

Abstract
Genetic changes during tumorigenesis are usually acquired sequentially. However, a recent study showed that in 2 to 3% of all cancers a single catastrophic event, termed chromothripsis, can lead to massive genomic rearrangements confined to one or a few chromosomes. In order to explore whether the degree of genomic instability and chromothripsis influences prognosis in cancer, we retrospectively applied array comparative genomic hybridization (aCGH) to 20 malignant melanomas (MM) that showed, despite comparable conventional clinical and pathological parameters, a profoundly different clinical course.
We compared 10 patients who died of MM 3.7 years (median, range 0.9 to 7.6 years) after diagnosis with 10 patients who survived MM and had a median disease-free survival of 14.8 years (range 12.5 to 16.7 years; P = 0.00001). We observed a striking association between the degree of chromosomal instability, both numerical and structural, and outcome. MM associated with good prognosis showed only few chromosomal imbalances (mean 1.6 alterations per case), predominantly whole chromosome or chromosome arm gains and losses while MM with poor prognosis harbored significantly more chromosomal aberrations (13.9 per case; P = 0.008). Array-based CGH demonstrated that these aberrations were mostly focal events, culminating in two cases in a pattern consistent with the phenomenon of chromothripsis, which was confirmed by paired-end sequencing. This is the first description of chromothripsis in primary MM. Our study therefore links focal copy number alterations and chromothripsis with poor outcome in MM patients (P = 0.0002) and provides a genetic approach to predict outcome in MM.
Keywords: Chromothripsis, malignant melanoma, copy number alterations
Introduction
The clinical course of malignant melanoma (MM), a tumor with increasing incidence, is difficult to predict. The diagnosis of MM is based on histology, and disease prognosis depends mainly on mitotic rate, Breslow tumor thickness and ulceration. Recent studies showed that fluorescence in situ hybridization (FISH) with a set of specific probes or array comparative genomic hybridization (aCGH) can help to classify patients into low- and high-risk groups (1, 2). Despite this progress, available data are limited and often problematic to interpret because of short follow-up observation periods. Initial findings of our group, however, revealed considerably more chromosomal aberrations in MM with metastases than in MM without metastases (3).
In most cancers, chromosomal alterations that define invasive disease are acquired sequentially during disease progression (4). Recently, however, a phenomenon termed chromothripsis was reported (5). Chromothripsis describes a single catastrophic cellular event, in which one or a few chromosomes, chromosome arms or chromosomal subregions are shattered into tens to hundreds of pieces and reassembled incorrectly with the consequence of defined copy number changes. This pulverization of parts of the genome might result from defective and asynchronous DNA replication in micronuclei, which are a consequence of chromosome segregation errors in mitosis (5, 6). Chromothripsis occurs in approximately 2 to 3% of all cancers, yet more frequently in osteosarcoma and chordoma (up to 25%) while MM is affected in 7.8%, deduced from the analysis of MM-derived cell lines (5, 7). Chromothripsis was associated with a more aggressive clinical course in multiple myeloma, colorectal cancer, medulloblastoma, and neuroblastoma (8–11).
However, to date the significance of chromothripsis for prognosis in patients with MM is entirely unclear. We therefore investigated whether the pattern and degree of chromosomal instability might have an influence on the clinical course of MM.
In order to address this question, we designed a retrospective case control study using aCGH to map chromosomal gains and losses in two groups of MM patients with profoundly different survival after long-term follow-up. We confirmed the chromothripsis-like genomic aberration patterns that we suspected by aCGH analysis in MM with poor prognosis by paired-end sequencing, which revealed complex inter- and intrachromosomal rearrangements consistent with chromothripsis.
Materials and Methods
Tumor material and clinical data
Twenty formalin-fixed paraffin-embedded (FFPE) MM, which had been diagnosed between 1992 and 2006 with available detailed clinical long-term follow-up data, were collected from the archive of the Institute of Pathology, Paracelsus Medical University Salzburg, Austria. The study was performed in accordance with the guidelines of the local research ethics committee of the Paracelsus Medical University Salzburg and with preoperative patient’s informed consent. The diagnosis of all 20 tumors was established based on 4-µm thick hematoxylin and eosin (HE) stained sections by two board certified pathologists (RK and TG). Tumor staging was determined according to the latest American Joint Committee on Cancer staging system (7th edition) (12). The 20 selected MM samples comprised 10 MM with good prognosis and 10 MM with poor prognosis. Good prognosis was inferred when MM patients were still alive during the observation interval and had a minimum follow-up of >10.0 years (median 14.8 years ranging from 12.5 to 16.7 years) with clear documentation of neither local relapse nor the occurrence of metastases based on regular aftercare examination. In contrast, patients with MM were assigned to the poor prognosis group when MM related death occurred (median 3.7 years ranging from 0.9 to 7.6 years) as proven by either autopsy or by data from the Salzburg tumor registry. The two different prognostic groups were matched in terms of age at diagnosis, sex, Breslow thickness, Clark level, ulceration, mitotic rate, disease stage at diagnosis and localization. The groups differed significantly in terms of survival (Table 1).
Table 1.
Comparison of clinical and histopathological characteristics of MM patients with good and poor prognosis
| Variable | MM with good prognosis (n = 10) |
MM with poor prognosis (n = 10) |
P value |
|---|---|---|---|
| Age at diagnosis [a] | |||
| mean ± SEM | 55 ± 3 | 62 ± 5 | 0.22 |
| median (range) | 53 (40 – 70) | 63.5 (35 – 83) | 0.17 |
| Sex | |||
| male | 6 | 8 | 0.63 |
| female | 4 | 2 | |
| Breslow thickness [mm] | |||
| mean ± SEM | 2.2 ± 0.2 | 2.9 ± 0.5 | 0.20 |
| median (range) | 2.1 (1.4 – 3.4) | 3.0 (0.9 – 6.0) | 0.40 |
| Clark level | |||
| II | 1 | 0 | 0.37 |
| III | 5 | 3 | |
| IV | 4 | 7 | |
| Ulceration | |||
| present | 4 | 3 | 1 |
| absent | 6 | 7 | |
| Mitoses [No./mm2] | |||
| mean ± SEM | 1.3 ± 0.5 | 2.5 ± 0.6* | 0.15 |
| median (range) | 0.6 (0.4 – 4.6) | 2.0 (0.9 – 5.7)* | 0.06 |
| AJCC stage at diagnosis | |||
| IB | 5 | 4 | 0.53 |
| IIA | 2 | 4 | |
| IIB | 3 | 2 | |
| Locus | |||
| Head/neck | 1 | 0 | 0.38 |
| Trunk | 5 | 3 | |
| Extremity | 3 | 5 | |
| N/A | 1 | 2 | |
| Follow-up/Survival [a] | |||
| mean ± SEM | 14.5±0.5 | 4.0±0.7 | 0.00000002 |
| median (range) | 14.8 (12.5 – 16.7) | 3.7 (0.9 – 7.6) | 0.000001 |
n = 9 as in one sample massive pigmentation prevented mitotic count
MM, malignant melanoma.
DNA isolation
Seven consecutive sections (first and seventh section: 4-µm thick, HE stained; second to sixth section: 20-µm thick, unstained) were prepared from each of the FFPE blocks of the 20 MM cases. On sections one and seven, regions with >80% of tumor cells were marked by two board certified pathologists (RK and TG), and tissue was dissected from sections two to six for DNA preparation as described previously (13).
Array comparative genomic hybridization
Isolated FFPE DNA was labeled using the Genomic DNA ULS Labeling Kit (Agilent Technologies, Inc., Santa Clara, CA) and subsequently hybridized on Agilent SurePrint G3 Human CGH Microarrays 4×180K (Agilent) according to the manufacturer’s protocol version 3.1. Briefly, 500 ng of tumor DNA and 500 ng of sex-matched human genomic DNA (Promega, Madison, WI) as reference were differentially labeled with ULS-Cy3 and ULS-Cy5 (both Agilent), respectively. After hybridizing and washing according to the manufacturer’s instructions, slides were scanned with microarray scanner G2565BA (Agilent) and images were analyzed by Feature Extraction software version 10.7.1.1 (Agilent). The aCGH data were visualized and analyzed using CGH Analytics software 4.0.76 (Agilent) and Nexus Copy Number™ software version 5 (BioDiscovery, El Segundo, CA). The quality of the slides was assessed with control metrics provided by CGH Analytics (Agilent).
Paired-end sequencing
Illumina paired-end sequencing libraries were prepared following the manufacturer’s protocol with modifications. To increase sequence fragment diversity, two libraries were prepared in parallel and pooled prior to sequencing. For each library, 1 µg of DNA was sheared on a Covaris S1 to a mean fragment size of 350 bp. The sheared DNA was end repaired and phosphorylated with T4 DNA Polymerase, T4 Polynucleotide Kinase and Klenow, followed by adenylation using exo- Klenow Fragment. Illumina paired-end adapters were ligated to the prepared DNA fragments with DNA ligase. Adapter ligated products were size-selected on a Caliper LabChip XT, retaining fragments of 450bp +/− 20%. The resultant libraries were amplified with Illumina PCR Primer InPE1.0, PCR Primer InPE2.0 and PCR Primer Indexes. The two indexed, amplified libraries were pooled and sequenced on a single lane of an Illumina HiSeq2000. Reads were aligned to the reference human genome (hg19) using bwa-0.6.2 and converted to BAM format using samtools (14, 15). Duplicates were removed using the Picard MarkDuplicates software. The Genomic Analysis of Structural Variants software was then applied to the 114 million uniquely mapped read pair to define potential structural variant breakpoints (16). Potential cancer structural variants were filtered by removing candidates with fewer than 5 supporting read pairs or that had a breakpoint overlapping those found in a database of normal samples. Data were visualized using Circos software (17).
Statistical analysis
Differences in clinical and pathological parameters between the two different prognostic groups of MM were estimated by Student’s t-test for the mean, Wilcoxon rank sum test for the median, and Fisher’s exact or Freeman-Halton test for categorical variables. Differences in terms of survival and the indices of average number of copy alterations (ANCA, calculated as the number of aberrations divided by the number of samples, see (4)) between both groups of MM patients were estimated by Student’s t-test. The association of the incidence of genomic imbalances with outcome as well as the association of chromothripsis and focal copy number alterations with outcome was determined by Fisher’s exact test. P values lower than 0.05 were considered significant.
Results and Discussion
We were able to analyze 18 of the collected 20 MM samples by aCGH (8 MM with poor prognosis and 10 MM with good prognosis). Two samples were not included because of insufficient FFPE DNA quality.
Of the eight analyzed MM cases with poor prognosis, all eight (100%) were found to have copy number changes by aCGH, a proportion differing significantly from the MM cases with good prognosis (P = 0.004), where only three of 10 (30%) samples showed aberrations (Figure 1A, Supplemental Table S1). Independent of prognosis, genomic imbalances characteristic for MM, such as losses of chromosome arms 9p, especially of 9p21.3 (CDKN2A/p16 locus), and 10q, were detectable in both groups (18, 19). Yet, we were surprised of the low incidence of genomic imbalances in MM with good prognosis since these lesions were definitively malignant tumors and not benign melanocytic lesions as determined by histopathological examination. The lesions did not differ from MM with poor prognosis in terms of age at diagnosis, sex, histopathology, disease stage at diagnosis and location (Table 1). Notably, the few aberrations found in MM with good prognosis were all whole chromosome or chromosome arm gains or losses; this was in strong contrast to MM with poor prognosis which mostly displayed focal copy number gains or losses. This resulted in a far higher number of breakpoints and in an increased average number of copy alterations (ANCA) index (Figure 1B) (4). The ANCA index, which was calculated by dividing the sum of observed copy number imbalances by the respective number of cases, was 1.6 in MM associated with good prognosis and was significantly higher in MM with poor prognosis (13.9; P = 0.008). This indicates that increased genomic instability in MM was associated with poor prognosis, consistent with previous evidence from our own laboratory (3). Focal copy number alterations were present in all (8 of 8, 100%) MM with poor prognosis, but were observed only once in the MM with good prognosis (gain of 1q21.1-23.3 in case number 11; P = 0.0002). In two of eight (25%) MM with poor prognosis (case numbers 14 and 20), these focal copy number alterations culminated in aberration patterns consistent with the recently discovered phenomenon of chromothripsis (Figure 2) (5). These aberration patterns presented as complex genomic rearrangements whose positions next to one another markedly differed from chromosomal aberrations previously described in primary MM by us and others (3, 18, 19). These patterns were confined to segmental chromosomal regions, rapidly alternating between two or three distinct copy number states, including high level amplifications. To infer the occurrence of chromothripsis from copy number profiles, Rausch and colleagues required at least 10 changes in copy number involving up to three distinct copy number states on a single chromosome (11). When applied to our samples, these criteria were fulfilled by both MM with poor prognosis harboring regions of complex aberrations, while the vast majority of chromosomes displayed considerably fewer than 10 copy number changes per chromosome. Although one fundamental characteristic of chromothripsis is a series of clustered focal events along a chromosome, we wished to confirm our interpretation by identifying the breakpoints of the rearrangements. To this end, we performed whole genome paired-end sequencing on case number 14. This revealed genomic aberration patterns that were similar to the ones shown by Stephens and colleagues and included both inter- and intrachromosomal rearrangements (Figure 3) (5).
Figure 1. Chromosomal aberrations in MM with good and poor prognosis.
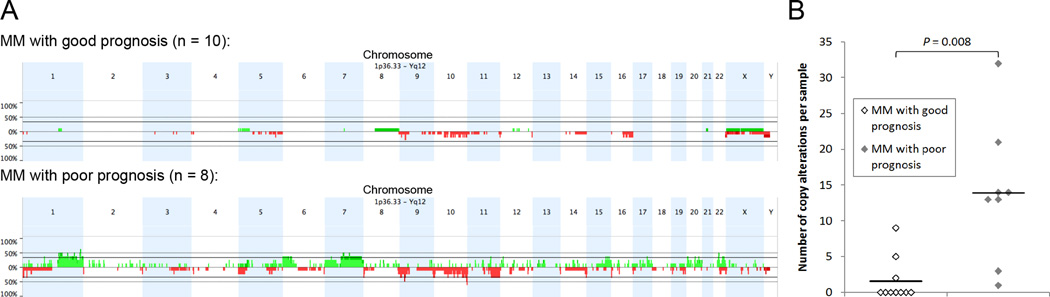
(A) Frequency plot of copy number gains (green) and losses (red) identified by array comparative genomic hybridization in MM with good and poor prognosis. (B) Number of copy alterations in each sample as a function of prognosis. Horizontal lines show mean number of copy alterations, which differs significantly between MM with good and poor prognosis (P = 0.008). MM, malignant melanoma.
Figure 2. Array-based CGH ideograms of chromosomal gains and losses for MM samples with poor prognosis.

(A,C) Genome views of case numbers 14 (A) and 20 (C) show the presence of genome-wide distributed chromosomal aberrations and localized regions of chromothripsis-like aberration patterns. (B,D) Chromosome views of the chromosomes affected by these chromothripsis-like aberration patterns. (B), case number 14, (D), case number 20. MM, malignant melanoma.
Figure 3. Whole genome circos plots of case number 14. The outer ring shows ideograms of the chromosomes, the inner ring represents the respective copy number changes. The arcs in the center indicate chromosomal rearrangements joining the two relevant genomic regions of each rearrangement.

(A) A whole genome circos plot, showing massive inter- and intrachromosomal rearrangements, predominantly involving regions of chromosomes 5 and 20. (B) A zoomed in version of the circos plot depicting the most affected regions of chromosomes 5p and 20q.
However, chromothripsis-positive MM did not only harbor the characteristic massive local genomic rearrangements but also several chromosomal aberrations, which revealed a recurrent pattern of chromosomal aberrations typical for MM (Figure 2A,C) (18, 19). This finding is in line with Stephens and colleagues who also described the coexistence of chromothripsis with other types of chromosomal alterations, likely acquired at distinct time points.
Although the association of both number and structure of chromosomal aberrations with prognosis is intriguing, the small sample size and the low incidence of chromosomal aberrations in MM with good prognosis might conceal evidence for focal copy number alterations and chromothripsis in MM with good prognosis. MM is not a rare tumor, but we would like to emphasize that it is exceedingly difficult to identify a sample collection for which the clinical long-term follow-up has been as meticulously documented as in our samples.
The fact that chromothripsis is not only connected with poor outcome in MM but also in multiple myeloma, colorectal cancer, medulloblastoma, and neuroblastoma suggests chromothripsis as a possible genetic hallmark of particularly aggressive subtypes of various cancers; this could translate to a useful prognostic marker.
In conclusion, we could demonstrate for the first time that MM which were matched according to clinical and histopathological features, but differed profoundly in terms of prognosis, showed striking disparities in both numerical and structural chromosomal aberrations. MM with poor prognosis were associated with a significantly higher incidence of genomic imbalances and harbored significantly more copy number changes than MM associated with good prognosis. In addition, while genomic imbalances in MM with good prognosis, when present, virtually always affected whole chromosomes or chromosome arms, focal copy number alterations and a pattern consistent with chromothripsis were exclusively found in MM with poor prognosis.
Supplementary Material
Acknowledgments
The authors thank Mr. Buddy Chen for preparing figures and IT-related support, Dr. Maria R. Gaiser for critical reading of the manuscript, and Prof. Christoph A. Klein, Chair of Experimental Medicine and Therapy Research, Faculty of Medicine, University of Regensburg, Germany, for providing expert advice. Daniela Hirsch is grateful for a stipend from the RISE program of the German Academic Exchange Service (DAAD).
Financial support
The study was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute. Daniela Hirsch was supported by the RISE program of the German Academic Exchange Service.
Footnotes
Conflict of interest statement
No conflict of interest has to be declared for any contributing author in this study.
References
- 1.Conway C, Beswick S, Elliott F, Chang YM, Randerson-Moor J, Harland M, et al. Deletion at chromosome arm 9p in relation to BRAF/NRAS mutations and prognostic significance for primary melanoma. Genes Chromosomes Cancer. 2010;49:425–438. doi: 10.1002/gcc.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jonsson G, Busch C, Knappskog S, Geisler J, Miletic H, Ringner M, et al. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin Cancer Res. 2010;16:3356–3367. doi: 10.1158/1078-0432.CCR-09-2509. [DOI] [PubMed] [Google Scholar]
- 3.Gaiser T, Kutzner H, Palmedo G, Siegelin MD, Wiesner T, Bruckner T, et al. Classifying ambiguous melanocytic lesions with FISH and correlation with clinical long-term follow up. Mod Pathol. 2010;23:413–419. doi: 10.1038/modpathol.2009.177. [DOI] [PubMed] [Google Scholar]
- 4.Ried T, Heselmeyer-Haddad K, Blegen H, Schrock E, Auer G. Genomic changes defining the genesis, progression, and malignancy potential in solid human tumors: a phenotype/genotype correlation. Genes Chromosomes Cancer. 1999;25:195–204. doi: 10.1002/(sici)1098-2264(199907)25:3<195::aid-gcc1>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 5.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–58. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kloosterman WP, Hoogstraat M, Paling O, Tavakoli-Yaraki M, Renkens I, Vermaat JS, et al. Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol. 2011;12:R103. doi: 10.1186/gb-2011-12-10-r103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Magrangeas F, Avet-Loiseau H, Munshi NC, Minvielle S. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood. 2011;118:675–678. doi: 10.1182/blood-2011-03-344069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012 doi: 10.1038/nature10910. [DOI] [PubMed] [Google Scholar]
- 11.Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edge SB. AJCC Cancer Staging Manual. 7th Edition ed. New York: Springer; 2010. [Google Scholar]
- 13.Gaiser T, Camps J, Meinhardt S, Wangsa D, Nguyen QT, Varma S, et al. Genome and transcriptome profiles of CD133-positive colorectal cancer cells. Am J Pathol. 2011;178:1478–1488. doi: 10.1016/j.ajpath.2010.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sindi S, Helman E, Bashir A, Raphael BJ. A geometric approach for classification and comparison of structural variants. Bioinformatics. 2009;25:i222–i230. doi: 10.1093/bioinformatics/btp208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome research. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bastian BC, LeBoit PE, Hamm H, Brocker EB, Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 1998;58:2170–2175. [PubMed] [Google Scholar]
- 19.Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.