

Abstract
For more than 50 years, it has been recognized that immunity contributes to hypertension. Recent data have defined an important role of T cells and various T cell-derived cytokines in several models of experimental hypertension. These studies have shown that stimuli like angiotensin II, DOCA-salt and excessive catecholamines lead to formation of effector like T cells that infiltrate the kidney and perivascular regions of both large arteries and arterioles. There is also accumulation of monocyte/macrophages in these regions. Cytokines released from these cells, including IL-17, IFN-γ, TNFα and IL-6 promote both renal and vascular dysfunction and damage, leading to enhanced sodium retention and increased systemic vascular resistance. The renal effects of these cytokines remain to be fully defined, but include enhanced formation of angiotensinogen, increased sodium reabsorption and increased renal fibrosis. Very recent experiments have defined a link between oxidative stress and immune activation in hypertension. These have shown that hypertension is associated with formation of reactive oxygen species in dendritic cells that lead to formation of gamma ketoaldehydes, or isoketals. These rapidly adduct to protein lysines and are presented by dendritic cells as neoantigens that activate T cells and promote hypertension. Thus, cells of both the innate and adaptive immune system contribute to end-organ damage and dysfunction in hypertension. Therapeutic interventions to reduce activation of these cells may prove beneficial in reducing end-organ damage and preventing consequences of hypertension including myocardial infarction, heart failure, renal failure and stroke.
Keywords: cytokines, effector T cell, antigen presenting cell, nitric oxide synthase, angiotensin II, sodium
Introduction
Hypertension affects one-third of Western populations and increases in frequency with age, such that 70% of adults develop this disease by age 70. Hypertension is also a major risk factor for stroke, myocardial infarction, renal failure, and heart failure, and therefore is an enormous health care burden. Despite its prevalence, the etiology of most cases of adult hypertension, or essential hypertension, remains unknown. Perturbations of the kidneys, vasculature, and central nervous system have all been implicated in hypertension. In the past several years, it has become increasingly evident that hypertension is an inflammatory process that involves the transmigration and accumulation of both innate and adaptive immune cells into the interstitium of affected tissues where they release cytokines and promote oxidative stress. In this review, we will discuss how these cells contribute to dysfunction of the kidney and vasculature, promoting blood pressure elevation and end-organ damage.
Historical perspectives
The concept that immune cells contribute to hypertension is not new. Almost one-half century ago, Grollman and White showed that immunosuppression lowers blood pressure in rats with partial renal infarction,1 and found that these animals develop antibodies to renal tissue. Importantly, these pioneering investigators showed that transfer of lymph node cells from rats with renal infarction raised blood pressure in normal recipient rats.2 In 1970, Finn Olsen described an inflammatory reaction of blood vessels in response to angiotensin II infusion in rats.3 He noted “The cellular reaction was predominantly composed of mononuclear cells derived from the blood. The majority … looked like lymphocytes, and the rest like typical monocytes.” He went on to describe the time course and location of the cellular infiltration. “The reaction began as a sticking phenomenon corresponding to the damaged endothelium followed by a penetration of mononuclear cells into the arteriolar walls…. A marked periarteriolar cellular infiltration like that seen in cases of chronic hypertensive vascular disease in different experimental animals was produced…” In a subsequent paper published in 1972,4 Dr. Olsen showed that vascular inflammation occurs in humans with a variety of causes of hypertension. Again, he noted “The cellular infiltration was composed of mononuclear cells exclusively which adhered to the surface of the endothelium of the vessels or had penetrated into the tunica media or the adventitia.” Indeed, subsequent studies as described below have identified the adventitia and perivascular adipose tissue of both large and small vessels as sites of immune cell accumulation in hypertension.
Following the early observations by Grollman, White, and Olsen, a number of studies appeared supporting the role of immune cells in hypertension. These described perturbations of antibodies in the Spontaneously Hypertensive Rat (SHR)5–7 and reduced hypertensive responses in athymic nude mice. Bendich et al found that treatment with anti-thymocyte serum lowers blood pressure in the SHR,8 and the immunosuppressant cyclophosphamide was also found to have anti-hypertensive effects.9 Subsequent experiments by Finn Olsen showed that transfer of splenocytes from rats with deoxycorticosterone (DOCA)-salt hypertension raises blood pressure in recipient rats.10
Thus, by the 1980s, a substantial body of data suggested that immune cells participate in hypertension, although the mechanisms were poorly understood. Unfortunately, this field seemed to stagnate for nearly two decades after these initial observations. This may partly have been due to a lack of understanding of the immune system and a paucity of tools available to further study this topic. Fortunately, the field of immunology has dramatically expanded in recent years. Our immunologist colleagues have defined subsets of innate and adaptive immune cells and gained insights into mechanisms by which the innate and adaptive immune systems interact. Myriad cytokines and chemokines have been identified that orchestrate inflammatory reactions, as have the signaling pathways that guide their synthesis. There has been an explosion of therapeutic interventions for immune/autoimmune diseases that can be used to probe events in hypertension, not only in animals but also in humans. Numerous mouse models have been produced that have provided enormous insight into the role of immune cells, cytokines, and cell trafficking in hypertension. These models have facilitated our ability to study hypertension in mice and rats.
In this review, we will discuss the more recent observations that support a role of the immune system in hypertension. We will consider the contribution of immunity and inflammation in blood pressure elevation and its parallel roles in producing end-organ damage. We will also consider mechanisms by which hypertensive stimuli activate the immune system and finally discuss therapeutic options that might prove useful in the treatment of hypertension.
A brief primer of the immune system
The immune system has two major components, the innate and the adaptive systems, which closely interact. Innate immunity represents the first line of defense against invading organisms and is comprised of epithelial cells that provide a barrier to invasion, phagocytes that engulf and destroy foreign organisms, and the complement system that helps to kill and clear pathogens. Innate immune cells contain pattern recognition receptors and Toll-like receptors (TLRs) that recognize pathogen associated molecular motifs such as lipopolysaccharide (LPS), lipotechoic acid, flagellin, double stranded RNA and unmethylated CpG. Upon binding, these signal a series of cellular events including cytokine and chemokine production, expression of the inducible NO synthase, and production of reactive oxygen species (ROS). Importantly, the effects of TLR ligation can be influenced by the autonomic nervous system. As an example, Tracey and colleagues have described an inflammatory reflex, in which locally released cytokines and prostaglandins activate vagal afferent nerves that transmit information to the brainstem, the hypothalamus, and higher centers.11 Reflex signals from the brainstem alter behavior, reduce heart rate variability, increase vagal efferent activity and increase sympathetic outflow. Vagal and sympathetic nerves promote acetylcholine release from a unique population of T cells, which in turn acts on nicotinic receptors on nearby macrophages to dampen cytokine production and reduce the inflammatory response. A recent study from Harwani et al has shown that while TLR priming normally reduces production of IL-6 and IL-1β in response to nicotinic stimulation, these responses are paradoxically increased in young SHR, even before the onset of hypertension.12
Monocytes and macrophages are components of the innate immune system that are particularly relevant to cardiovascular diseases. Uptake of oxidized LDL by macrophages leads to foam cell formation and represents an early step in atherogenesis. Macrophages are also a source of ROS that alter vascular reactivity, promote inflammation, and together with matrix metalloproteinases, contribute to vascular remodeling. These cells are also potent sources of cytokines, including IL-6 and TNFα, which are discussed in depth later in this review. Historically, macrophages were thought to derive from circulating monocytes; however it is now clear that some tissues contain resident macrophages that derive from a unique lineage.13 Moreover, recent evidence indicates that monocytes can traffic in and out of tissues without becoming macrophages.14
In contrast to the innate immune response, the adaptive immune response is designed to respond specifically to foreign antigens. In the case of T cell activation, antigen-presenting cells (APCs), including dendritic cells (DCs), macrophages, and B cells process foreign proteins to small peptides that are presented in major histocompatibility complexes (MHC). There is division of duty, so that MHC-I activates CD8+ T cells, while MHC-II activates CD4+ T cells. The peptide/MHC complex is recognized by specific T cell receptors (TCRs), created by random recombinations of the TCR genes, lending a high degree of specificity to T cell activation, and is often referred to as signal 1 (Figure 1).
Figure 1.
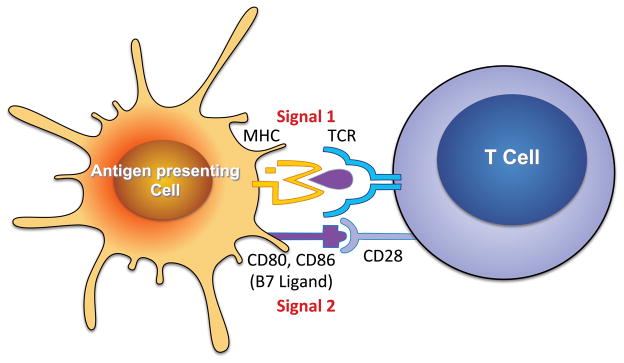
Classical pathway for T cell activation. Antigen presenting cells, including dendritic cells, B cells, macrophages and others process foreign antigens to peptides and present these in major histocompatibility complexes (MHC). MHC-I present to CD8+ T cells, while MHC-II present to CD4+ T cells. The peptide/MHC complex is recognized by a unique T cell receptor (TCR), representing signal 1. In addition, other receptor/ligand interactions occur which together with the TCR/MHC interaction for the immunological synapse. One such signal is the co-stimulatory interaction between CD28 and the B7 ligands, referred to as Signal 2. In the absence of this interaction full T cell activation does not occur. T cells possess numerous accessory receptors that modify response to Signals 1 and 2, proliferation and cytokine production (Signal 3).
In addition to the TCR/MHC signal, T cell co-stimulation, often referred to as signal 2, is necessary for full T cell activation. Classically this is mediated by interaction between CD28 on T cells and B7 ligands, CD80 and CD86, on APCs (Figure 1). The B7 ligands are upregulated as APCs mature after antigen processing. In the absence of co-stimulation, full T cell activation is prevented and T cell apoptosis often occurs. As the immune reaction proceeds, T cells begin to elaborate the cytotoxic lymphocyte antigen 4 (CTLA4), which binds to the B7 ligands and prevents their interaction with CD28, ultimately inhibiting further T cell activation. Chimeric molecules composed of CTLA-4 linked to immunoglobulin have proven effective in treatment of autoimmune diseases and transplant rejection. We have used one such agent, abatacept, to show that T cell co-stimulation is essential for hypertension, and that treatment of mice with this agent after onset of either angiotensin II- or DOCA-salt-mediated hypertension lowered blood pressure.15 We also found that B7-deficient mice are resistant to blood pressure elevation in response to angiotensin II. Of note, hypertension was associated with an increase in CD86 in spleen and lymph node DCs. This study defined an important role of co-stimulation, and more importantly, DCs in hypertension.
T cells develop in the thymus from bone marrow derived precursors and migrate to secondary lymphoid organs such as the spleen and lymph nodes, where they reside in a quiescent (naïve) state until activated by an APC. TCR ligation and co-stimulation lead to a cascade of signaling events that cause T cell oligoclonal expansion, cytokine production, and an alteration in surface proteins that allow exodus of cells from secondary lymphoid organs to peripheral tissues. These effector T cells are guided to sites of infection or tissue damage by chemokines and cytokines released from the affected tissues. Once present in the peripheral tissues, effector T cells produce mediators that further orchestrate an inflammatory response. Importantly, this response is limited in duration, and resolution signals such as CTLA-4, programmed cell death protein 1 (PD-1), and others lead to apoptosis of most effector T cells. A few of these cells remain as memory cells, which can either return to the secondary lymphoid organs as central memory cells or remain in the periphery as effector memory cells. These memory cells can rapidly respond to a second challenge when the original antigen is re-encountered.
Four distinct effector phenotypes of CD4+ cells exist, three of which seem to have evolved to respond to specific pathogen challenges. TH1 cells, which produce IFN-γ, are classically associated with intracellular bacteria and viruses. TH2 cells respond to helminthes and other allergens. The newest T helper subclass, TH17 cells, protect against extracellular bacteria and fungi. A fourth T helper subset, termed T regulatory cells (Tregs), are immunosuppressive. The cytokine milieu and the nature of antigen presentation that guide naïve T cells to these various subsets have been reviewed extensively elsewhere,16, 17 and will be mentioned in the context of hypertension and cardiovascular disease in this review. Of note, CD8+ T cells, while generally considered to function by releasing cytotoxic molecules like perforin and granzyme B, can also produce cytokines and can be classified in a manner similar to CD4+ T cells (i.e. TC1, TC2, TC17, and cytotoxic T reg cells). These aspects of T cell function have been reviewed in depth.18
A concept relevant to cardiovascular disease and hypertension is immune senescence. Following involution of the thymus in early adulthood, naïve T cells decline and memory cells, particularly effector memory CD8+ T cells expand.19 This is in part driven by recurrent and/or persistent viral infections.20 After repeated divisions, these cells assume a senescent phenotype characterized by shortened telomeres, loss of the co-stimulatory factors CD27 and CD28 and an increase in the surface marker CD57. Due to the absence of co-stimulatory receptors, senescent T cells are incapable of classical activation by TCR engagement. Nevertheless, these cells exhibit a state of persistent pro-inflammatory activation, producing IFN-γ, IL-6 and TNFα. Senescent CD8+ T cells also produce large amounts of cytotoxic granzyme. Senescent T cells have been recovered from atherosclerotic plaques of humans with unstable angina,21 and rheumatoid arthritis has been associated with premature T cell aging and accumulation of senescent T cells in the synovium.22 Recently, Youn et al found that relatively young hypertensive humans have increased circulating CD8+ T cells that are deficient in CD28 and produce excessive IFN-γ, perforin and granzyme.23 The contribution of these cells in human hypertension has yet to be defined, but their profile of cytokine production indicates that they might play a critical role.
Role of the immune system in blood pressure elevation
As mentioned above, the immunology field has generated several genetically altered animal models that have proven extremely useful for studies of hypertension. The recombinase activating genes (RAG) 1 and 2 are responsible for recombining the genetic sequences that encode immunoglobulins and the T cell receptor. Mice lacking either of these fail to develop B and T lymphocytes. Several years ago, our group found that RAG-1−/− mice develop blunted hypertension in response to either angiotensin II or DOCA-salt challenge. In these studies, we found that hypertension was associated with accumulation of T cells with an effector phenotype in the perivascular adipose tissue and adventitia. Adoptive transfer of T (but not B) cells into RAG-1−/− mice restored hypertension and its attendant end-organ dysfunction24. Subsequently, we found that stress-induced hypertension is also reduced in RAG-1−/− mice and restored by adoptive transfer of T cells. Others have confirmed these findings. Crowley and coworkers showed that mice with severe combined immune deficiency (SCID) are protected against hypertension.25 In parallel with our findings in mice, Mattson et al deleted the RAG-1 gene in Dahl salt sensitive rats using zinc finger DNAse technology and showed that this not only blunted hypertension but also reduced renal damage caused by salt feeding.26 In more recent studies, this group has deleted CD247, the CD3 ζ chain, selectively removing T cells in Dahl-salt sensitive rats. This led to a very similar phenotype to that observed with RAG-1 deletion, blunting the blood pressure elevation and renal damage caused by salt feeding.
In keeping with these experimental findings, several studies have shown that the T cell suppressing agent mycophenolate mofetil reduces blood pressure and renal inflammation in experimental models of hypertension.27–29 This agent has also reduced blood pressure in a small number of hypertensive patients with rheumatoid arthritis or psoriasis.30
There is also evidence that innate immune cells have an important role in hypertension. De Ciuceis et al studied mice lacking the macrophage colony stimulating factor (m-CSF), also referred to as Op/Op mice, which exhibit severe osteoporosis and growth retardation and are deficient in monocytes and macrophages.31 These animals exhibited minimal elevation of blood pressure in response to chronic angiotensin II infusion and had preserved endothelium-dependent vasodilatation of the resistance mesenteric vessels. While angiotensin II stimulated an increase in NADPH oxidase activity in the vessels of normal mice, this response was markedly blunted in the Op/Op mice. Subsequent studies further demonstrated that Op/Op mice are also resistant to DOCA-salt hypertension.32 Wenzel et al, employed LysM-targeting of the diphtheria toxin (DT) receptor to delete monocytes from mice33. The authors showed that angiotensin II-induced hypertension increased aortic monocyte/macrophages and markers of aortic inflammation, characterized by increased mRNA for the vascular cell adhesion molecule-1, cyclooxygenase 2, and the inducible nitric oxide synthase. Hypertension was also associated with increase vascular superoxide production and endothelial dysfunction. DT treatment prevented virtually all of these vascular perturbations and the hypertension caused by angiotensin II infusion. Hypertension could be restored by adoptively transferring monocytes but not granulocytes to these mice. As discussed below, we have identified a critical role of monocyte-derived DCs in hypertension, and it is conceivable that loss of these cells in the Op/Op mice and the elimination of monocytes in the study by Wenzel et al reduced the contribution of these important cells.
Cytokines contributing to hypertension
At first glance, it is difficult to understand how the immune system could contribute to hypertension. Studies from our group and others have shown that hypertension is associated with accumulation T cells and monocyte/macrophages in vessels and the kidney. In keeping with Olsen’s observations 40 years ago, the vascular accumulation is predominantly in the adventitia and the perivascular fat. In the kidney, T cells increase in both the medulla and renal cortex. We have found that these cells have markers suggestive of effector memory cells. Data from our group and others indicate that these cells produce potent cytokines that affect vascular and renal function. In the past several years, important roles of at least 5 cytokines have been identified in hypertension, which will be discussed below.
Interleukin 17
A particularly important cytokine is IL-17, produced by a unique set of CD4+ T cells, referred to as TH17 cells. In addition to CD4+ T cells, γ/δ T cells, the TC17 subset of CD8+ T cells, some B cells and natural killer T cells, and miscellaneous other cells produce IL-17.34 There are 6 isoforms of IL-17, classified as A-F, which share varying degrees of sequence homology. IL-17A and IL-17F are the most closely related isoforms (sharing 50% sequence homology) and are generally produced by the same cell types. These isoforms are encoded on the same chromosome and can bind as homo- or heterodimers to the IL17 receptor complex composed of IL17RA and IL17RC. These receptor chains are thought to heterodimerize upon ligand binding and signal through the adaptor molecule Act1 and tumor necrosis receptor associated factor-6 (TRAF6) leading to NFκB, MAP kinase and C/EBPδ activation35. Importantly, IL-17A seems to act synergistically with other cytokines, in particular TNFα, to stimulate these signals. As an example, Sharma et al have shown that IL-17 and TNFα synergistically enhance lung inflammation and CXCL-1, CCL-2 and CCL-5 production following ischemia/reperfusion in an NADPH oxidase-dependent fashion.36 We also demonstrated a marked synergism of these two cytokines on altering gene expression in human vascular smooth muscle cells.37 IL-17A has been implicated in a variety of autoimmune diseases, including psoriasis, experimental autoimmune encephalomyelitis, asthma, Crohn’s disease, and rheumatoid arthritis.34 The anti-IL-17A antibodies, Ixekizumab and Secukinumab, as well as the anti-IL17RA receptor antibody, Brodalumab, have proven effective in phase II/III human trials for the treatment of psoriasis,38, 39 and are being studied in other inflammatory diseases.
In recent years, several reports indicate that IL-17A contributes to hypertension. We found that angiotensin II infusion increases IL-17A production from mouse T cells, and that plasma levels of IL-17A are increased in humans with hypertension.37 Importantly, mice lacking IL-17A develop blunted hypertension and do not develop endothelial dysfunction in response to angiotensin II infusion. In keeping with this, the increase in vascular superoxide production generally observed in hypertension does not occur in mice lacking IL-17A. A particularly striking finding was that the vascular infiltration of total leukocytes and T cells was markedly reduced in IL-17A−/− mice. In keeping with this finding, we found that when co-administered with TNFα to human vascular smooth muscle cells in culture, IL-17A increased expression of a variety of cytokines and chemokines, including CCL8, CSF3, CXCL2, and CCL7. Thus, it seems that IL-17A might coordinate an inflammatory response leading to accumulation of multiple immune cell subtypes in hypertension.
Subsequent studies have confirmed a role of IL-17A in hypertension. Nguyen et al made the fascinating discovery that IL-17A can induce phosphorylation of threonine 495 on the endothelial nitric oxide synthase (eNOS) in a Rho kinase-dependent manner. Phosphorylation at this site prevents calmodulin binding and leads to conformational changes of eNOS, reducing production of nitric oxide (NO).40, 41 These investigators also showed that infusion of IL-17A in mice caused a modest elevation of blood pressure in the absence of other hypertensive stimuli.
Recently, Amador et al found that rats with DOCA-salt hypertension have a striking increase in circulating TH17 cells and reduced Tregs and that treatment with spironolactone reverses this pattern.42 The investigators also found a marked increase in the mRNA for IL-17A in the heart and kidney and these values were normalized by spironolactone. Interestingly, “triple therapy” with hydralazine, reserpine and hydrochlorothiazide, which normalized blood pressure, did not reduce these elevated IL-17 mRNA levels, suggesting that the increase in this cytokine was not purely a consequence of blood pressure elevation. The investigators also found that treating rats with an antibody against IL-17A reduced blood pressure and levels of collagen1 in the heart and kidneys.
In keeping with a role of IL-17A in collagen deposition, we have recently discovered that this cytokine plays a critical role in aortic stiffening.43 A normal compliant aorta expands during systole and collapses during diastole, respectively, storing and then ejecting a portion of the cardiac stroke volume during these two phases of the cardiac cycle. Aortic stiffening occurs in a variety of disease conditions, including aging and hypertension, leading to loss of this “Windkessel” or capacitance function. This results in rapid transmittance of blood volume to the periphery, an increase in systolic blood pressure and a decrease in diastolic blood pressure. Recently, we found that both angiotensin II and DOCA-salt induced hypertension lead to a striking deposition of collagen in the adventitia of mice and a marked loss of aortic compliance. Interestingly, this aortic stiffening was found to be due to the presence of T cells because it did not occur in RAG-1−/− mice, and was restored by adoptive transfer of T cells to these animals. Moreover, our studies implicated IL-17A as a causative cytokine in aortic stiffening, as collagen deposition and aortic stiffening did not occur in IL-17A−/− mice. Subsequent studies of cultured aortic fibroblasts showed that IL-17A induced mRNA for collagens I, III and V in a p38MAP kinase dependent fashion.
Of interest, TGFβ alone promotes formation of T regulatory cells (Tregs) and there is substantial plasticity of these cells, such that exposure to IL-6 in conjunction with TGFβ can convert Tregs to TH17 cells. Pre-eclampsia has been associated with an imbalance of Tregs and TH17 cells.44 Toldi et al found a modest increase in circulating T cells containing IL-17A in patients with pre-eclampsia.45 In keeping with this, Cornelius et al found a striking increase in circulating TH17 cells in a placental ischemia model of pre-eclampsia and showed that IL-17 inhibition using a soluble form of the IL-17 receptor C ameliorated many aspects of pre-eclampsia in this model, including the blood pressure elevation.46 An important cause of pre-eclampsia is the production of agonistic antibodies against the angiotensin type 1 receptor, and these were markedly reduced by this novel treatment strategy.
There is one study that has shown a worsening of renal damage in mice lacking either IL-17 or IL-23.47 These authors, however, subjected the experimental animals to uninephrectomy, DOCA-pellet implantation, salt-feeding and an extraordinarily high dose of angiotensin II infusion. Other investigators, including our group, have found that either lower doses of angiotensin II or the DOCA-salt challenge alone is sufficient to cause hypertension, and their combined use of high dose angiotensin II with DOCA-salt hypertension likely induced severe vascular and renal injury. Given the multiple other papers showing a role of IL-17 in promoting hypertension, it is likely the paradoxical finding of this report was due to the unusual model employed by these authors.
Interferon gamma
The type II interferon, IFN-γ, is the signature cytokine of TH1 cells, but is also produced in high amounts by CD8+ T cells (TC1 cells) and natural killer T cells. We have consistently found an increase in IFN-γ-forming CD4+ and CD8+ T cells in hypertensive mice, however it appears that this cytokine might have mixed effects on the response to various hypertensive stimuli. Ishimitsu et al. found that subcutaneous injections of IFN-γ attenuated hypertension, proteinuria, and glomerular injury in Dahl salt sensitive rats, while having no effect in spontaneously hypertensive rats.48 This study and others where the cytokine was administered may not reflect the role of endogenously produced IFN-γ in hypertension. More recently, Garcia et al demonstrated a blunted hypertensive response to chronic aldosterone infusion (combined with uninephrectomy and salt-feeding) in IFN-γ−/− mice compared to wild-type animals.49 However, IFN-γ−/− mice exhibited exaggerated left ventricular (LV) hypertrophy, reduced LV cavity size, and worse diastolic dysfunction.
We have recently shown that mice deficient in the lymphocyte adaptor protein, LNK, exhibit severe hypertension and renal/vascular dysfunction. Interestingly, these mice have elevated numbers of CD4+ and particularly CD8+ T cells that produce IFN-γ. In addition, we showed that IFN-γ deficiency results in blunted hypertension in response to angiotensin II infusion (ref to come out today).
Marko et al50 recently examined a role of IFN-γ in hypertension by studying mice lacking IFN-γ receptor 1. While these animals did not exhibit an alteration in the hypertensive response to high dose angiotensin II infusion, they developed less renal fibrosis and maintained their glomerular filtration rate. These animals also demonstrated reduced cardiac fibronectin and collagen and fewer inducible arrhythmias than observed in wild-type mice subjected to angiotensin II infusion.
An important mechanism by which IFN-γ might promote hypertension relates to its capacity to induce angiotensinogen expression in both hepatocytes and renal proximal tubular cells.51, 52 While angiotensinogen is not considered rate limiting for the systemic production of angiotensin II, its role in the tubular production of angiotensin II seems more critical. Navar and colleagues have shown that proximal tubular cells produce angiotensinogen, which is subsequently converted to angiotensin I and angiotensin II within the tubule or within epithelial cells.53 This locally formed angiotensin II promotes sodium and volume reabsorption in both the proximal and distal nephron. In the proximal tubule, this is mediated by actions on apical sodium hydrogen exchanger 3 (NHE3), basolateral Na/HCO3 co-transport, and the Na-K ATPase. In the distal nephron, renin produced by collecting duct cells act on proximally derived angiotensinogen to form additional intratubular angiotensin II which has multiple actions including activation of the epithelial sodium channel and the sodium chloride cotransporter. Of particular interest, recently Satou et al have shown that prolonged exposure of renal proximal tubule cells to IFN-γ increases angiotensinogen protein production more than 2-fold in a JAK2/STAT3-dependent fashion. Thus, it is conceivable that infiltrating T cells that release IFN-γ might modulate the local production of angiotensinogen, enhance sodium reabsorption, and worsen hypertension in a feed-forward fashion. In keeping with this, Bravo et al demonstrated that mycophenolate mofetil reduced the presence of angiotensin II-producing cells of the kidney, some of which were proximal tubular epithelial cells.28
Interestingly, Kamat et al recently showed that mice deficient in IL-17A or IFN-γ exhibit alterations in proximal and/or distal tubule sodium transporters such as NHE3 and the sodium chloride co-transporter (NCC) leading to enhanced pressure natriuresis and decreased distal sodium reabsorption, respectively54. Thus, in addition to affecting the local renin-angiotensin system, pro-inflammatory cytokines may have direct effects on the expression of renal sodium transporters and thus on salt and water balance.
Tumor necrosis factor α
TNFα is produced by a variety of cells including T cells, macrophages, endothelial cells, fibroblasts and neuronal cells. It acts on two receptors TNFR1 and TNRF2, which are ubiquitously expressed and form homotrimers upon TNFα binding. These receptors in turn activate multiple signals including death and survival pathways, NADPH oxidase activation, JNK and NFκB.55 NFκB and NADPH oxidase activation contribute to several of the cardiovascular and renal effects of TNFα, including chemokine and adhesion molecule expression, vascular remodeling, and sodium retention by the kidney.56 TNFα has a multitude of untoward effects on endothelial nitric oxide (NO) production that could contribute to hypertension. Superoxide production by the NADPH oxidase rapidly reacts with NO, forming the strong oxidant peroxynitrite. TNFα also inhibits the eNOS promoter and causes destabilization of the endothelial nitric oxide synthase (eNOS) mRNA, ultimately reducing eNOS protein levels and the ability of the endothelium to produce NO.57, 58 Recent studies have shown that microRNA 155 contributes to destabilizing the eNOS mRNA and reducing endothelium-dependent vasodilatation in response to TNFα.59 Thus, like IL-17A, TNFα impairs the ability of the endothelium to produce NO, promoting vasoconstriction.
In addition to its vascular effects, TNFα has renal effects that could affect blood pressure. Ramseyer and Garvin have recently reviewed this topic.60 As in the case of the endothelium, TNFα decreases eNOS expression in medullary thick ascending limb cells within 24 hours of exposure in a Rho-kinase dependent manner.61 NO inhibits sodium reabsorption at several sites along the renal tubule including the mTAL and collecting duct, and its loss would therefore lead to sodium retention.62 Loss of NO in the vasa vasorum could also promote sodium reabsorption in the renal medulla by perturbing tubulovascular crosstalk.63 Prolonged and sustained exposure of renal parenchymal cells to TNFα could promote renal injury and thus shift the pressure natriuresis curve to favor blood pressure elevation.
In keeping with a role of TNFα in hypertension, we found that angiotensin II infusion stimulates T cells to produce TNFα, and that the TNFα antagonist etanercept blunts the blood pressure elevation and vascular superoxide production caused by angiotensin II.24 TNFα blockade has also proven effective in either reducing blood pressure or protecting against renal injury in various models of hypertension including pre-eclampsia, a model of lupus erythematosus, angiotensin II infusion in rats, transgenic rats and fructose feeding.27, 64–67 Despite the fact that TNFα antagonists have been used extensively to treat humans with a variety of autoimmune diseases, there is no evidence that they have blood pressure lowering effects. There are conflicting reports that these agents affect vascular stiffness in humans.
Sriramula et al demonstrated a virtual absence of blood pressure elevation and a reduction of left ventricular hypertrophy in response to chronic angiotensin II infusion in TNFα−/− mice.68 An interesting finding in this study was that the water drinking behavior induced by angiotensin II was markedly reduced in the TNFα-deficient mice, indicating a central role of this cytokine. In keeping with this, activation of NFκB in the hypothalamus occurred in wild-type, but not TNFα−/− mice. These findings are in keeping with a role of hypothalamic inflammation in hypertension.
TNFα inhibitors are widely used in the treatment of humans with autoimmune diseases. To date, there are no reports that these agents lower blood pressure in humans, however mixed effects on arterial stiffness have been described.69, 70
Interleukin 6
IL-6 is a small, 21 kDa glycoprotein produced by numerous cells, including DCs, macrophages, monocytes, subsets of TH1 T cells and vascular cells.71 IL-6 binds to its receptor, IL-6R, which dimerizes with the transmembrane protein gp130, which is ubiquitously expressed and has a large intracellular signaling domain that activates the Janus kinase (JAK) tyrosine kinases and the downstream signal transducer and activator of transcription (STAT)3. Cells not expressing IL-6R can be transactivated by IL-6 binding to soluble IL-6R, which in turn binds to gp130. Via these signaling pathways and others, IL-6 induces multiple effects on target tissues, stimulating bone resorption, neutrophil chemotaxis and polarization of helper T cells. Because it stimulates hepatocyte production of C-reactive protein (CRP), elevations of this acute phase reactant often reflect the actions of IL-6. As mentioned above, IL-6 is a major signal to promote polarization of CD4+ T cells to produce IL-17. Experimental studies have implicated IL-6 in diverse inflammatory conditions including malignancies, autoimmune diseases, atherosclerosis and hypertension. Circulating levels of IL-6 are increased in humans with polymyalgia rheumatica and giant cell arteritis, and are suppressed by corticosteroid therapy.72 The humanized anti-IL-6R antibody Tocilizumab has proven effective in treatment of rheumatoid and juvenile arthritis, and is being evaluated for treatment of several other inflammatory diseases, including Crohn’s disease, systemic sclerosis and ankylosing spondylitis.73
There is substantial evidence that IL-6 contributes to hypertension. Levels of IL-6 correlate with blood pressure in hypertensive subjects, and are reduced by treatment with angiotensin II-receptor blockade.74 In keeping with this, angiotensin II infusion in humans increases IL-6 levels, and this is blocked by treatment with spironolactone, implicating activation of the mineralocorticoid receptor.75 Lee et al found that IL-6−/− mice develop blunted hypertension and albuminuria in response to high salt and angiotensin II infusion compared to wild-type mice.76 In cultured cortical collecting duct cells, IL-6 increases the protein levels and activity of the epithelial sodium channel,77 and therefore has the potential to enhance sodium and volume resorption in vivo. This direct effect of IL-6, together with its ability to skew T cells from a regulatory phenotype to IL-17-producing cells is likely important in hypertension.
Interleukin 10/T regulatory cells
Interleukin 10 is an anti-inflammatory cytokine that was originally discovered based on its ability to inhibit IL-2 and IFN-γ production.78 A variety of cells, almost all lymphocytes, monocytes, macrophages, DCs and endothelial cells, can produce IL-10. IL-10 inhibits cytokine production by T cells and monocyte/macrophages and reduces DC maturation, diminishing antigen presentation, MHC, and the co-stimulatory B7 molecule expression. In T cells, ligation of the TCR stimulates IL-10 production in a feedback loop fashion.79
There is evidence that IL-10 has protective functions in hypertension. The -627 polymorphism of the IL-10 promoter is associated with a reduced incidence of essential hypertension in Russian Tatars.80 Didion et al have defined a critical role of IL-10 in modulating endothelial function in hypertension.81 These investigators found that direct application of angiotensin II doubled superoxide production in carotid arteries of IL-10−/− mice, but not in WT mice. These investigators also found that direct application of angiotensin II markedly impaired vasodilatation caused by acetylcholine in IL-10−/− mice, while having no effect in vessels of WT mice. This defect in vascular function was corrected by treatment with a cell permeable form of superoxide dismutase, linking increased oxidative stress to vascular dysfunction in IL-10 deficient vessels. The hypertensive response to 10 days of angiotensin II infusion was similar between WT and IL-10−/− mice. Of interest, the aortic levels of IL-6 mRNA were markedly increased, and TNFα mRNA slightly increased by angiotensin II in IL-10−/− mice compared to values observed in WT aortas.
There is also evidence that IL-10 ameliorates hypertension associated with pregnancy. Placentas from women with pre-eclampsia exhibit reduced staining for IL-10 compared to women with normal gestation and serum levels of IL-10 are reduced in pre-eclampsia.82 Tinsley et al demonstrated that daily IP injections of recombinant IL-10 normalized blood pressure and endothelial function in pregnant rats with DOCA-salt hypertension.83 This model was associated with proteinuria and increased circulating levels of endothelin-1 and IFN-γ, all of which were reduced by IL-10. In addition, placental levels of IFN-γ and PECAM-1 were elevated by DOCA-salt hypertension and corrected by IL-10 treatment. Likewise, Lai et al showed that exposure of wild-type pregnant mice to hypoxia led to hypertension, proteinuria, reduced fetal weight and renal injury, and that these parameters were more severe in IL-10−/− mice.84 Treatment with recombinant IL-10 corrected hypertension, proteinuria and fetal weight in the IL-10 deficient animals. Of note, increased circulating levels of the soluble vascular endothelial cell growth factor-1 receptor sFlt have been implicated in the pathogenesis of pre-eclampsia.85 The investigators found that hypoxia also increased sFlt in this model and that IL-10 treatment normalized these levels.
While numerous cells can produce IL-10, it is a key product of T regulatory cells (Tregs). Adoptive transfer of Tregs has been shown to lower angiotensin II- and aldosterone-induced hypertension, cardiac fibrosis, coronary inflammation, and electrical remodeling.86–89 These effects are likely mediated at least in part by release of IL-10 from Tregs, As Kassan et al showed, IL-10−/− mice exhibit enhanced hypertension, endothelial dysfunction, and increased NADPH oxidase activity in response to angiotensin II, and these effects were ameliorated by adoptive transfer of Tregs from WT but not IL-10−/− mice.90 In keeping with this concept, Viel et al studied Dahl salt sensitive rats that had undergone transfer of chromosome 2 from normotensive Brown Norway rats.91 These animals exhibited enhanced T regulatory cell markers, increased IL-10 levels and reduced parameters of vascular inflammation. Taken together, these studies support an anti-inflammatory and anti-hypertensive role of T regulatory cells in response to multiple stimuli.
A novel mechanism of T cell activation in hypertension – role of oxidative stress
Despite the studies above, it has remained unclear as to how and why T cells are activated in hypertension. Recently, we have established a new mechanism of T cell activation related to oxidative stress.92 More than 15 years ago, we discovered that angiotensin II and DOCA-salt hypertension lead to an increase in vascular superoxide production due to activation of the NADPH oxidase.93, 94 Studies from others have shown that the NADPH oxidase plays a role in hypertension at many levels. In the subfornical organ of the brain, ROS produced by this enzyme complex stimulate neuronal firing and in the kidney, this enzyme stimulates sodium reabsorption. We now have discovered that the NADPH oxidase is activated in DCs in hypertension, and this leads to formation of gamma-ketoaldehydes, also known as isoketals. These are products of fatty acid oxidation, which rapidly react with protein lysines. Recent evidence indicate that these oxidatively modified proteins are immunogenic,95 and we find that isoketal protein adducts abundantly accumulate in DCs of mice made hypertensive by either angiotensin II or DOCA-salt hypertension. This is particularly evident in CD11chi/CD11b+ DCs, which are stimulated to express high levels of CD80 and CD86 when they form isoketal adducts. DCs affected in this manner produce large amounts of IL-6, IL-23 and IL-1β, known to drive T cell polarization. Indeed, we find that these DCs drive T cell proliferation and production of IL-17A, TNFα and IFN-γ, which as noted above contribute to hypertension. Adoptive transfer of these DCs to recipient mice primes severe hypertension in response to low dose angiotensin II. Moreover, induction of oxidative stress in DCs by addition of tert-butyl hydroperoxide (t-BHP) promotes formation of isoketal-adducted proteins and hypertension when these t-BHP-treated DCs are transferred to recipient mice.
In these studies, we also examined the efficacy of 2-hydroxybenzylamine (2-HOBA), a compound known to scavenge isoketals before they adduct proteins. We found 2-HOBA virtually eliminated isoketal-adducted proteins in DCs, prevented DC expression of CD80 and CD86 and production of polarizing cytokines. Importantly, 2-HOBA prevented the capacity of DCs to drive T cell proliferation and the ability of DCs to convey hypertension to recipient mice when administered to the donor mice. We studied several related compounds, some of which were known to scavenge isoketal and others modified to be inactive, and found that the active compounds had anti-hypertensive properties.
These recent studies from our group define a new mechanism of hypertension and T cell activation as depicted in Figure 2. Given that oxidative stress contributes to a variety of diseases including atherosclerosis, diabetes and obesity, it is possible that isoketal adducts promote immune mechanisms in these conditions as well and that drugs like 2-HOBA might be beneficial in these conditions.
Figure 2.

T cell activation by isoketal protein modification. Hypertension induces production of reactive oxygen species (ROS) in dendritic cells (DCs), leading to oxidation of arachidonic acid and formation of gamma ketoaldehydes or isoketals. Isoketals rapidly ligate to protein lysines in the DC, forming proteins that are recognized as non-self. Peptides from these are presented to T cells, leading to T cell proliferation. Isoketal formation also promotes DC production of cytokines including IL-1β, IL-6 and IL-23, which polarize T cells to produce specific cytokines.
Concluding remarks
Classic teachings from Guyton and co-workers indicate sustained hypertension requires an alteration in renal function followed by an increase in systemic vascular resistance.96 Dr. Guyton described several physiological and pathophysiological phenomena that alter the renal pressure natriuresis curve and proposed that this renal event is followed by systemic autoregulation, which causes an increase in systemic vascular resistance and a further increase in blood pressure. It is now clear that in addition to these events, immune cell entry into the kidney and perivascular regions contribute to blood pressure elevation (Figure 3).
Figure 3.

Cytokines and end-organ dysfunction in hypertension. A) In vessels T cells infiltrate the adventitia and perivascular fat through the vasa vasorum. T cell-derived IL-17A acts on smooth muscle cells and adventitial fibroblasts to increase eNOS phosphorylation, reactive oxygen species (ROS) production, collagen synthesis, and chemokine production leading to a decrease in bioavailable nitric oxide (NO) and impaired vasodilation, increased vascular stiffness, and increased recruitment of immune cells, propagating the inflammatory response. These effects result in vascular dysfunction. B) In the renal medulla and cortex, activated T cells produce cytokines such as IL-6 and IFNγ that stimulate production of angiotensinogen. Angiotensinogen is converted to angiotensin I (Ang I) by intrarenal renin and subsequently to angiotensin II (Ang II) by intrarenal angiotensin converting enzyme. Angiotensin II upregulates and stimulates transport channels in the proximal and distal convoluted tubules including the sodium hydrogen exchanger 3 (NHE3) and sodium chloride co-transporter (NCC). In conjunction with salt and water retention, T cell activation causes an increase in renal ROS production, and renal injury and fibrosis, all of which lead to renal dysfunction. The culmination of vascular and renal dysfunction caused by T cell derived cytokines exacerbates hypertension. (Illustration credit: Ben Smith).
While the number of immune cells is modest, the cytokines released are extremely potent in eliciting changes in the kidney such as fibrosis, glomerular damage and alterations in sodium transport. In the vasculature, these mediators promote oxidative injury, endothelial dysfunction, vascular remodeling with luminal narrowing and fibrosis, and arterial stiffness which contribute to elevations in systemic vascular resistance. In addition, activation of microglial cells, which are specialized macrophages of the brain, promote hypertension by increasing sympathetic outflow.97 These immune mediated events therefore contribute to sustained elevations of blood pressure.
We devote substantial effort to defining blood pressure goals in the treatment of hypertension, but the major reason to treat this disease is to prevent end-organ damage. If hypertension did not cause strokes, myocardial infarctions, heart failure, renal failure and dementia, it is unlikely that physicians would even measure blood pressure. The data discussed in this review supports the notion that an important component of the end-organ damage associated with hypertension is mediated by inflammation. Thus, while blood pressure lowering is important, an underlying goal should clearly be prevention of the local inflammation that accompanies this disease. We currently have no drugs to specifically accomplish this, except by indirect means. Moreover, our ability to detect local inflammation is limited to assays of circulating markers. Substantial additional research is needed to understand this field in detail and to develop diagnostic and therapeutic tools for management of this widespread health problem.
Acknowledgments
We would like to thank L. Gabriel Navar for helpful discussions regarding the influence of IFN-γ and other cytokines on renal production of angiotensinogen and the downstream consequences of this.
Sources of Funding: American Heart Association Post-Doctoral Fellowship (13POST14440041), National Institutes of Health grants R01HL039006, P01HL058000, P01HL095070, P01GM015431, R01HL108701, K08HL121671, R01HL105294 and T32 HL69765.
Non-standard abbreviations
- IL
Interleukin
- IFN
interferon
- TNF
turmor necrosis factor
- DOCA
deoxycorticosterone acetate
- SHR
spontaneously hypertensive rat
- TLR
Toll-like receptor
- LPS
lipopolysaccharide
- CpG
cytosine and guanine triphosphate deoxynucleotides with phospodiester linkage
- MHC
Major histocompatibility complex
- CTLA4
cytotoxic lymphocyte antigen 4
- CD
Cluster of differentiation
- DC
Dendritic cells
- APC
Antigen presenting cell
- TH
T helper
- Tregs
T regulatory cells
- PD-1
programmed cell death protein 1
- RAG
recombinase activating gene
- SCID
severe combined immune deficiency
- mCSF
macrophage stimulating factor
- DT
diphtheria toxin
- Op/Op mice
osteoporosis spontaneous mutation mice
- LysM
lysozyme M
- TRAF6
tumor necrosis receptor associated factor-6
- CXCL
chemokine C-X-C motif
- NFκB
nuclear factor kappa B
- MAP
mitogen activated kinease
- C/EBP
CCAAT-enhancer-binding protein
- CCL
ligands for chemokines with two adjacent cysteines
- eNOS
endothelial nitric oxide synthase
- TGF
transforming growth factor
- NHE
sodium hydrogen exchanger
- Na
sodium
- K
potassium
- JAK
Janus kinase
- STAT
signal transducer and activator of transcription
- JNK
c-Jun N-terminal kinases
- gp
glycosylated protein
- sFLT
soluble fms-like tyrosine kinase
- tBHP
tert butyl hydroperoxide
- 2-HOBA
2-hydroxybenzylamine
Footnotes
Disclosures: The authors have no conflicts of interest to disclose.
Literature Cited
- 1.White FN, Grollman A. Autoimmune Factors Associated with Infarction of the Kidney. Nephron. 1964;204:93–102. doi: 10.1159/000179322. [DOI] [PubMed] [Google Scholar]
- 2.Okuda T, Grollman A. Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex Rep Biol Med. 1967;25:257–64. [PubMed] [Google Scholar]
- 3.Olsen F. Type and course of the inflammatory cellular reaction in acute angiotensin-hypertensive vascular disease in rats. Acta Pathol Microbiol Scand A. 1970;78:143–50. doi: 10.1111/j.1699-0463.1970.tb00249.x. [DOI] [PubMed] [Google Scholar]
- 4.Olsen F. Inflammatory cellular reaction in hypertensive vascular disease in man. Acta Pathol Microbiol Scand A. 1972;80:253–6. [PubMed] [Google Scholar]
- 5.Takeichi N, Hamada J, Takimoto M, Fujiwara K, Kobayashi H. Depression of T cell-mediated immunity and enhancement of autoantibody production by natural infection with microorganisms in spontaneously hypertensive rats (SHR) Microbiol Immunol. 1988;32:1235–44. doi: 10.1111/j.1348-0421.1988.tb01487.x. [DOI] [PubMed] [Google Scholar]
- 6.Takeichi N, Boone CW. Spontaneous rosette formation of rat thymus cells with guinea pig erythrocytes. Cell Immunol. 1976;27:52–9. doi: 10.1016/0008-8749(76)90153-2. [DOI] [PubMed] [Google Scholar]
- 7.Purcell ES, Wood GW, Gattone VH., 2nd Immune system of the spontaneously hypertensive rat: II. Morphology and function. Anat Rec. 1993;237:236–42. doi: 10.1002/ar.1092370211. [DOI] [PubMed] [Google Scholar]
- 8.Bendich A, Belisle EH, Strausser HR. Immune system modulation and its effect on the blood pressure of the spontaneously hypertensive male and female rat. Biochem Biophys Res Commun. 1981;99:600–7. doi: 10.1016/0006-291x(81)91787-3. [DOI] [PubMed] [Google Scholar]
- 9.Dzielak DJ. Immune mechanisms in experimental and essential hypertension. Am J Physiol. 1991;260:R459–67. doi: 10.1152/ajpregu.1991.260.3.R459. [DOI] [PubMed] [Google Scholar]
- 10.Olsen F. Transfer of arterial hypertension by splenic cells from DOCA-salt hypertensive and renal hypertensive rats to normotensive recipients. Acta Pathol Microbiol Scand C. 1980;88:1–5. doi: 10.1111/j.1699-0463.1980.tb00065.x. [DOI] [PubMed] [Google Scholar]
- 11.Olofsson PS, Rosas-Ballina M, Levine YA, Tracey KJ. Rethinking inflammation: neural circuits in the regulation of immunity. Immunol Rev. 2012;248:188–204. doi: 10.1111/j.1600-065X.2012.01138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harwani SC, Chapleau MW, Legge KL, Ballas ZK, Abboud FM. Neurohormonal Modulation of the Innate Immune System is Pro-inflammatory in the Pre-hypertensive Spontaneously Hypertensive Rat, a Genetic Model of Essential Hypertension. Circ Res. 2012 doi: 10.1161/CIRCRESAHA.112.277475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, Garcia-Sastre A, Stanley ER, Ginhoux F, Frenette PS, Merad M. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, Ivanov S, Duan Q, Bala S, Condon T, van Rooijen N, Grainger JR, Belkaid Y, Ma’ayan A, Riches DW, Yokoyama WM, Ginhoux F, Henson PM, Randolph GJ. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39:599–610. doi: 10.1016/j.immuni.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, Guzik TJ. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122:2529–37. doi: 10.1161/CIRCULATIONAHA.109.930446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boyman O, Purton JF, Surh CD, Sprent J. Cytokines and T-cell homeostasis. Curr Opin Immunol. 2007;19:320–6. doi: 10.1016/j.coi.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 17.Gutcher I, Becher B. APC-derived cytokines and T cell polarization in autoimmune inflammation. J Clin Invest. 2007;117:1119–27. doi: 10.1172/JCI31720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abbas A, Lichtman A, Pillai S. Cellular and Molecular Immunology. 7. Philadelphia, PA: Elsevier Saunders; 2010. [Google Scholar]
- 19.High KP, Akbar AN, Nikolich-Zugich J. Translational research in immune senescence: assessing the relevance of current models. Semin Immunol. 2012;24:373–82. doi: 10.1016/j.smim.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kared H, Camous X, Larbi A. T cells and their cytokines in persistent stimulation of the immune system. Curr Opin Immunol. 2014;29:79–85. doi: 10.1016/j.coi.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 21.Liuzzo G, Goronzy JJ, Yang H, Kopecky SL, Holmes DR, Frye RL, Weyand CM. Monoclonal T-cell proliferation and plaque instability in acute coronary syndromes. Circulation. 2000;101:2883–8. doi: 10.1161/01.cir.101.25.2883. [DOI] [PubMed] [Google Scholar]
- 22.Weyand CM, Yang Z, Goronzy JJ. T-cell aging in rheumatoid arthritis. Curr Opin Rheumatol. 2014;26:93–100. doi: 10.1097/BOR.0000000000000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY, Choi YS, Lee SH, Kang SM, Jang Y, Yoo OJ, Shin EC, Park S. Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension. 2013;62:126–33. doi: 10.1161/HYPERTENSIONAHA.113.00689. [DOI] [PubMed] [Google Scholar]
- 24.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–60. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1089–97. doi: 10.1152/ajpregu.00373.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol. 2013;304:R407–14. doi: 10.1152/ajpregu.00304.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, Fiebeler A, Ju X, Finckenberg P, Theuer J, Viedt C, Kreuzer J, Heidecke H, Haller H, Zenke M, Luft FC. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol. 2002;161:1679–93. doi: 10.1016/S0002-9440(10)64445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bravo Y, Quiroz Y, Ferrebuz A, Vaziri ND, Rodriguez-Iturbe B. Mycophenolate mofetil administration reduces renal inflammation, oxidative stress, and arterial pressure in rats with lead-induced hypertension. Am J Physiol Renal Physiol. 2007;293:F616–23. doi: 10.1152/ajprenal.00507.2006. [DOI] [PubMed] [Google Scholar]
- 29.Franco M, Martinez F, Quiroz Y, Galicia O, Bautista R, Johnson RJ, Rodriguez-Iturbe B. Renal angiotensin II concentration and interstitial infiltration of immune cells are correlated with blood pressure levels in salt-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol. 2007;293:R251–6. doi: 10.1152/ajpregu.00645.2006. [DOI] [PubMed] [Google Scholar]
- 30.Herrera J, Ferrebuz A, MacGregor EG, Rodriguez-Iturbe B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol. 2006;17:S218–25. doi: 10.1681/ASN.2006080918. [DOI] [PubMed] [Google Scholar]
- 31.De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol. 2005;25:2106–13. doi: 10.1161/01.ATV.0000181743.28028.57. [DOI] [PubMed] [Google Scholar]
- 32.Ko EA, Amiri F, Pandey NR, Javeshghani D, Leibovitz E, Touyz RM, Schiffrin EL. Resistance artery remodeling in deoxycorticosterone acetate-salt hypertension is dependent on vascular inflammation: evidence from m-CSF-deficient mice. Am J Physiol Heart Circ Physiol. 2007;292:H1789–95. doi: 10.1152/ajpheart.01118.2006. [DOI] [PubMed] [Google Scholar]
- 33.Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, Karbach SH, Schwenk M, Yogev N, Schulz E, Oelze M, Grabbe S, Jonuleit H, Becker C, Daiber A, Waisman A, Munzel T. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124:1370–81. doi: 10.1161/CIRCULATIONAHA.111.034470. [DOI] [PubMed] [Google Scholar]
- 34.Gu C, Wu L, Li X. IL-17 family: cytokines, receptors and signaling. Cytokine. 2013;64:477–85. doi: 10.1016/j.cyto.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–67. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma AK, Mulloy DP, Le LT, Laubach VE. NADPH oxidase mediates synergistic effects of IL-17 and TNF-alpha on CXCL1 expression by epithelial cells after lung ischemia-reperfusion. Am J Physiol Lung Cell Mol Physiol. 2014;306:L69–79. doi: 10.1152/ajplung.00205.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–7. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, Puig L, Nakagawa H, Spelman L, Sigurgeirsson B, Rivas E, Tsai TF, Wasel N, Tyring S, Salko T, Hampele I, Notter M, Karpov A, Helou S, Papavassilis C, Group ES, Group FS. Secukinumab in plaque psoriasis--results of two phase 3 trials. N Engl J Med. 2014;371:326–38. doi: 10.1056/NEJMoa1314258. [DOI] [PubMed] [Google Scholar]
- 39.Mease PJ, Genovese MC, Greenwald MW, Ritchlin CT, Beaulieu AD, Deodhar A, Newmark R, Feng J, Erondu N, Nirula A. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N Engl J Med. 2014;370:2295–306. doi: 10.1056/NEJMoa1315231. [DOI] [PubMed] [Google Scholar]
- 40.Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. 2001;88:E68–75. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- 41.Piazza M, Taiakina V, Guillemette SR, Guillemette JG, Dieckmann T. Solution structure of calmodulin bound to the target peptide of endothelial nitric oxide synthase phosphorylated at Thr495. Biochemistry. 2014;53:1241–9. doi: 10.1021/bi401466s. [DOI] [PubMed] [Google Scholar]
- 42.Amador CA, Barrientos V, Pena J, Herrada AA, Gonzalez M, Valdes S, Carrasco L, Alzamora R, Figueroa F, Kalergis AM, Michea L. Spironolactone decreases DOCA-salt-induced organ damage by blocking the activation of T helper 17 and the downregulation of regulatory T lymphocytes. Hypertension. 2014;63:797–803. doi: 10.1161/HYPERTENSIONAHA.113.02883. [DOI] [PubMed] [Google Scholar]
- 43.Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, Madhur MS, Chen W, Harrison DG. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res. 2014;114:616–25. doi: 10.1161/CIRCRESAHA.114.302157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santner-Nanan B, Peek MJ, Khanam R, Richarts L, Zhu E, Fazekas de St Groth B, Nanan R. Systemic increase in the ratio between Foxp3+ and IL-17-producing CD4+ T cells in healthy pregnancy but not in preeclampsia. J Immunol. 2009;183:7023–30. doi: 10.4049/jimmunol.0901154. [DOI] [PubMed] [Google Scholar]
- 45.Toldi G, Rigo J, Jr, Stenczer B, Vasarhelyi B, Molvarec A. Increased prevalence of IL-17-producing peripheral blood lymphocytes in pre-eclampsia. American journal of reproductive immunology. 2011;66:223–9. doi: 10.1111/j.1600-0897.2011.00987.x. [DOI] [PubMed] [Google Scholar]
- 46.Cornelius DC, Hogg JP, Scott J, Wallace K, Herse F, Moseley J, Wallukat G, Dechend R, Lamarca B. Administration of Interleukin-17 Soluble Receptor C Suppresses TH17 Cells, Oxidative Stress, and Hypertension in Response to Placental Ischemia During Pregnancy. Hypertension. 2013 doi: 10.1161/HYPERTENSIONAHA.113.01514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krebs CF, Lange S, Niemann G, Rosendahl A, Lehners A, Meyer-Schwesinger C, Stahl RA, Benndorf RA, Velden J, Paust HJ, Panzer U, Ehmke H, Wenzel UO. Deficiency of the interleukin 17/23 axis accelerates renal injury in mice with deoxycorticosterone acetate+angiotensin ii-induced hypertension. Hypertension. 2014;63:565–71. doi: 10.1161/HYPERTENSIONAHA.113.02620. [DOI] [PubMed] [Google Scholar]
- 48.Ishimitsu T, Uehara Y, Numabe A, Tsukada H, Ogawa Y, Iwai J, Ikeda T, Matsuoka H, Sugimoto T, Yagi S. Interferon gamma attenuates hypertensive renal injury in salt-sensitive Dahl rats. Hypertension. 1992;19:804–8. doi: 10.1161/01.hyp.19.6.804. [DOI] [PubMed] [Google Scholar]
- 49.Garcia AG, Wilson RM, Heo J, Murthy NR, Baid S, Ouchi N, Sam F. Interferon-gamma ablation exacerbates myocardial hypertrophy in diastolic heart failure. Am J Physiol Heart Circ Physiol. 2012;303:H587–96. doi: 10.1152/ajpheart.00298.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marko L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ, Bowman EP, Kleinewietfeld M, Fokuhl V, Dechend R, Muller DN. Interferon-gamma signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension. 2012;60:1430–6. doi: 10.1161/HYPERTENSIONAHA.112.199265. [DOI] [PubMed] [Google Scholar]
- 51.Jain S, Shah M, Li Y, Vinukonda G, Sehgal PB, Kumar A. Upregulation of human angiotensinogen (AGT) gene transcription by interferon-gamma: involvement of the STAT1-binding motif in the AGT promoter. Biochim Biophys Acta. 2006;1759:340–7. doi: 10.1016/j.bbaexp.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 52.Satou R, Miyata K, Gonzalez-Villalobos RA, Ingelfinger JR, Navar LG, Kobori H. Interferon-gamma biphasically regulates angiotensinogen expression via a JAK-STAT pathway and suppressor of cytokine signaling 1 (SOCS1) in renal proximal tubular cells. FASEB J. 2012;26:1821–30. doi: 10.1096/fj.11-195198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59:251–87. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 54.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, Delpire E, Harrison DG, McDonough AA. Renal Transporter Activation During Angiotensin-II Hypertension is Blunted in Interferon-gamma−/− and Interleukin-17A−/− Mice. Hypertension. 2015 doi: 10.1161/HYPERTENSIONAHA.114.04975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kleinbongard P, Heusch G, Schulz R. TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacology & therapeutics. 2010;127:295–314. doi: 10.1016/j.pharmthera.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 56.Landry DB, Couper LL, Bryant SR, Lindner V. Activation of the NF-kappa B and I kappa B system in smooth muscle cells after rat arterial injury. Induction of vascular cell adhesion molecule-1 and monocyte chemoattractant protein-1. Am J Pathol. 1997;151:1085–95. [PMC free article] [PubMed] [Google Scholar]
- 57.Neumann P, Gertzberg N, Johnson A. TNF-alpha induces a decrease in eNOS promoter activity. Am J Physiol Lung Cell Mol Physiol. 2004;286:L452–9. doi: 10.1152/ajplung.00378.2002. [DOI] [PubMed] [Google Scholar]
- 58.Alonso J, Sanchez de Miguel L, Monton M, Casado S, Lopez-Farre A. Endothelial cytosolic proteins bind to the 3′ untranslated region of endothelial nitric oxide synthase mRNA: regulation by tumor necrosis factor alpha. Mol Cell Biol. 1997;17:5719–26. doi: 10.1128/mcb.17.10.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun HX, Zeng DY, Li RT, Pang RP, Yang H, Hu YL, Zhang Q, Jiang Y, Huang LY, Tang YB, Yan GJ, Zhou JG. Essential role of microRNA-155 in regulating endothelium-dependent vasorelaxation by targeting endothelial nitric oxide synthase. Hypertension. 2012;60:1407–14. doi: 10.1161/HYPERTENSIONAHA.112.197301. [DOI] [PubMed] [Google Scholar]
- 60.Ramseyer VD, Garvin JL. Tumor necrosis factor-alpha: regulation of renal function and blood pressure. Am J Physiol Renal Physiol. 2013;304:F1231–42. doi: 10.1152/ajprenal.00557.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramseyer VD, Hong NJ, Garvin JL. Tumor necrosis factor alpha decreases nitric oxide synthase type 3 expression primarily via Rho/Rho kinase in the thick ascending limb. Hypertension. 2012;59:1145–50. doi: 10.1161/HYPERTENSIONAHA.111.189761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garvin JL, Herrera M, Ortiz PA. Regulation of renal NaCl transport by nitric oxide, endothelin, and ATP: clinical implications. Annu Rev Physiol. 2011;73:359–76. doi: 10.1146/annurev-physiol-012110-142247. [DOI] [PubMed] [Google Scholar]
- 63.Dickhout JG, Mori T, Cowley AW., Jr Tubulovascular nitric oxide crosstalk: buffering of angiotensin II-induced medullary vasoconstriction. Circ Res. 2002;91:487–93. doi: 10.1161/01.res.0000035243.66189.92. [DOI] [PubMed] [Google Scholar]
- 64.Irani RA, Zhang Y, Zhou CC, Blackwell SC, Hicks MJ, Ramin SM, Kellems RE, Xia Y. Autoantibody-mediated angiotensin receptor activation contributes to preeclampsia through tumor necrosis factor-alpha signaling. Hypertension. 2010;55:1246–53. doi: 10.1161/HYPERTENSIONAHA.110.150540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Venegas-Pont M, Manigrasso MB, Grifoni SC, LaMarca BB, Maric C, Racusen LC, Glover PH, Jones AV, Drummond HA, Ryan MJ. Tumor necrosis factor-alpha antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension. 2010;56:643–9. doi: 10.1161/HYPERTENSIONAHA.110.157685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Elmarakby AA, Quigley JE, Imig JD, Pollock JS, Pollock DM. TNF-alpha inhibition reduces renal injury in DOCA-salt hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2008;294:R76–83. doi: 10.1152/ajpregu.00466.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tran LT, MacLeod KM, McNeill JH. Chronic etanercept treatment prevents the development of hypertension in fructose-fed rats. Mol Cell Biochem. 2009;330:219–28. doi: 10.1007/s11010-009-0136-z. [DOI] [PubMed] [Google Scholar]
- 68.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51:1345–51. doi: 10.1161/HYPERTENSIONAHA.107.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maki-Petaja KM, Elkhawad M, Cheriyan J, Joshi FR, Ostor AJ, Hall FC, Rudd JH, Wilkinson IB. Anti-tumor necrosis factor-alpha therapy reduces aortic inflammation and stiffness in patients with rheumatoid arthritis. Circulation. 2012;126:2473–80. doi: 10.1161/CIRCULATIONAHA.112.120410. [DOI] [PubMed] [Google Scholar]
- 70.Mathieu S, Pereira B, Couderc M, Rabois E, Dubost JJ, Soubrier M. No significant changes in arterial stiffness in patients with ankylosing spondylitis after tumour necrosis factor alpha blockade treatment for 6 and 12 months. Rheumatology (Oxford) 2013;52:204–9. doi: 10.1093/rheumatology/kes272. [DOI] [PubMed] [Google Scholar]
- 71.Rincon M. Interleukin-6: from an inflammatory marker to a target for inflammatory diseases. Trends Immunol. 2012;33:571–7. doi: 10.1016/j.it.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 72.Roche NE, Fulbright JW, Wagner AD, Hunder GG, Goronzy JJ, Weyand CM. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis Rheum. 1993;36:1286–94. doi: 10.1002/art.1780360913. [DOI] [PubMed] [Google Scholar]
- 73.Ogata A, Tanaka T. Tocilizumab for the treatment of rheumatoid arthritis and other systemic autoimmune diseases: current perspectives and future directions. Int J Rheumatol. 2012;2012:946048. doi: 10.1155/2012/946048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vazquez-Oliva G, Fernandez-Real JM, Zamora A, Vilaseca M, Badimon L. Lowering of blood pressure leads to decreased circulating interleukin-6 in hypertensive subjects. J Hum Hypertens. 2005;19:457–62. doi: 10.1038/sj.jhh.1001845. [DOI] [PubMed] [Google Scholar]
- 75.Luther JM, Gainer JV, Murphey LJ, Yu C, Vaughan DE, Morrow JD, Brown NJ. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension. 2006;48:1050–7. doi: 10.1161/01.HYP.0000248135.97380.76. [DOI] [PubMed] [Google Scholar]
- 76.Lee DL, Sturgis LC, Labazi H, Osborne JB, Jr, Fleming C, Pollock JS, Manhiani M, Imig JD, Brands MW. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol. 2006;290:H935–40. doi: 10.1152/ajpheart.00708.2005. [DOI] [PubMed] [Google Scholar]
- 77.Li K, Guo D, Zhu H, Hering-Smith KS, Hamm LL, Ouyang J, Dong Y. Interleukin-6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am J Physiol Regul Integr Comp Physiol. 2010;299:R590–5. doi: 10.1152/ajpregu.00207.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170:2081–95. doi: 10.1084/jem.170.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sabat R, Grutz G, Warszawska K, Kirsch S, Witte E, Wolk K, Geginat J. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010;21:331–44. doi: 10.1016/j.cytogfr.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 80.Timasheva YR, Nasibullin TR, Zakirova AN, Mustafina OE. Association of interleukin-6, interleukin-12, and interleukin-10 gene polymorphisms with essential hypertension in Tatars from Russia. Biochemical genetics. 2008;46:64–74. doi: 10.1007/s10528-007-9130-x. [DOI] [PubMed] [Google Scholar]
- 81.Didion SP, Kinzenbaw DA, Schrader LI, Chu Y, Faraci FM. Endogenous interleukin-10 inhibits angiotensin II-induced vascular dysfunction. Hypertension. 2009;54:619–24. doi: 10.1161/HYPERTENSIONAHA.109.137158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hennessy A, Pilmore HL, Simmons LA, Painter DM. A deficiency of placental IL-10 in preeclampsia. J Immunol. 1999;163:3491–5. [PubMed] [Google Scholar]
- 83.Tinsley JH, South S, Chiasson VL, Mitchell BM. Interleukin-10 reduces inflammation, endothelial dysfunction, and blood pressure in hypertensive pregnant rats. Am J Physiol Regul Integr Comp Physiol. 2010;298:R713–9. doi: 10.1152/ajpregu.00712.2009. [DOI] [PubMed] [Google Scholar]
- 84.Lai Z, Kalkunte S, Sharma S. A critical role of interleukin-10 in modulating hypoxia-induced preeclampsia-like disease in mice. Hypertension. 2011;57:505–14. doi: 10.1161/HYPERTENSIONAHA.110.163329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP, Karumanchi SA. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350:672–83. doi: 10.1056/NEJMoa031884. [DOI] [PubMed] [Google Scholar]
- 86.Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension. 2011;57:469–76. doi: 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- 87.Matrougui K, Abd Elmageed Z, Kassan M, Choi S, Nair D, Gonzalez-Villalobos RA, Chentoufi AA, Kadowitz P, Belmadani S, Partyka M. Natural regulatory T cells control coronary arteriolar endothelial dysfunction in hypertensive mice. Am J Pathol. 2011;178:434–41. doi: 10.1016/j.ajpath.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, Rahn HP, Plehm R, Wellner M, Elitok S, Gratze P, Dechend R, Luft FC, Muller DN. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation. 2009;119:2904–12. doi: 10.1161/CIRCULATIONAHA.108.832782. [DOI] [PubMed] [Google Scholar]
- 89.Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, Neves MF, Laurant P, Paradis P, Schiffrin EL. T Regulatory Lymphocytes Prevent Aldosterone-Induced Vascular Injury. Hypertension. 2011 doi: 10.1161/HYPERTENSIONAHA.111.181123. [DOI] [PubMed] [Google Scholar]
- 90.Kassan M, Galan M, Partyka M, Trebak M, Matrougui K. Interleukin-10 released by CD4(+)CD25(+) natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler Thromb Vasc Biol. 2011;31:2534–42. doi: 10.1161/ATVBAHA.111.233262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Viel EC, Lemarie CA, Benkirane K, Paradis P, Schiffrin EL. Immune regulation and vascular inflammation in genetic hypertension. Am J Physiol Heart Circ Physiol. 2010;298:H938–44. doi: 10.1152/ajpheart.00707.2009. [DOI] [PubMed] [Google Scholar]
- 92.Kirabo A, Fontana V, de Faria APC, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, DWT, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen SC, Mernaugh RL, Laffer CL, Elijovich F, Davies SL, Moreno LH, Madhur MS, Roberts J, II, Harrison DG. Dendritic cell isoketal-modified proteins activate T cells and promote hypertension. Journal of Clinical Investigation. 2014 doi: 10.1172/JCI74084. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–23. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Somers MJ, Mavromatis K, Galis ZS, Harrison DG. Vascular superoxide production and vasomotor function in hypertension induced by deoxycorticosterone acetate-salt. Circulation. 2000;101:1722–8. doi: 10.1161/01.cir.101.14.1722. [DOI] [PubMed] [Google Scholar]
- 95.Miyashita H, Chikazawa M, Otaki N, Hioki Y, Shimozu Y, Nakashima F, Shibata T, Hagihara Y, Maruyama S, Matsumi N, Uchida K. Lysine pyrrolation is a naturally-occurring covalent modification involved in the production of DNA mimic proteins. Sci Rep. 2014;4:5343. doi: 10.1038/srep05343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guyton AC. Abnormal renal function and autoregulation in essential hypertension. Hypertension. 1991;18:III49–53. doi: 10.1161/01.hyp.18.5_suppl.iii49. [DOI] [PubMed] [Google Scholar]
- 97.Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, Sriramula S, Francis J, Sumners C, Raizada MK. Brain microglial cytokines in neurogenic hypertension. Hypertension. 2010;56:297–303. doi: 10.1161/HYPERTENSIONAHA.110.150409. [DOI] [PMC free article] [PubMed] [Google Scholar]