

Abstract
Oncogenic conversion of the RET (rearranged during transfection) tyrosine kinase is associated with several cancers. A fragment-based chemical screen lead to the identification of a novel RET inhibitor, Pz-1. Modeling and kinetic analysis identified Pz-1 as a Type-II tyrosine kinase inhibitor, able to bind the DFG-out conformation of the kinase. Importantly, from a single-agent polypharmacology standpoint, Pz-1 was shown active on VEGFR2, which can block blood supply required for RET-stimulated growth. In cell based assays, 1.0 nM of Pz-1 strongly inhibited phosphorylation of all tested RET oncoproteins. At 1.0 mg/kg/day per os, Pz-1 abrogated formation of tumors induced by RET-mutant fibroblasts and blocked phosphorylation of both RET and VEGFR2 in tumor tissue. Pz-1 featured no detectable toxicity up to 100.0 mg/kg, which indicated a large therapeutic window. This study validates the effectiveness and usefulness of a medicinal chemistry polypharmacology approach to obtain an inhibitor capable of targeting multiple oncogenic pathways
Keywords: RET, Polypharmacology, VEGFR2, Synergistic Medicinal Chemistry, Kinase inhibitor
Goals of targeted treatments are to provide cancer patients with agents, such as tyrosine kinase inhibitors (TKI), designed to target tumor cells or the tumor micro-environment. Unfortunately, drug resistance with TKIs invariably develops due to an inability to sustainably knock out tumor-survival pathways or to maintain activity on the target as additional mutations form.[1–3] Resistance can be reduced by identifying, through medicinal chemistry polypharmacology[4] (MCP), personalized TKIs with optimized inhibitory profiles for critical disease-promoting kinases, including crucial mutant targets. A single agent with an appropriate inhibitory profile can possess the benefits of combination therapy and effectively target tumor growth while preventing resistance formation.[5] Herein, we have applied MCP using a novel fragment-based approach to identify a pan-RET/VEGFR2 dual inhibitor. The inhibitor is capable of effectively treating both parenchyma (RET) and stroma (VEGFR2) of RET-driven tumors, while maintaining activity on all tested RET oncogene mutants
RET is a transmembrane tyrosine kinase (TK) receptor that has emerged as a molecular target for the treatment of several cancer types, primarily medullary thyroid carcinoma (MTC).[6–8] In a tumor environment, VEGFR2-mediated angiogenesis provides oxygen and nutrients necessary for RET oncogenic signaling. Vandetanib (Caprelsa®) and cabozantinib (Cometriq®) were originally discovered as VEGFR2 inhibitors and have been approved to treat MTC, based on their capability to prolong progression-free survival compared to placebo.[9–10] Both agents possess activity on wild type and oncogenic RET mutants, but not on RET gatekeeper (V804) mutants. Therefore, our goal was to implement the concept of single-agent polypharmacology (SAP) in order to design a RET/VEGFR2 dual inhibitor selective and equipotent on both kinases, including clinically relevant mutants, to provide a new targeted agent for RET-driven cancers.
Compound design was largely guided by computational modeling and the use of a modified fragment-based approach. For the modeling studies, the RET tyrosine kinase domain has previously been crystallized in complex with small, Type-I inhibitors, which bind to the DFG-in conformation of the kinase.[11] In order to search for more selective RET TKIs, a DFG-out homology model capable of interacting with Type-II inhibitors was generated by utilizing an available RET sequence (PDB# 2IVU) and a VEGFR2 DFG-out template (PDB# 2OH4) (Supplemental Figure 1). The model displayed the appropriate shift in the DFG loop, which opened the allosteric pocket and was used for molecular docking and structure-activity relationship (SAR) studies.
Kinase fragment libraries are traditionally composed of small hinge binders of one or two fused aryl and/or heteroaryl ring systems with hydrogen-bond donors or acceptors. Such fragments are highly polar and can aggregate at millimolar concentrations that are often necessary to achieve activity in a biochemical fragment screen. As a result, achieving a good noise to signal ratio is a major challenge with this methodology. A kinase-directed fragment (KDF) library was designed to enhance sensitivity and more effectively interrogate binding functionalities at a kinase hinge (Figure 1).[12] This KDF library contains a diverse set of heterocyclic, hinge-region binders along with moieties that can engage lipophilic pockets or the ribose sugar pocket. Accordingly, KDFs have larger molecular weights and are generally more active than fragments contained in traditional libraries, permitting screening in the micromolar range. From such a library, a benzimidazole analogue was identified as a promising starting platform for a RET TKI (compound 1, Figure 2A, Table 1). Compound 1 effectively inhibited RET at both 100.0 and 20.0 μM (Table 1). Molecular modeling of compound 1 in the RET kinase showed that heterocyclic substitution at the bromine would be tolerated (compound 1a, Figure 2A, D). This region is solvent exposed and is occupied by the adenine ring system when ATP is bound. Compound 1a displayed improved RET potency with an IC50 of 18.2 ± 0.81 μM (Table 1). Further modeling displayed that the DFG pocket of the RET kinase could be accessed with the addition of a bridge connected to an allosteric group such as meta-trifluromethylphenyl (compound 2, Figure 2A, C); compound 2 showed improved potency against the RET kinase with an IC50 of 2.1 ± 0.24 μM (Table 1). The combination of SAR information from compounds 1, 1a, and 2 resulted in compound 3 with RET, VEGFR2, and RETV804M Kds of 0.006, 0.018, and 0.006 μM, respectively (Table 1). Compound 3 was further optimized on RET and VEGFR2 at both the allosteric region and position 5 of benzimidazole, which lead to compound 4 (Pz-1) (Figure 2A, E, Supplemental Figure 2). Specifically, pyrazole at the solvent region and isoxazole in the allosteric pocket were highly favored for strong RET inhibition. Pz-1 was found active on RET, RET mutants, and VEGFR2 at similar IC50 values, all < 1.0 nM (Table 1). The equal potency was later confirmed in cell-based assays. Pz-1 is expected to simultaneously block VEGFR2-mediated nutrient accumulation and RET/RET-mutant cell proliferation.
Figure 1.
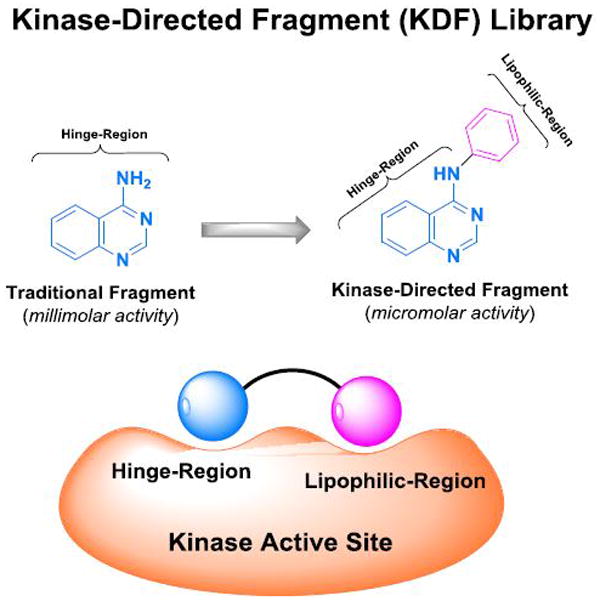
Figure 2.

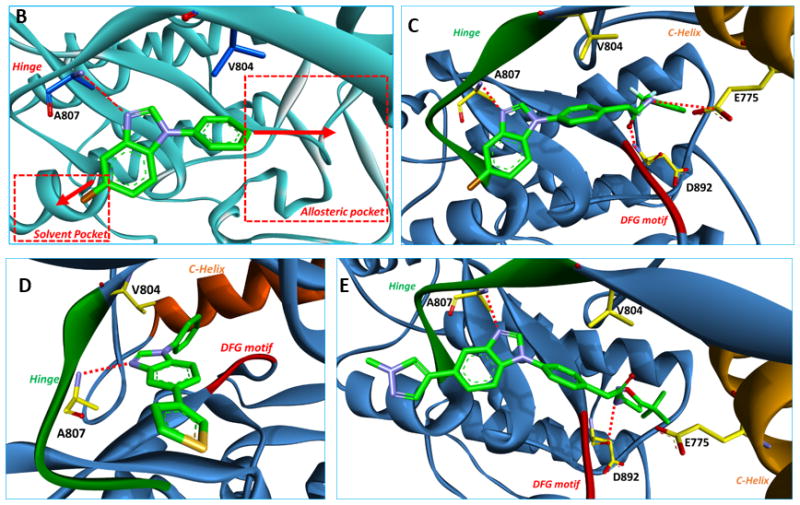
Table 1.
Kinase activities on RET, VEGFR2, and RETV804M.
| Compd. | RET | VEGFR2 | RETV804M |
|---|---|---|---|
| 1 | 28% ± 3% [b], 63% ± 5%[c] | 59% ± 3%[c] | / |
| 1a | 18.2 ± 0.8 μM[a] | 89% ± 4%[c] | / |
| 2 | 2.1 ± 0.2 μM[a] | 93% ± 2%[c] | / |
| 3 | 0.006 μM[d] | 0.018 μM[d] | 0.006 μM[d] |
| 3b | 0.047 ± 0.007 μM[a] | 97% ± 2%[c] | / |
| Pz-1 (4) | < 0.001 μM[a] | < 0.001 μM[a] | < 0.001 μM[a] |
IC50 values were determined using a microfluidics separation based assay. The data were means from at least three independent experiments. [ATP] = 190 μM.
Residual activity at 100.0 μM in triplicate. [ATP] = 190 μM.
Residual activity at 20.0 μM in triplicate. [ATP] = 190 μM.
Kd values were determined using an active site-directed competition binding assay that was outsourced to KINOMEScan®.
Molecular modeling demonstrated that Pz-1 could bind both RET and VEGFR2 with different binding geometries from free rotation of the methylene linker that bridges the hinge region to the allosteric pocket (Supplemental Figure 3). In complex with RET, the linker amide is positioned ‘up’, while with VEGFR2 the linker amide is positioned ‘down’. Such free rotation of Pz-1 presumably helps the inhibitor achieve equipotency on RET, RET mutants, and VEGFR2. Accordingly, Pz-1 exhibited an IC50 of < 1.0 nM for both wild type RET and VEGFR2 kinases (Table 1). Computational modeling of Pz-1 within the RET kinase showed that binding occurred > 3.5 Å away from the gatekeeper residue (V804) (Figure 2E). As a result of the large binding distance from the gatekeeper region, Pz-1 exhibited strong affinity for the gatekeeper mutant RETV804M (Table 1). RETV804M is refractory to both vandetanib and cabozantinib,[13] suggesting Pz-1 has a novel RET binding mode. Vandetanib is ~2.8 Å away from the gatekeeper residue (V804), almost 1.0 Å closer than Pz-1; vandetanib affinity for RETV804M and RETV804L is likely abrogated because of steric hindrance with the larger mutant residues at the gatekeeper region.
To identify the type of inhibition exhibited, the ATP Km for RET was determined and found to be 12.00 ± 0.26 μM (Supplemental Figure 4). Since the utilized RET kinase assay is not suitable to precisely determine activity of highly potent inhibitors such as Pz-1 (IC50 < 1.0 nM), compound 3b was selected for inhibition studies because its activity was within the limits of assay reliability. The IC50 of 3b was determined at ATP concentrations of 6.2, 12.5, 50.0, and 100.0 μM. Although there was a slight increase in IC50 in parallel to an increase in ATP concentration, compound 3b kept strong inhibition at highly saturating ATP concentrations (Table 2, Supplemental Figure 5). This suggested that the benzimidazole scaffold of Pz-1 is somewhat competitve with ATP for binding since inhibtion was relieved but not completely aborgated.
Table 2.
Determination of inhibition for compound 3b.[a]
| ATP
|
6.2 μM | 12.5 μM | 50.0 μM | 100.0 μM |
| IC50 | 20.6 ± 4.3 nM | 27.4 ± 1.1 nM | 33.7 ± 3.18 nM | 46.5 ± 6.9 nM |
IC50 values were determined using a microfluidics separations based assay. The data were means from at least three independent experiments.
In cell-based assays, consistent with the biochemical SAR, Pz-1 featured increased RET inhibitory activity towards oncogenic RETC634R compared with predecessor compounds (1, 2 and 3a) (Figure 3A). Pz-1 was also highly effective against other common oncogenic RET point mutants, including RETM918T, the most common mutant associated with inherited multiple endocrine neoplasia type 2B (MEN2B) syndrome and sporadic MTC, as well as RETV804M and RETV804L mutants (Figure 3B). Moreover, Pz-1 also confirmed to be equally potent on VEGFR2, inhibiting VEGFA-induced VEGFR2 phosphorylation at a 1.0 nM dose in transfected HEK293 cells and, at the endogenous level, in HUVEC cells (Figure 3C, Supplemental Figure 6).
Figure 3.

Pz-1 was subjected to kinome screening to determine global kinase selectivity. At a concentration of 50.0 nM, Pz-1 was screened against a 91 kinase panel representing each kinome cluster (Supplemental Figure 7). Pz-1 featured good global kinase selectivity displaying activity on only 7 additonal kinases (TRKB, TRKC, GKA, FYN, SRC, TAK1, MUSK), which was consistent with low toxicity observed in the cell-based assays (Supplemental Figure 9).
In order to address in vivo RET-driven effects, Pz-1 was tested on tumors induced by oncogenic RET compared to a constitutively-active control Ras oncogene. To this aim, NIH3T3 fibroblasts transformed by either RETC634Y or HRasG12V were used. RETC634Y in vitro cell proliferation was inhibited by Pz-1 with an IC50 of 0.5 nM, a dose significantly lower than that required to inhibit NIH3T3 HRasG12V cell proliferation (IC50 = 34.4 nM) (Supplemental Figure 8B). This was paralleled by efficient inhibition of RETC634Y tyrosine phosphorylation (Supplemental Figure 8A). At up to 100.0 nM, Pz-1 did not exert detectable growth inhibition in parental NIH3T3 cells (Supplemental Figure 9).
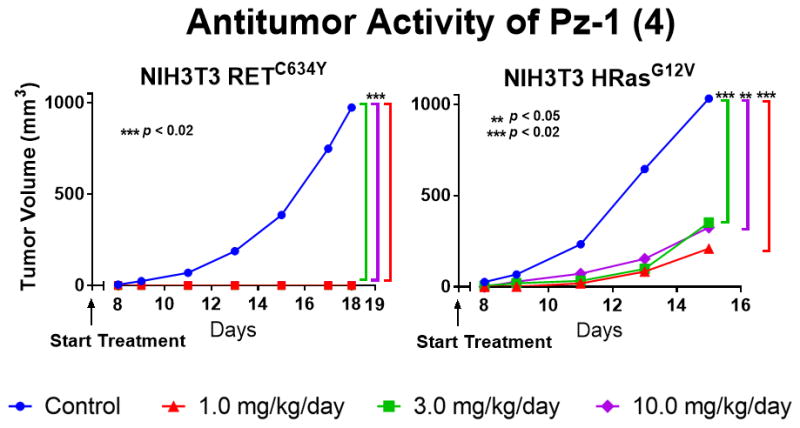
Immunodeficient (nu/nu) mice were then injected with NIH3T3 RETC634Y or NIH3T3 HRasG12V cells and, before tumors had appeared, treated PO with Pz-1 (1.0, 3.0 or 10.0 mg/kg/day) or left untreated (Figure 4, Supplemental Table 1). Pz-1 preferentially inhibited RET with respect to Ras driven tumors (Figure 4). Indeed, while treatment completely prevented formation of tumors induced by RETC634Y, treatment reduced, but did not abrogate, formation of tumors driven by HRasG12V (Figure 4, Supplemental Table 1). Only in RET-driven tumors was Pz-1 able to inhibit MAPK and mTOR mitogenic singling cascades (Supplemental Figure 10). Even at a dose of 10.0 mg/kg, Pz-1 had no effect on MAPK signaling in Ras tumors. However, in both RET and Ras driven tumors, Pz-1 (1.0 mg/kg) inhibited VEGFR2 phosphorylation. The preferential efficacy of Pz-1 for RET-driven tumors may be explained by its dual effect on both parenchyma (RET) and stroma (VEGFR2) supporting the polypharmacological strategy to target these tumor types. Upon one week in mice, Pz-1 was highly tolerated at daily doses up to 100.0 mg/kg (Supplemental Figure 11). Alanine transaminase (ALT) increased in a linear fashion parallel to Pz-1 dose, though remaining within a normal range, which behaved as a monitorable toxicity marker (Supplemental Figure 11). Further, Pz-1 displayed highly favorable pharmacokinetic properties (Supplimental Table 2).
Figure 4.
In summary, we identified Pz-1 using KDF library screening in conjunction with computational modeling. In cell-based assays, 1.0 nM of Pz-1 strongly inhibited tyrosine phosphorylation of VEGFR2 and clinically relevant RET mutants, including those refractory to vandetanib and cabozantinib (RETV804M and RETV804L). Pz-1 completely blocked RET-driven tumor formation at 1.0 mg/kg with no detectable toxicity up to a dose of 100.0 mg/kg. The high activity and low toxicity of Pz-1 can be explained by the selective, dual inhibition of both RET and VEGFR2. In conclusion, this study validates medicinal chemistry and single agent polypharmacology methods to guide development of compounds with well-defined and balanced activities against multiple cancer relevant targets for synergistic outcomes; the clinical development of Pz-1 may offer a promising therapeutic approach for patients afflicted with RET-driven malignancies and represents a paradigm shift for targeted therapies.
Supplementary Material
Acknowledgments
RAT1 cells expressing RET mutants were kindly donated by M. Billaud, and VEGFR2/KDR expressing vector by S. De Falco. This study was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC), by the MERIT grant and grants from MIUR, Italy. Additionally, this work was supported by a training grant from The National Institutes of Health (T32 GM008804), University of Arizona startup funding, and The Caldwell Health Sciences Research Fellowship. This was also supported by the ACS IRG grant.
Footnotes
Part 1 of the Synergistic Medicinal Chemistry Seires.
References
- 1.Capdeville R, Buchdunger E, Zimmermann J, Matter A. Nat Rev Drug Disc. 2002;1:493–502. doi: 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- 2.a) Settleman J. Drug Disco Today: Disease Mechanisms. 2005;2:139–144. [Google Scholar]; b) Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, Perl AE, Travers KJ, Wang S, Hunt JP, Zarrinkar PP, Schadt EE, Kasarskis A, Kuriyan J, Shah NP. Nature. 2012;485:260–263. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lito P, Rosen N, Solit DB. Nat Med. 2013;19:1401–9. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 4.a) Dar AC, Das TK, Shokat KM, Cagan RL. Nature. 2012;486:80–4. doi: 10.1038/nature11127. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gujral TS, Peshkin L, Kirschner LM. Proc Natl Acad Sci USA. 2014;111:5048–53. doi: 10.1073/pnas.1403080111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Shah NP, Tran C, Lee FY, Norris D, Chen P, Sawyers CL. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]; b) Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Gary N. Nature. 2009;462:1070–4. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, Sawyer CL. eLife. 2013;2:e00499. doi: 10.7554/eLife.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Mulligan LM. Nat Rev Cancer. 2014;14:173–186. doi: 10.1038/nrc3680. [DOI] [PubMed] [Google Scholar]; b) Carlomagno F, Santoro M. Nat Rev Endo. 2011;7:65–67. doi: 10.1038/nrendo.2010.232. [DOI] [PubMed] [Google Scholar]; c) Takahashi M, Ritz J, Cooper GM. Cell. 1985;42:581–588. doi: 10.1016/0092-8674(85)90115-1. [DOI] [PubMed] [Google Scholar]; d) Santoro M, Carlomagno F, Romano A, Bottaro DP, Dathan NA, Grieco M, Fusco A, Vecchio G, Matoskova B, Kraus MH, Di Fiore PP. Science. 1995;267:381–383. doi: 10.1126/science.7824936. [DOI] [PubMed] [Google Scholar]
- 7.a) Hutchinson KE, Pao W. Nat Med. 2012;18:349–351. doi: 10.1038/nm.2697. [DOI] [PubMed] [Google Scholar]; b) Ballerini P, Struski S, Cresson C, Prade N, Toujani S, Deswarte C, Dobbelstein S, Petit A, Lapillonne H, Gautier EF, Demur C, Lippert E, Pages P, Mansate Mas V, Donadieu J, Huguet F, Dastugue N, Broccardo C, Perot C, Delabesse E. Leukemia. 2012;26:2384–2389. doi: 10.1038/leu.2012.109. [DOI] [PubMed] [Google Scholar]; d) Wiesner T, He J, Yelensky R, Esteve-Puig R, Botton T, Yeh I, Lipson D, Otto G, Brennan K, Murali R, Garrido M, Miller VA, Ross JS, Berger MF, Sparatta A, Palmedo G, Cerroni L, Busam KJ, Kutzner H, Cronin MT, Stephens PJ, Bastian BC. Nat Commun. 2014;5:3116–3122. doi: 10.1038/ncomms4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Rizzi E, Cassinelli G, Dallavalle S, Lanzi C, Cincinelli R, Nannei R, Cuccuru G, Zunino F. Bioorg Med Chem Lett. 2007;17:3962–3968. doi: 10.1016/j.bmcl.2007.04.091. [DOI] [PubMed] [Google Scholar]; b) Robinett RG, Freemerman AJ, Skinn MA, Shewchuk L, Lackey K. Bioorg Med Chem Lett. 2007;17:5886–5893. doi: 10.1016/j.bmcl.2007.07.104. [DOI] [PubMed] [Google Scholar]; c) Cincinelli R, Cassinelli G, Dallavalle S, Lanzi C, Merlini L, Botta M, Tuccinardi T, Martinelli A, Penco S, Zunino F. J Med Chem. 2008;51:7777–7787. doi: 10.1021/jm8007823. [DOI] [PubMed] [Google Scholar]
- 9.a) [Accessed 1/26/2015];Press Announcement #UCM330143. http://www.fda.gov.; b) [Accessed 1/26/2015];Press Announcement #UCM250168. http://www.fda.gov. (archived); c) Wells SA, Jr, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, Baudin E, Elisei R, Jarzab R, Vasselli JR, Read J, Langmuir P, Ryan AJ, Schlumberger MJ. J Clin Oncol. 2012;30:134–141. doi: 10.1200/JCO.2011.35.5040. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Carlomagno F, Vitagliano D, Guida T, Ciardiello F, Tortora G, Vecchio G, Ryan AJ, Fontanini G, Fusco A, Santoro M. Cancer Res. 2002;62:7284–7290. [PubMed] [Google Scholar]
- 10.a) Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, Orf J, You A, Laird AD, Engst S, Lee L, Lesch J, Chou YC, Joly AH. Mol Cancer Ther. 2011;10:2298–2308. doi: 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]; b) Elisei R, Schlumberger MJ, Müller SP, Schöffski P, Brose MS, Shah MH, Licitra L, Jarzab B, Medvedev V, Kreissl MC, Niederle B, Cohen EE, Wirth LJ, Ali H, Hessel C, Yaron Y, Ball D, Nelkin B, Sherman SI. J Clin Oncol. 2013;31:3639–46. doi: 10.1200/JCO.2012.48.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Kufareva I, Abagyan R. J Med Chem. 2008;51:7921–7932. doi: 10.1021/jm8010299. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Knowles PP, Murray-Rust J, Kjer S, Scott RP, Hanrahan S, Santoro M, Ibanez CF, McDonald NQ. J Biol Chem. 2006;281:33577–33587. doi: 10.1074/jbc.M605604200. [DOI] [PubMed] [Google Scholar]; c) Trott O, Olson AJ. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hanrahan S, Totty N, McDonald NQ. Nat Struct Mol Biol. 2010;17:726–731. doi: 10.1038/nsmb.1808. [DOI] [PubMed] [Google Scholar]; e) Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, Graham AG, Grob PM, Hickey ER, Moss N, Pav S, Regan J. Nat Struct Biol. 2002;9:268–272. doi: 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- 12.De Kloe GE, Bailey D, Leurs RR, de Esch IJP. Drug Discovery Today. 2009;14:630–646. doi: 10.1016/j.drudis.2009.03.009. [DOI] [PubMed] [Google Scholar]; b) Joseph-McCarthy D. J Comput Aided Mol Des. 2009;23:449–451. doi: 10.1007/s10822-009-9293-0. [DOI] [PubMed] [Google Scholar]; c) Rees DC, Congreve M, Murray CW, Carr RR. Nat Rev Drug Disc. 2004;3:660–672. doi: 10.1038/nrd1467. [DOI] [PubMed] [Google Scholar]; d) Hajduk PJ, Greer J. Nat Rev Drug Disc. 2007;6:221–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 13.a) Carlomagno F, Guida T, Anaganti S, Provitera L, Kjaer S, McDonald NQ, Ryan AJ, Santoro M. Endocr Relat Cancer. 2009;16:233–241. doi: 10.1677/ERC-08-0213. [DOI] [PubMed] [Google Scholar]; b) Carlomagno F, Guida T, Anaganti S, Vecchio G, Fusco A, Ryan AJ, Billaud M, Santoro M. Oncogene. 2004;23:6056–6063. doi: 10.1038/sj.onc.1207810. [DOI] [PubMed] [Google Scholar]; Mologni L, Redaelli S, Morandi A, Plaza-Menacho I, Gambacorti-Passerini C. Mol Cell Endocrinol. 2013;377:1–6. doi: 10.1016/j.mce.2013.06.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.