been induced with a 45-mg/kg dose of kainic acid. Treatment was initiated after 5 min of seizure activity. Both treatments caused a cessation of electrographic and behavioral seizure activity but only GYKI 52466 was associated with sustained suppression of electrographic seizures. In the experiment shown, subtle behavioral seizure activity (slight head nodding or forelimb clonus) was apparent in the diazepam-treated animal at 1 h post treatment.
To quantitatively compare the response to the treatments administered after 5 min of status epilepticus, the latency to seizure termination was determined from EEG records in groups of six mice treated with vehicle, GYKI 52466 or diazepam. As illustrated in Fig. 2A, electrographic seizure activity stopped rapidly with both active treatments. There was no significant difference between the latency values in the two active drug groups (GYKI 52466, 14.5 ± 6.2 min; diazepam, 20.1 ± 14.9 min). The total EEG seizure duration during a 5 h monitoring period was also determined (Fig. 2B). Because in the absence of active treatment, seizure activity was continuous during the 5 h observation period, the latency and cumulative seizure duration values for the vehicle treated group are identical (280.0 ± 77.4 min). For the GYKI 52466 treated group, the latency and cumulative seizure duration values are also similar because once seizure activity had stopped in this group, it did not tend to recur during the observation period. The cumulative duration for the GYKI 42466 treated group was 13.5 ± 5.3 min; this value is slightly less than the latency value because there were short breaks in seizure activity before termination. In contrast, for the diazepam group, the cumulative duration value (148.3 ± 24.3) is markedly greater than the latency value because seizure activity recurred in long stretches.
Figure 2.
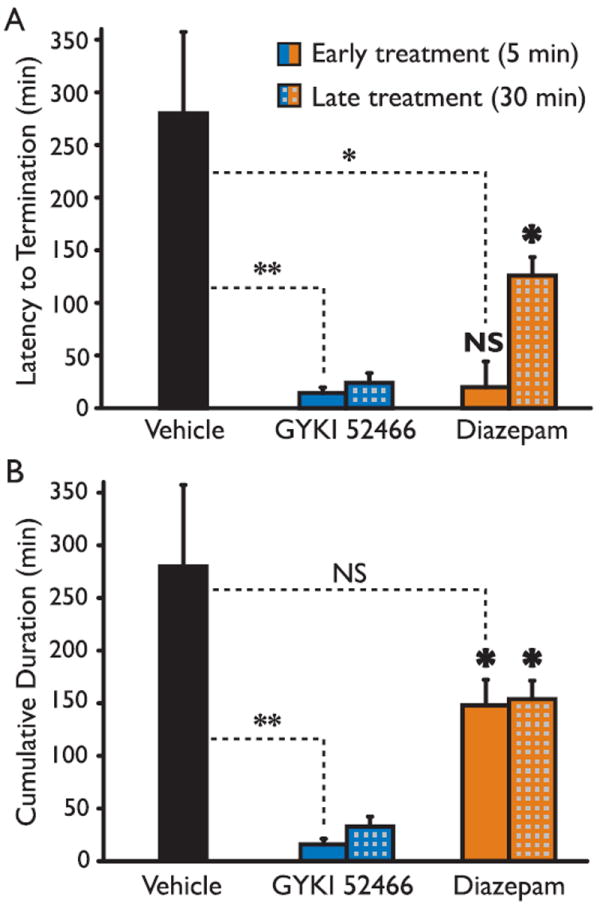
Effects of early and late treatment with GYKI 52466 and diazepam on the duration of EEG seizure activity. Status epilepticus was induced by i.p. injection of kainic acid (vehicle and 5 min treatment group: 45 mg/kg, i.p.; 30 min treatment group: 40 mg/kg, i.p.). GYKI 52466 and diazepam treatment was initiated either 5 min or 30 min after onset of status epilepticus. (A) Mean time to the first termination of continuous seizure activity. (B) Mean cumulative time of seizure activity during the 5 h monitoring period. None of the animals in the vehicle treated group exhibited a break in seizure activity so that the time to first termination and total duration are identical. Each group consisted of 4–6 animals; the total number of mice was 26. Bars indicate mean ± - SEM. Dashed lines indicate comparisons with vehicle: NS, not significant; *p < 0.05; **p < 0.01. Bold symbols indicate comparisons between corresponding GYKI 52466 and diazepam groups.
Epilepsia © ILAE
In a separate series of experiments, the treatments were administered 25 min after an initial 5 min period of continuous electrographic seizure activity. Such late administration of GYKI 52466 was still highly effective in terminating electrographic status epilepticus (latency, 24.5 ± 3.4 min) with only minimal recurrence as is apparent since the latency value and cumulative seizure duration value (33.1 ± 9.3 min) are similar (Fig. 2A,B). Cumulative recurrence durations in the four mice were (in minutes) 0, 0.4, 2.4 and 32.3. As is apparent in Fig. 2A, late administration of diazepam was slower to terminate electrographic status epilepticus (126.3 ± 17.9 min) but once terminated with this treatment protocol, there was only modest recurrence (cumulative, 154.1 ± 17.5 min). Overall, diazepam produced only a modest reduction in the total seizure duration when administered early or late whereas GYKI 52466 was more effective in suppressing seizure activity.
Neurological responsiveness
In clinical settings, the reliable assessment of a patient’s neurological status becomes problematic under the influence of benzodiazepines or barbiturates due to their sedative and muscle relaxant effects. AMPA antagonists are also known to have mild sedative and muscle relaxant side effects (Szabados et al., 1999). In order to determine whether diazepam or GYKI 52466 administered under the treatment conditions used in these experiments produced impairment of neurological responsiveness, we assessed the responsiveness of the mice to mechanical stimulation of the vibrissae 15 min after the second treatment bolus. At this time, GYKI 52466-treated mice exhibited clear sedation and immobility. Nevertheless, all 10 mice showed movement of all four limbs upon mechanical stimulation of the vibrissae (Fig. 3A). All diazepam treated animals were comparably sedated and immobile; only one of 10 showed a motor reaction upon mechanical stimulation. Mice in the vehicle treated group continued to exhibit motor seizures and were not tested for responsiveness to mechanical stimulation as seizure-related and reflex movements were not distinguishable.
Mortality
As illustrated in Fig. 3B, vehicle treated animals that had received a 45 mg/kg dose of kainic acid exhibited 33% mortality. Animals that received GYKI 52466 at 5 or 30 min all survived. The mortality rate in animals receiving diazepam at 5 min (45 mg/kg kainic acid) was greater than in the vehicle group whereas the animals receiving diazepam at 30 min (40 mg/kg kainic acid) did not show mortality.
Body weight
The general condition of the surviving animals was assessed by comparing their weight 24 h after status epilepticus to the weight immediately before kainic acid treatment. None of the groups of surviving animals exhibited statistically significant weight differences. The vehicle and 5 min GYKI 52466 groups exhibited slight weight gain (0.18 ± 0.7 and 0.8 ± 0.4 g, respectively) whereas the 5 min diazepam group exhibited a slight weight loss (0.23 ± 0.3 g). Similarly, the 30 min GYKI 52466 group exhibited a slight weight gain (1.9 ± 0.5 g) whereas the 30 min diazepam group had a slight weight loss (0.4 ± 0.3 g).
Acute effects of GYKI 52466 and diazepam on cardiovascular function
To assess the propensity for the treatments to influence cardiovascular function, pulse rate and blood pressure measurements were made in groups of 10 non–kainic acid treated mice that received either 50 mg/kg GYKI 52466 or 25 mg/kg diazepam (Fig. 4). The average pretreatment pulse rate was comparable in both treatment groups (GYKI 52466, 640.0 ± 10.7 beats/min; diazepam, 623.5 ± 14.6 beats/min). GYKI 52466 and diazepam induced a rapid sustained drop in pulse rate (respectively, 204.4 ± 12.1 beats/min and 126.2 ± 20.2 beats/min), which was significantly greater in GYKI 52466 treated mice (p < 0.01).
Figure 4.
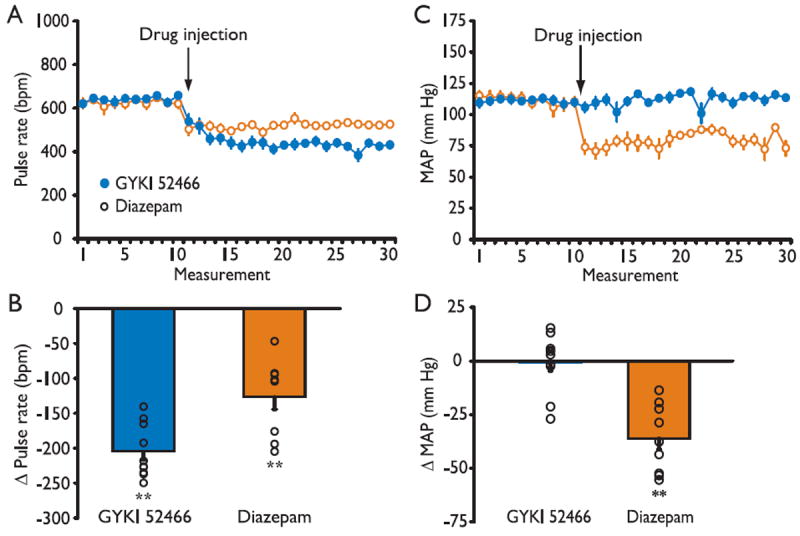
Effects of GYKI 52466 and diazepam on pulse rate (A, B) and mean arterial blood pressure (MAP) (C, D). (A, C) Data points indicate the mean ± SEM of 10 baseline measurements and 20 measurements following treatment with 50 mg/kg GYKI 52466 or 25 mg/kg diazepam. The measurements were made at ~1 min intervals. There were 10 mice in each treatment group. (B, D) Circles indicate percentage change in the mean of all after-treatment pulse rate or MAP values with respect to mean of all baseline values for each animal. Bars indicate mean ± SEM of the mean percentage change values for the 10 animals in each group. The overall reductions in pulse rate for the GYKI 52466 and diazepam groups were significant by the Wilcoxon matched-pairs signed-rank test (p < 0.01). The overall reduction in MAP for the diazepam but not the GYKI 52466 group was significant (p < 0.01). The overall reduction in pulse rate was significantly greater in the GYKI 52466 group than the diazepam group by the Wilcoxon two sample test (p < 0.01). The overall reduction in MAP was significantly greater in the diazepam group than in the GYKI 52466 group (p < 0.001).
Epilepsia © ILAE
Average pretreatment baseline tail-cuff mean arterial pressure (MAP) also did not differ between treatment groups (GYKI 52466, 110.8 ± 2.4 mmHg; diazepam, 111.8 ± 3.4 mmHg). Despite the significant drop in pulse rate, GYKI 52466 treatment did not affect the MAP, whereas diazepam caused a rapid, sustained decrease. Figure 4D shows the change from the mean baseline value of the mean of all of the after-treatment MAP values for each diazepam and GYKI 52466 treated animal. The aggregate after-treatment MAP values are significantly reduced in the diazepam group (p < 0.01) whereas there is no significant change in the GYKI 52466 group. There is a significant difference between the diazepam and GYKI 52466 groups (p < 0.001).
Discussion
AMPA receptor antagonists, including GYKI 52466, have broad-spectrum anticonvulsant activity in a variety of acute rodent seizure models and they are also effective in the amygdala kindling model of chronic epilepsy (Chapman et al., 1991; Yamaguchi et al., 1993; Dürmüller et al., 1994). We now show for the first time that GYKI 52466 can terminate ongoing status epilepticus, providing long-lasting inhibition of electrographic and behavioral seizure activity. Following GYKI 52466 administration, seizure activity stopped rapidly and did not recur, so that all treated animals survived, even in the group that had received 45 mg/kg kainic acid where there was 33% mortality in untreated animals. At the dose used, GYKI 52466 caused sedation but neurological responding to sensory stimulation was maintained. In contrast, diazepam at a more deeply sedative dose was slower to terminate seizure activity and seizures recurred, so that there was high mortality. In experiments to assesses the cariovascular liability of the treatment agents, GYKI 52466 failed to affect blood pressure whereas diazepam caused a sustained reduction in MAP.
GYKI 52466, like other structurally related 2,3-benzodiazepine selective AMPA receptor antagonists, exhibits excellent bioavailability and blood brain barrier penetration (Vizzi et al., 1996). These agents inhibit AMPA receptor function in a noncompetitive fashion with respect to agonists of the receptor and are described as negative allosteric modulators (Donevan and Rogawski, 1998). Recently, by domain swapping and site-directed mutagenesis, it has been possible to localize the binding site at the interface between the S1 and S2 glutamate binding core and channel transmembrane domains (Balannik et al., 2005). Binding stabilizes the resting state of the channel and disrupts the transduction of agonist binding into channel opening. It is noteworthy that noncompetitive AMPA receptor antagonists may have a wider anticonvulsant spectrum in seizure models than competitive antagonists, possibly because their blocking action cannot be overcome by high synaptic glutamate levels (Yamaguchi et al., 1993).
A recent study demonstrated that systemic administration of the competitive (glutamate recognition site) AMPA receptor antagonist NS1209 also was efficacious in treating experimental status epilepticus in rats and that it provided faster and more complete seizure protection than diazepam (Pitkänen et al., 2007). An additional previous study demonstrated that intrahippocampal injection of the competitive AMPA receptor antagonist CNQX transiently suppressed seizures in the rat self-sustaining status epilepticus model (Mazarati & Wasterlain, 1999). Our study does not provide direct evidence that noncompetitive AMPA receptor antagonists are superior to competitive antagonists in the treatment of experimental status epilepticus. However, it is noteworthy that the study with NS1209 utilized a loading bolus injection followed by long-term intravenous infusion (Pitkänen et al., 2007). Continuous infusion was necessary to prevent seizure recurrence. Similarly, seizure recurrence occurred with intrahippocampal CNQX (Mazarati & Wasterlain, 1999). The duration of the anticonvulsant action of NS1209 on electroshock seizure threshold in mice is approximately fivefold longer than the competitive AMPA receptor antagonist NBQX (Nielsen et al., 1999), which has a comparable duration of action as GYKI 52466 in several seizure models, including the kindling model (Chapman et al., 1991; Smith et al., 1991). Therefore, it is surprising that the longer-acting NS1209 required application as continuous infusion whereas in our study GYKI 52466 was effective for many hours after only two bolus injections given 15 min apart. It is plausible that the sustained efficacy of GYKI 52466 is due to its improved bioavailability and the superiority of its noncompetitive mechanism of action.
It is important to terminate generalized convulsive status epilepticus promptly to prevent life threatening systemic complications, including pulmonary congestion and edema, elevation of body temperature, hypoglycemia, acidosis and rhabdomyolysis, which tend to occur as the duration of seizures lengthens (Lothman, 1990; Scholtes et al., 1994). There is also growing evidence from experimental animal data that the extent of status epilepticusrelated neuronal injury correlates with increased seizure duration (Meldrum & Horton, 1973; Fujikawa et al., 2000). Furthermore, refractoriness of status epilepticus to treatment is known to increase with the passage of time (Walton & Treiman, 1988; Mazarati et al., 1998; Treiman et al., 1998; Mayer et al., 2002). In our study, early administration of GYKI 52466 successfully terminated ongoing seizure activity and seizures did not recur, suggesting that AMPA receptor antagonists may be appropriate for the initial treatment of status epilepticus.
As status epilepticus continues, it becomes refractory to therapy with conventional treatment approaches. This was confirmed in the present study, where we found that late administration of diazepam was slow to inhibit seizures and failed to prevent recurrence. Unlike diazepam, administration of GYKI 52466 at the late time point was able to terminate seizures nearly as rapidly as when administered at the early time point and, as in the case of early treatment, there was only minimal seizure recurrence. Therefore, our results suggest that AMPA receptor blockade may be effective in the treatment of late, treatment refractory status epilepticus. As status epilepticus continues, synaptic benzodiazepine-sensitive GABAA receptors are internalized whereas ionotropic glutamate receptors, including AMPA receptors, are trafficked to synaptic sites (Goodkin et al., 2005; Naylor et al., 2005; Chen et al., 2007; Goodkin et al., 2008). It seems plausible that these molecular events underlie the greater efficacy of GYKI 52466 compared with diazepam when administered at the late time point. Our results indicate that the refractoriness to benzodiazepines that accompanies prolonged status epilepticus does not occur to AMPA receptor antagonists, indicating that AMPA receptors are a promising drug target for late status epilepticus treatment.
Like AMPA receptors, NMDA receptors are also externalized during persistent seizure activity suggesting that they could represent a treatment target for status epilepticus. Several studies have demonstrated that NMDA receptor antagonists control status epilepticus activity at late stages, but may lack efficacy during early and intermediate stages (Ormandy et al., 1989; Bertram & Lothman, 1990; Clifford et al., 1990; Mazarati et al., 1999; Borris et al., 2000). In the control of status epilepticus, noncompetitive NMDA receptor antagonists tend to be more effective than those that act competitively (Yen et al., 2004), as our results suggest may be the case for AMPA receptor antagonists. However, the available clinical data indicates that NMDA receptor antagonists may not be effective in controlling seizures in human epilepsy (Löscher & Rogawski, 2002) whereas preliminary evidence from at least one clinical trial of a 2,3-benzodiazepine analog of GYKI 52466 suggests that AMPA receptor antagonists may be more promising (Chappell et al., 2002). Moreover, NMDA receptor antagonists may have greater neurobehavioral toxicity than AMPA receptor antagonists (Löscher & Rogawski, 2002). Therefore, NMDA receptors seem to be a less favorable target for the development of antiepileptic drugs in general and specifically for the treatment of status epilepticus.
The dose of GYKI 52466 used in our study (100 mg/kg) is at the upper range of the ED50 values (3.6–100 mg/kg, i.p.) that have been found to confer seizure protection in a diverse group of rodent seizure models (Yamaguchi et al., 1993; Szabados et al., 2001). Therefore, although the dose was chosen empirically, it is in line with the protective doses in the acute seizure models, recognizing that the objective of the present experiments was to obtain complete and persistent seizure protection. It is noteworthy that many conventional antiepileptic drugs including phenytoin, carbamazepine, ethosuximide, valproate, topiramate and vigabatrin are inactive against seizures induced by systemic kainic acid, even when they are administered at high doses (Clifford et al., 1982; Cramer et al., 1994; Klitgaard et al., 1998; Kubera et al., 2004). Phenobarbital has been found to inhibit kainic acidinduced behavioral seizures and epileptiform discharges, but only at highly sedating doses that depress blood pressure and may cause lethality (Clifford et al., 1982; Klitgaard et al., 1998). Similarly, as confirmed in the present study, diazepam protects against kainic acid–induced electrographic activity but only at sedating doses that depress blood pressure (Clifford et al., 1982).
Although GYKI 52466 seems to be the most effective and least toxic treatment for kainic acid–induced status epilepticus described to date, it was not without side effects at the dose required to eliminate seizures. Mice receiving this dose appeared to be sedated, however, in contrast to the animals that had received diazepam that failed to respond to sensory stimulation, slight mechanical manipulation of the snout elicited movements of all limbs towards the site of stimulation, indicating only moderate sedation. Such a level of sedation would allow some assessment of neurological status and would not prohibit use of anAMPA receptor antagonist in an intensive care clinical setting. Overall good tolerability of GYKI 52466 was reflected by the observation that animals gained weight normally after they had recovered from the status epilepticus episode.
Respiratory and cardiovascular depression are common complications of standard treatment approaches for status epilepticus (Tomson & Kennebäck, 1997; Olkkola & Ahonen, 2008). Generally, cerebrovascular autoregulatory mechanisms are unable to maintain cerebral blood flow (CBF) if MAP falls below 70 mmHg (Drummond, 1997). When MAP drops below this lower limit during status epilepticus treatment, reductions in CBF may lead to irreversible brain damage (Chillon & Baumbach, 1995), which compounds damage due to ongoing seizure activity. As noted previously, diazepam treatment of status epilepticus in animal models is associated with blood pressure reduction. Intravenous bolus injection of diazepam, as administered in the treatment of status epilepticus, is similarly recognized to decrease MAP in human volunteers (Sunzel et al., 1988; Kitajima et al., 2004). Phenytoin (or phosphenytoin), which along with benzodiazepines is part of the standard treatment regimen for status epilepticus, also has significant cardiovascular side effects, including bradyarrhythmia and rare asystole (Tomson & Kennebäck, 1997; Adams et al., 2006). It has been observed that up to 59% of patients treated with the combination of diazepam (or lorazepam) and phenytoin experience a drop in blood pressure (Treiman et al., 1998). Using a noninvasive approach for measurement of blood pressure, we found that the dose of diazepam used in this study, which incompletely protected against status epilepticus, caused a significant drop in MAP. In contrast, GYKI 52466 at a dose that was more efficacious in eliminating seizure activity caused a pronounced fall in pulse rate but did not affect MAP, indicating that cardiovascular autoregulatory mechanisms are unimpaired. Although we did not assess the action of GYKI 52466 on respiratory function, it has previously been reported that much higher doses of GYKI 52466 (up to 200 mg/kg) do not cause serious respiratory complications even in mice that are completely paralyzed for several hours (Vizzi et al., 1996).
In conclusion, the results of the present study support the potential utility of AMPA receptor antagonists in the treatment of status epilepticus (Pitkänen et al., 2007), and suggest that 2,3-benzodiazepine noncompetitive AMPA receptor antagonists may be superior in this application to agents that act by a competitive mechanism. The prototype AMPA receptor antagonist we studied was highly effective in stopping seizure activity when administered early in the course of status epilepticus and equally efficacious in the treatment of late, benzodiazepine-refractory status epilepticus. The AMPA receptor antagonist not only rapidly terminated status epilepticus but recurrences, as occurred with diazepam, were avoided. Our results indicate that AMPA receptor antagonists may allow single agent therapy for status epilepticus, which would avoid the complexity and cardiovascular liability of the current standard two-agent regimen – a benzodiazepine followed by phenytoin (or phosphenytoin) – both of which can cause cardiovascular side effects. There is little evidence that AMPA receptors play a significant functional role in the peripheral vasomotor nervous system, heart or smooth muscle (Erdö, 1991) and this likely accounts for the benign cardiovascular profile of AMPA receptor antagonists. Overall, AMPA receptor antagonists have promise for the acute treatment of status epilepticus.
Acknowledgments
We thank Dr. Jean-Pierre Thierry for providing the noninvasive blood-pressure system and for his assistance with the acquisition of the data; Messrs. David Ide and Danny Trang for technical support; Dr. Janine Reis for commenting on the manuscript; and Den-Mat Holdings, especially Ms. Amber Touchstone, for making the Geristore® resin available. Dr. Brita Fritsch was supported by the NINDS Intramural Research Program.
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Footnotes
Disclosure: None of the authors has any conflict of interest to disclose.
References
- Abend NS, Dlugos DJ. Treatment of refractory status epilepticus: literature review and a proposed protocol. Pediatr Neurol. 2008;38:377–390. doi: 10.1016/j.pediatrneurol.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Adams BD, Buckley NH, Kim JY, Tipps LB. Fosphenytoin may cause hemodynamically unstable bradydysrhythmias. J Emerg Med. 2006;30:75–79. doi: 10.1016/j.jemermed.2005.01.034. [DOI] [PubMed] [Google Scholar]
- Alldredge BK, Gelb AM, Isaacs SM, Corry MD, Allen F, Ulrich S, Gottwald MD, O’Neil N, Neuhaus JM, Segal MR, Lowenstein DH. A comparison of lorazepam, diazepam, and placebo for the treatment of out-of-hospital status epilepticus. N Engl J Med. 2001;345:631–637. doi: 10.1056/NEJMoa002141. [DOI] [PubMed] [Google Scholar]
- Balannik V, Menniti FS, Paternain AV, Lerma J, Stern-Bach Y. Molecular mechanism of AMPA receptor noncompetitive antagonism. Neuron. 2005;48:279–288. doi: 10.1016/j.neuron.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Bertram EH, Lothman EW. NMDA receptor antagonists and limbic status epilepticus: a comparison with standard anticonvulsants. Epilepsy Res. 1990;5:177–184. doi: 10.1016/0920-1211(90)90036-u. [DOI] [PubMed] [Google Scholar]
- Bolme P, Fuxe K. Possible involvement of GABA mechanisms in central cardiovascular and respiratory control. Studies on the interaction between diazepam, picrotoxin and clonidine. Med Biol. 1977;55:301–309. [PubMed] [Google Scholar]
- Borris DJ, Bertram EH, Kapur J. Ketamine controls prolonged status epilepticus. Epilepsy Res. 2000;42:117–122. doi: 10.1016/s0920-1211(00)00175-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman AG, Smith SE, Meldrum BS. The anticonvulsant effect of the non-NMDA antagonists, NBQX and GYKI 52466, in mice. Epilepsy Res. 1991;9:92–96. doi: 10.1016/0920-1211(91)90018-b. [DOI] [PubMed] [Google Scholar]
- Chappell AS, Sander JW, Brodie MJ, Chadwick D, Lledo A, Zhang D, Bjerke J, Kiesler GM, Arroyo S. A crossover, add-on trial of talampanel in patients with refractory partial seizures. Neurology. 2002;58:1680–1682. doi: 10.1212/wnl.58.11.1680. [DOI] [PubMed] [Google Scholar]
- Chen JW, Naylor DE, Wasterlain CG. Advances in the pathophysiology of status epilepticus. Acta Neurol Scand. 2007;115(4 Suppl):7–15. doi: 10.1111/j.1600-0404.2007.00803.x. [DOI] [PubMed] [Google Scholar]
- Chillon JM, Baumbach GL. Autoregulation of cerebral blood flow. In: Welch KMA, Caplan LR, Reis DJ, Siesjö BK, Weir B, editors. Primer on cerebrovascular diseases. Academic Press; San Diego: 1995. p. 51. [Google Scholar]
- Clifford DB, Lothman EW, Dodson WE, Ferrendelli JA. Effect of anticonvulsant drugs on kainic acid-induced epileptiform activity. Exp Neurol. 1982;76:156–167. doi: 10.1016/0014-4886(82)90109-1. [DOI] [PubMed] [Google Scholar]
- Clifford DB, Olney JW, Benz AM, Fuller TA, Zorumski CF. Ketamine, phencyclidine, and MK-801 protect against kainic acid-induced seizure-related brain damage. Epilepsia. 1990;31:382–390. doi: 10.1111/j.1528-1157.1990.tb05492.x. [DOI] [PubMed] [Google Scholar]
- Cramer CL, Stagnitto ML, Knowles MA, Palmer GC. Kainic acid and 4-aminopyridine seizure models in mice: evaluation of efficacy of anti-epileptic agents and calcium antagonists. Life Sci. 1994;54:PL271–PL275. doi: 10.1016/0024-3205(94)00845-0. [DOI] [PubMed] [Google Scholar]
- De Sarro G, Rizzo M, Sinopoli VA, Gitto R, De Sarro A, Zappala M, Chimirri A. Relationship between anticonvulsant activity and plasma level of some 2,3-benzodiazepines in genetically epilepsyprone rats. Pharmacol Biochem Behav. 1998;61:215–220. doi: 10.1016/s0091-3057(98)00036-7. [DOI] [PubMed] [Google Scholar]
- Donevan SD, Rogawski MA. GYKI 52466, a 2,3-benzodiazepine, is a highly selective, noncompetitive antagonist of AMPA/kainate receptor responses. Neuron. 1993;10:51–59. doi: 10.1016/0896-6273(93)90241-i. [DOI] [PubMed] [Google Scholar]
- Donevan SD, Rogawski MA. Allosteric regulation of α-amino-3-hydroxy-5-methyl-4-isoxazole-propionate receptors by thiocyanate and cyclothiazide at a common modulatory site distinct from that of 2,3-benzodiazepines. Neuroscience. 1998;87:615–629. doi: 10.1016/s0306-4522(98)00109-2. [DOI] [PubMed] [Google Scholar]
- Drummond JC. The lower limit of autoregulation: time to revise our thinking? Anesthesiology. 1997;86:1431–1433. doi: 10.1097/00000542-199706000-00034. [DOI] [PubMed] [Google Scholar]
- Dürmüller N, Craggs M, Meldrum BS. The effect of the non-NMDA receptor antagonist GYKI 52466 and NBQX and the competitive NMDA receptor antagonist D-CPPene on the development of amygdala kindling and on amygdala-kindled seizures. Epilepsy Res. 1994;17:167–174. doi: 10.1016/0920-1211(94)90016-7. [DOI] [PubMed] [Google Scholar]
- Erdö SL. Excitatory amino acid receptors in the mammalian periphery. Trends Pharmacol Sci. 1991;12:426–429. doi: 10.1016/0165-6147(91)90622-y. [DOI] [PubMed] [Google Scholar]
- Friedman H, Abernethy DR, Greenblatt DJ, Shader RI. The pharmacokinetics of diazepam and desmethyldiazepam in rat brain and plasma. Psychopharmacology (Berl) 1986;88:267–270. doi: 10.1007/BF00180822. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Itabashi HH, Wu A, Shinmei SS. Status epilepticus-induced neuronal loss in humans without systemic complications or epilepsy. Epilepsia. 2000;41:981–991. doi: 10.1111/j.1528-1157.2000.tb00283.x. [DOI] [PubMed] [Google Scholar]
- Goodkin HP, Yeh JL, Kapur J. Status epilepticus increases the intracellular accumulation of GABAA receptors. J Neurosci. 2005;25:5511–5520. doi: 10.1523/JNEUROSCI.0900-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, Joshi S, Mtchedlishvili Z, Brar J, Kapur J. Subunit-specific trafficking of GABAA receptors during status epilepticus. J Neurosci. 2008;28:2527–2538. doi: 10.1523/JNEUROSCI.3426-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci. 1997;17:7532–7540. doi: 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima T, Kanbayashi T, Saito Y, Takahashi Y, Ogawa Y, Sugiyama T, Kaneko Y, Aizawa R, Shimizu T. Diazepam reduces both arterial blood pressure and muscle sympathetic nerve activity in human. Neurosci Lett. 2004;355:77–80. doi: 10.1016/j.neulet.2003.10.054. [DOI] [PubMed] [Google Scholar]
- Klitgaard H, Matagne A, Gobert J, Wülfert E. Evidence for a unique profile of levetiracetam in rodent models of seizures and epilepsy. Eur J Pharmacol. 1998;353:191–206. doi: 10.1016/s0014-2999(98)00410-5. [DOI] [PubMed] [Google Scholar]
- Kokate TG, Cohen AL, Karp E, Rogawski MA. Neuroactive steroids protect against pilocarpine- and kainic acid-induced limbic seizures and status epilepticus in mice. Neuropharmacology. 1996;35:1049–1056. doi: 10.1016/s0028-3908(96)00021-4. [DOI] [PubMed] [Google Scholar]
- Kubera M, Budziszewska B, Jaworska-Feil L, Basta-Kaim A, Leśkiewicz M, Tetich M, Maes M, Kenis G, Marciniak A, Czuczwar SJ, Jagła G, Nowak W, Lasoń W. Effect of topiramate on the kainateinduced status epilepticus, lipid peroxidation and immunoreactivity of rats. Pol J Pharmacol. 2004;56:553–561. [PubMed] [Google Scholar]
- Leppik IE, Derivan AT, Homan RW, Walker J, Ramsay RE, Patrick B. Double-blind study of lorazepam and diazepam in status epilepticus. JAMA. 1983;249:1452–1454. [PubMed] [Google Scholar]
- Löscher W, Rogawski MA. Epilepsy. In: Lodge D, Danysz W, Parsons CG, editors. Ionotropic glutamate receptors as therapeutic targets. F.P. Graham Publishing Co.; Johnson City, TN: 2002. pp. 91–132. [Google Scholar]
- Lothman E. The biochemical basis and pathophysiology of status epilepticus. Neurology. 1990;40:13–23. [PubMed] [Google Scholar]
- Martin BS, Kapur J. A combination of ketamine and diazepam synergistically controls refractory status epilepticus induced by cholinergic stimulation. Epilepsia. 2008;49:248–255. doi: 10.1111/j.1528-1167.2007.01384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer SA, Claassen J, Lokin J, Mendelsohn F, Dennis LJ, Fitzsimmons BF. Refractory status epilepticus: frequency, risk factors, and impact on outcome. Arch Neurol. 2002;59:205–210. doi: 10.1001/archneur.59.2.205. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Wasterlain CG. N-methyl-d-asparate receptor antagonists abolish the maintenance phase of self-sustaining status epilepticus in rat. Neurosci Lett. 1999;265:187–190. doi: 10.1016/s0304-3940(99)00238-4. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Baldwin RA, Sankar R, Wasterlain CG. Time-dependent decrease in the effectiveness of antiepileptic drugs during the course of self-sustaining status epilepticus. Brain Res. 1998;814:179–185. doi: 10.1016/s0006-8993(98)01080-4. [DOI] [PubMed] [Google Scholar]
- Meldrum BS, Horton RW. Physiology of status epilepticus in primates. Arch Neurol. 1973;28:1–9. doi: 10.1001/archneur.1973.00490190019001. [DOI] [PubMed] [Google Scholar]
- Meldrum BS, Rogawski MA. Molecular targets for antiepileptic drug development. Neurotherapeutics. 2007;4:18–61. doi: 10.1016/j.nurt.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor DE, Liu H, Wasterlain CG. Trafficking of GABAA receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci. 2005;25:7724–7733. doi: 10.1523/JNEUROSCI.4944-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen EØ, Varming T, Mathiesen C, Jensen LH, Møller A, Gouliaev AH, Wätjen F, Drejer J. SPD 502: a water-soluble and in vivo long-lasting AMPA antagonist with neuroprotective activity. J Pharmacol Exp Ther. 1999;289:1492–1501. [PubMed] [Google Scholar]
- Olkkola KT, Ahonen J. Midazolam and other benzodiazepines. Handb Exp Pharmacol. 2008;182:335–360. doi: 10.1007/978-3-540-74806-9_16. [DOI] [PubMed] [Google Scholar]
- Ormandy GC, Jope RS, Snead OC., III Anticonvulsant actions of MK-801 on the lithium-pilocarpine model of status epilepticus in rats. Exp Neurol. 1989;106:172–180. doi: 10.1016/0014-4886(89)90091-5. [DOI] [PubMed] [Google Scholar]
- Pitkänen A, Mathiesen C, Rønn LC, Møller A, Nissinen J. Effect of novel AMPA antagonist, NS1209, on status epilepticus. An experimental study in rat. Epilepsy Res. 2007;74:45–54. doi: 10.1016/j.eplepsyres.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Prasad K, Al-Roomi K, Krishnan PR, Sequeira R. Anticonvulsant therapy for status epilepticus. Cochrane Database Syst Rev. 2005;4 doi: 10.1002/14651858.CD003723.pub2. CD003723. [DOI] [PubMed] [Google Scholar]
- Rogawski MA, Kurzman PS, Yamaguchi SI, Li H. Role of AMPA and GluR5 kainate receptors in the development and expression of amygdala kindling in the mouse. Neuropharmacology. 2001;40:28–35. doi: 10.1016/s0028-3908(00)00112-x. [DOI] [PubMed] [Google Scholar]
- Rutecki PA, Sayin U, Yang Y, Hadar E. Determinants of ictal epileptiform patterns in the hippocampal slice. Epilepsia. 2002;43(Suppl 5):179–183. doi: 10.1046/j.1528-1157.43.s.5.34.x. [DOI] [PubMed] [Google Scholar]
- Scholtes FB, Renier WO, Meinardi H. Generalized convulsive status epilepticus: causes, therapy, and outcome in 346 patients. Epilepsia. 1994;35:1104–1112. doi: 10.1111/j.1528-1157.1994.tb02562.x. [DOI] [PubMed] [Google Scholar]
- Sheth RD, Gidal BE. Refractory status epilepticus: response to ketamine. Neurology. 1998;51:1765–1766. doi: 10.1212/wnl.51.6.1765. [DOI] [PubMed] [Google Scholar]
- Smith SE, Dürmüller N, Meldrum BS. The non-N-methyl-D-aspartate receptor antagonists, GYKI 52466 and NBQX are anticonvulsant in two animal models of reflex epilepsy. Eur J Pharmacol. 1991;201:179–183. doi: 10.1016/0014-2999(91)90342-n. [DOI] [PubMed] [Google Scholar]
- Sunzel M, Paalzow L, Berggren L, Eriksson I. Respiratory and cardiovascular effects in relation to plasma levels of midazolam and diazepam. Br J Clin Pharmacol. 1988;25:561–569. doi: 10.1111/j.1365-2125.1988.tb03346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabados T, Gigler G, Gyertyán I, Gacsályi I, Lévay G. Duration of action of GYKI 52466 and its analogues in antiepileptic, anti-ischemic and muscle relaxant tests. Neurobiology. 1999;7:87–88. [PubMed] [Google Scholar]
- Szabados T, Gigler G, Gacsályi I, Gyertyán I, Lévay G. Comparison of anticonvulsive and acute neuroprotective activity of three 2,3-benzodiazepine compounds, GYKI 52466, GYKI 53405, and GYKI 53655. Brain Res Bull. 2001;55:387–391. doi: 10.1016/s0361-9230(01)00516-0. [DOI] [PubMed] [Google Scholar]
- Tomson T, Kennebäck G. Arrhythmia, heart rate variability, and antiepileptic drugs. Epilepsia. 1997;38(Suppl 11):s48–s51. doi: 10.1111/j.1528-1157.1997.tb06128.x. [DOI] [PubMed] [Google Scholar]
- Treiman DM, Meyers PD, Walton NY, Collins JF, Colling C, Rowan AJ, Handforth A, Faught E, Calabrese VP, Uthman BM, Ramsay RE, Mamdani MB. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med. 1998;339:792–798. doi: 10.1056/NEJM199809173391202. [DOI] [PubMed] [Google Scholar]
- Ubogu EE, Sagar SM, Lerner AJ, Maddux BN, Suarez JI, Werz MA. Ketamine for refractory status epilepticus: a case of possible ketamine-induced neurotoxicity. Epilepsy Behav. 2003;4:70–75. doi: 10.1016/s1525-5050(02)00643-1. [DOI] [PubMed] [Google Scholar]
- Vizzi ES, Mike A, Tarnawa I. 2,3-benzodiazepines (GYKI 52466 and analogs): Negative allosteric modulators of AMPA receptors. CNS Drug Rev. 1996;2:91–126. [Google Scholar]
- Walton NY, Treiman DM. Response of status epilepticus induced by lithium and pilocarpine to treatment with diazepam. Exp Neurol. 1988;101:267–275. doi: 10.1016/0014-4886(88)90010-6. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Donevan SD, Rogawski MA. Anticonvulsant activity of AMPA/kainate antagonists: comparison of GYKI 52466 and NBOX in maximal electroshock and chemoconvulsant seizure models. Epilepsy Res. 1993;15:179–184. doi: 10.1016/0920-1211(93)90054-b. [DOI] [PubMed] [Google Scholar]
- Yen W, Williamson J, Bertram EH, Kapur J. A comparison of three NMDAreceptor antagonists in the treatment of prolonged status epilepticus. Epilepsy Res. 2004;59:43–50. doi: 10.1016/j.eplepsyres.2004.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]